
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vanadia–titania multilayer nanodecoration of carbon onions via atomic layer deposition for high performance electrochemical energy storage†
Simon
Fleischmann
a,
Aura
Tolosa
ab,
Marco
Zeiger
ab,
Benjamin
Krüner
ab,
Nicolas J.
Peter
c,
Ingrid
Grobelsek
b,
Antje
Quade
d,
Angela
Kruth
d and
Volker
Presser
 *ab
*ab
aDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany
cMax-Planck-Institut für Eisenforschung GmbH, 40237 Düsseldorf, Germany
dLeibniz Institute for Plasma Science and Technology, 17489 Greifswald, Germany
First published on 21st December 2016
Abstract
Atomic layer deposition has proven to be a particularly attractive approach for decorating mesoporous carbon substrates with redox active metal oxides for electrochemical energy storage. This study, for the first time, capitalizes on the cyclic character of atomic layer deposition to obtain highly conformal and atomically controlled decoration of carbon onions with alternating stacks of vanadia and titania. The addition of 25 mass% TiO2 leads to expansion of the VO2 unit cell, thus greatly enhancing lithium intercalation capacity and kinetics. Electrochemical characterization revealed an ultrahigh discharge capacity of up to 382 mA h g−1 of the composite electrode (554 mA h g−1 per metal oxide) with an impressive capacity retention of 82 mA h g−1 (120 mA h g−1 per metal oxide) at a high discharge rate of 20 A g−1 or 52C. Stability benchmarking showed stability over 3000 cycles when discharging to a reduced potential of −1.8 V vs. carbon. These capacity values are among the highest reported for any metal oxide system, while in addition, supercapacitor-like power performance and longevity are achieved. At a device level, high specific energy and power of up to 110 W h kg−1 and 6 kW kg−1, respectively, were achieved when employing the hybrid material as anode versus activated carbon cathode.
1. Introduction
Electrochemical energy storage devices are typically divided into two categories: (1) supercapacitors, including electrical double-layer capacitors (EDLCs), which store energy by fast and reversible electrosorption of ions at the charged interface of high surface area electrodes and the electrolyte1,2 and (2) batteries that utilize redox reactions in the bulk of the electrodes for energy storage. The non-faradaic charge storage mechanism of EDLCs is characterized by a rapid response and high achievable power ratings, yet they suffer from a relatively low energy density, compared to state-of-the-art lithium ion batteries.3 Typical electrode materials for EDLCs are carbons with a high specific surface area (SSA), such as activated carbon,4,5 carbide-derived carbon,6,7 carbon onions (onion-like carbon, OLC),8,9 carbon nanotubes (CNTs),10,11 or carbon nanofibers.12,13 The double-layer capacitance of these materials is limited to about 0.1 F m−2,14,15 which is why the implementation of faradaic charge transfer into the supercapacitor system is explored as a way to further increase their energy capacity.16 For the latter, a common strategy is the creation of hybrid electrodes by nanoscopically decorating carbon with redox-active materials, such as metal oxides,17 or surface functional groups.18,19 Various processing methods have been employed to coat carbon surfaces with metal oxides, such as chemical vapor deposition,20 atomic layer deposition (ALD),21,22 or wet-chemical approaches.23 Especially ALD has been found to be a very promising technique to fabricate nanoscopic films on various carbon surfaces because of the highly controllable film thickness at a sub-nanometer level, the conformity of the layers, and the possibility of coating small nanopores that exhibit sizes above 1–2 nm.22,24 By utilizing nanoscopic layers of redox-active materials, faradaic reactions are localized near the electrode surface, avoiding long diffusion paths of ions to reach reactive sites, which results in a high power handling.25,26In a hybrid electrode, the carbon substrate is required to provide a sufficiently high SSA, good electrical conductivity, and accessible pore volume.22 In a previous study, we have investigated the influence of the carbon porosity on the resulting electrochemical performance of carbon/vanadia hybrid electrodes, concluding that carbon materials which exhibit exclusively external surface area, such as OLCs, yield beneficial properties compared to nanoporous carbons.22 OLCs are spherical carbon nanoparticles that consist of several concentric shells of sp2 carbon and that are typically smaller than 10 nm, depending on the synthesis method and precursor. They possess a SSA of up to 600 m2 g−1,8 a high electrical conductivity of up to 4 S cm−1,27 and form mainly interparticle and intercluster pores.28 Our study clearly demonstrated that the interparticle pore space of OLCs provides favorable conditions for a conformal and nanocrystalline vanadia film growth via ALD.22
Depending on the observed charge–voltage-profile, redox systems are categorized either as pseudocapacitors (“capacitor-like”, exhibition of constant charge-vs.-voltage profiles, e.g., MnO2,29–31 RuO2,32 MXene33), or as battery-like devices (showing well-defined redox peaks; e.g., V2O5,34,35 polyaniline36).37 In particular, vanadia has gained considerable attention as both a cathode material in lithium batteries38 and a redox-active component in hybrid electrodes.21 It features the ability to intercalate Li+ ions, which manifests in a battery-like response for crystalline V2O5 and VO2,39 and exhibits pseudocapacitive behavior in amorphous and nanocrystalline states.25 When discharging to 3.2 V vs. Li, crystalline V2O5 can reversibly facilitate 0.5 Li for each V by forming δ-LiV2O5, which yields a theoretical capacity of 147 mA h g−1.39 Higher degrees of lithiation are achieved by discharging up to 2.3 V vs. Li, yielding an irreversible phase transformation to γ-LixV2O5 with theoretical capacities as high as 300 mA h g−1.39 Monoclinic VO2(M) (rutile type) exhibits a performance of around 120 mA h g−1,40 while higher capacities have been reported for metastable VO2(B) phases due to the presence of V4O10-type double layers, which share corners to form tunnel-like structures, enabling rapid Li transport.39,41 Cycling of VO2(B) between 4 V and 1 V vs. Li leads to the formation of LiVO2, corresponding to a theoretical capacity of 320 mA h g−1.42 For nanostructured VO2(B) aerogels, initial discharge capacities as high as 500 mA h g−1 have been reported, yet a stable performance of 175 mA h g−1 over 20 cycles was only achieved when operating within a smaller voltage window between 4 V and 2.4 V vs. Li.43
Instead of using a single metal oxide, the addition of a second phase may further enhance the electrochemical properties of redox systems. In a study by Takahashi et al., the addition of anatase TiO2 to the V2O5 structure led to a significant rise in capacity from 120 mA h g−1 to 180 mA h g−1, compared to pure V2O5 nanorods.44 The authors ascribed the effect to possible modifications in the V2O5 lattice structure, yielding a more open space for Li intercalation, yet analytical evidence was not provided by the authors.44 Jampani et al. reported a similar capacity increase resulting from Ti-doping of the vanadia structure, which was correlated with an increase in electrical conductivity, caused by incorporation of titanium at vanadium lattice sites.20 Park et al. investigated the effect of lattice strain on the electrochemical performance of rutile-type VO2:Sb:SnO2 which was chosen as a substrate for VO2 deposition because of its appropriate lattice mismatch towards rutile VO2. As a result, an in-plane expansion of the rutile type structure was achieved at the interface of the two materials, yielding an enhanced capacity of 350 mA h g−1 per mass of VO2.45
In this study, we introduce a novel approach for creating hybrid electrodes combining both high specific energy and high power. Owing to the cyclic process characteristics of ALD that allow for ultra-precise deposition of sub-nanometer layers, highly conformal and atomically controlled decoration of carbon onions with alternating stacks of vanadia and titania was obtained. Due to excellent control over multilayer growth and composition, first conclusive analytical evidence of the origin of improved energy storage performance for the mixed vanadia/titania system was obtained. The addition of about 25 mass% TiO2 leads to an expansion of the VO2 unit cell, greatly enhancing lithium intercalation capacity and kinetics, as we demonstrate in both half-cell and asymmetrical full-cell setups.
2. Materials and methods
2.1 Materials
To obtain OLCs, a detonation nanodiamond precursor (diameter 4–6 nm, NaBond Technologies) was thermally annealed in a water-cooled high temperature furnace (Thermal Technology Inc.). The synthesis was carried out in an argon atmosphere at 1700 °C for 1 h, with a heating/cooling rate of 20 K min−1. From the resulting OLC powder, 50 μm thick electrodes were prepared by admixing 10 mass% of polytetrafluoroethylene (aqueous solution of 60 mass% of PTFE, Sigma Aldrich), a detailed description is given elsewhere.46Carbon onion electrodes were coated with an open-load atomic layer deposition system (Oxford Instruments) that was placed inside an argon filled glovebox (M. Braun Intertgas-Systeme) in order to ensure dry loading of the samples. Vanadium(V)-oxytriisopropoxide (VOTIP, SAFC Hitech) and tetrakis(dimethylamido)titanium(IV) (TDMAT, SAFC Hitech) were employed as metalorganic precursors for the deposition of vanadium oxide and titanium oxide, respectively, and deionized water vapor (Milli-Q, Merck) was chosen as reactant gas. The precursors were delivered by an argon carrier gas, with dosage over a period of 20 s during each reaction cycle, followed by purging of the reactor with pure argon over a period of 10 s after each precursor dosage step. After each ALD cycle, the reactor was evacuated for 8 s. During the ALD process, the reactor temperature was kept at 180 °C. In general, for each coating, 100 ALD cycles were applied. For synthesis of multilayer coatings, alternating sequences of vanadia and titania were deposited as indicated by the sample nomenclature; for example, OLC/VTiO6:2 was fabricated by repetitively alternating sequences of 6 and 2 ALD cycles of vanadia and titania, respectively, until 100 cycles were reached.
Thermal annealing of the coated samples was carried out in a quartz tube furnace under an argon atmosphere at a flow rate of 100 sccm. During the annealing procedure, all samples were heated at 5 °C min−1 to 500 °C, held for 0.5 h at that temperature, and cooled down to room temperature at 40 °C min−1. Thereby, the PTFE fully decomposes to volatile species, leaving a connected, binder-free network of OLC/metal oxide hybrid electrodes.
2.2 Materials characterization
Transmission electron microscopy (TEM) was carried out using a JEOL 2100F system operating at 200 kV. Sample preparation was performed by dispersion and sonication of the powder samples in isopropanol and deposition on a copper grid with a lacey carbon film (Gatan Inc.).X-ray diffraction (XRD) experiments were conducted employing a D8 Advance diffractometer (Bruker AXS) with a copper X-ray source (CuKα, 40 kV, 40 mA), a nickel filter and a LYNXEYE detector. All samples were placed on a sapphire single crystal and measured in a range from 10 to 80° 2θ with a step width of 0.01° 2θ and a step duration of 10 s. Calculation of the lattice parameters was carried out using the EVA Diffrac Suite software.
Raman spectroscopy was performed with a Renishaw inVia Raman Microscope using an Nd:YAG laser with an excitation wavelength of 532 nm and a power of about 0.25 mW. A 50× objective and a grating with 2400 lines per mm were used to reach a spectral resolution of about 1.2 cm−1. The acquisition time for each spectrum was 30 s with 10 accumulations.
The vanadium and titanium binding energies in different coating compositions were determined by X-ray photo-electron spectroscopy (XPS). The measurements were carried out on an AXIS Ultra DLD electron spectrometer (Kratos Analytical). For the excitation of the photoelectron spectra, monochromatic Al Kα was used. The spectra were acquired by setting the instrument to medium magnification (field of view 2) lens mode and by selecting the slot mode. Charge neutralization was implemented by low energy electrons injected into the magnetic field of the lens from a filament located directly above the sample. Three sets of measurements were performed. Survey scans and individual photo-electron lines were acquired using the X-ray source operating at 150 W power and 80 eV analyzer pass energy. Additionally, high resolution measurements of the C 1s, Ti 2p, and V 2p lines with the pass energy of 10 eV at the power of 225 W were performed. Three spots at different positions on each sample were analyzed and averaged. Data acquisition and processing were carried out using CasaXPS (Casa Software Ltd.). After subtraction of Shirley background, the peaks were fitted using the Gaussian Lorentzian GL(30) peak shape. The binding energy (BE) scale was corrected for charging using an electron BE of 285.0 eV for the C–Caliph component in the C 1s spectra.
Thermogravimetric analysis (TGA) was carried out using a TG 209 F1 Libra system (Netzsch). The samples were heated from room temperature to 650 °C (TiO2 to 750 °C) at a heating rate of 5 °C min−1 under a synthetic air atmosphere (80% N2, 20% O2). The change in mass during heating was used to determine the metal oxide content of the sample. For samples containing vanadia, the V4+/V5+ oxidation process and the associated increase in mass occurring during the transition from VO2 to V2O5 were considered.
2.3 Electrochemical benchmarking
The electrochemical performance was evaluated in a three-electrode setup (half-cell), which has been described in detail elsewhere,47 and in a two-electrode setup (full-cell). In half-cells, ALD-coated carbon onion electrodes with masses varying between 1.0 and 1.5 mg were employed as a working electrode, whereas an about five times oversized PTFE-bound activated carbon (type YP80-F, Kuraray) was used as a counter electrode. The potential was measured against an activated carbon reference electrode (type YP50-F, Kuraray). In full-cell measurements, OLC/VTiO6:2 electrodes with masses between 1.0 and 1.5 mg were used as the anode and PTFE-bound activated carbon (type YP80-F, 5 mass% PTFE) as the cathode with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5 to achieve charge-balance. In addition, the potential evolution at the cathode and anode was monitored with a separate, PTFE-bound activated carbon reference electrode (type YP50-F). Carbon-coated, 12 mm diameter aluminum foils (Zflo 2653, Exopack Technologies) and glass fiber mats (GF/D, Whatman) were employed as current collectors and separators, respectively. Prior to electrolyte filling in an argon filled glovebox (MBraun Labmaster 130, O2 and H2O < 1 ppm), the cells were dried in a vacuum oven at 120 °C overnight. 1 M LiClO4 (battery grade, Sigma Aldrich) in a 50:50 mixture of ethylene carbonate and dimethyl carbonate (EC/DMC; battery grade, BASF) was used as the electrolyte.
:3.5 to achieve charge-balance. In addition, the potential evolution at the cathode and anode was monitored with a separate, PTFE-bound activated carbon reference electrode (type YP50-F). Carbon-coated, 12 mm diameter aluminum foils (Zflo 2653, Exopack Technologies) and glass fiber mats (GF/D, Whatman) were employed as current collectors and separators, respectively. Prior to electrolyte filling in an argon filled glovebox (MBraun Labmaster 130, O2 and H2O < 1 ppm), the cells were dried in a vacuum oven at 120 °C overnight. 1 M LiClO4 (battery grade, Sigma Aldrich) in a 50:50 mixture of ethylene carbonate and dimethyl carbonate (EC/DMC; battery grade, BASF) was used as the electrolyte.
Electrochemical testing procedures were carried out using a potentiostat/galvanostat (VSP300, Bio-Logic). For half-cells, cyclic voltammetry (CV) was performed in a potential window from +1.0 V to −2.0 V vs. carbon, at a scanning rate of 1 mV s−1. Galvanostatic charge/discharge with potential limitation (GCPL) was carried out by cycling between +1.0 V and −2.0 V vs. carbon with specific currents ranging from 0.05 to 20 A g−1, normalized to the total composite electrode mass. The specific capacity Csp was calculated by integration of the discharge current I over the discharge time t accounting for the lithiation step from +1.0 V to −2.0 V vs. carbon, normalized to the full composite electrode mass, m:
 | (1) |
For investigation of the cycling stability, galvanostatic discharge was carried out at 1 A g−1 in the potential window from +1.0 V to −2.0 V and −1.8 V vs. carbon. In full-cells, CV experiments ranged from 0 V up to 3.5 V cell voltage at a scanning rate of 1 mV s−1. GCPL procedures were performed by cycling to 3.2 V cell voltage at different rates between 0.05 A g−1 and 2.5 A g−1, normalized to the total mass of both electrodes. The specific energy Esp was calculated by integration of the voltage profile:
 | (2) |
3. Results and discussion
3.1 Structure and composition
The morphology of the annealed hybrid materials was studied by TEM. The synthesized OLCs are spherical, non-porous nanoparticles with diameters of 5–7 nm, present as agglomerates in the size-range of several 100 nm. Their pore volume consists of interparticle voids in the agglomerate network,28 which is where the metal oxide coating is mostly located, as shown in Fig. 1A–E, and schematically illustrated in Fig. 1F. After deposition of 100 ALD cycles onto the OLCs, most of the pore volume throughout the agglomerates is homogeneously occupied by the metal oxide, with little remaining pore space still visible in TEM images. These observations are confirmed by gas sorption analysis, showing a decline in the specific surface area from 352 m2 g−1 to 50 m2 g−1 (SSABET) and in the total pore volume from 0.93 cm3 g−1 to 0.12 cm3 g−1 for the OLC/VO2 hybrids (ESI, Fig. S1†). The deposited single and bimetal oxides are mostly present in nanocrystalline form, with visible crystal sizes ranging between 5 and 20 nm. | ||
| Fig. 1 (A–E) Transmission electron micrographs of all fabricated hybrid electrodes; (F) schematic illustration of the homogenous metal oxide distribution in the mesopore volume of OLC agglomerates. | ||
The chemical composition of the hybrid material, especially regarding the metal oxide content, was analyzed by means of thermogravimetric analysis (TGA) and energy dispersive X-ray spectroscopy (EDX); the quantitative results are given in Table 1 and Fig. 2. Application of 100 ALD cycles resulted in a metal oxide content between 66 and 72 mass%, as measured by TGA, being in line with semi-quantitative EDX analysis, where 61–66% were observed.
| Elemental composition (EDX) | Metal oxide content | |||||
|---|---|---|---|---|---|---|
| C, mass% | V, mass% | Ti, mass% | O, mass% | EDX (mass%) | TGA (mass%) | |
| OLC/VTiO6:2 | 35 ± 4 | 25 ± 3 | 18 ± 3 | 22 ± 5 | 65 ± 11 | 69 |
| OLC/VTiO4:2 | 34 ± 5 | 23 ± 2 | 19 ± 2 | 24 ± 3 | 66 ± 7 | 67 |
| OLC/VTiO2:2 | 38 ± 5 | 17 ± 2 | 20 ± 2 | 25 ± 4 | 62 ± 8 | 72 |
| OLC/VO2 | 39 ± 3 | 43 ± 7 | — | 18 ± 5 | 61 ± 12 | 67 |
| OLC/TiO2 | 34 ± 4 | — | 37 ± 4 | 29 ± 2 | 66 ± 6 | 66 |
 | ||
| Fig. 2 Thermogravimetric analysis of all hybrid electrode samples. The measurements were carried out in synthetic air with a flow rate of 20 ml min−1 up to a temperature of 650 °C at a heating rate of 5 °C min−1. | ||
To characterize the crystalline structure of the deposited metal oxides in the hybrid materials, X-ray diffraction (XRD) was carried out (Fig. 3A). In the sample OLC/VO2, vanadia is present in the monoclinic VO2(M) structure in space group C2/m (JCPDS 65-7960), most concisely represented by the (001) and (200) reflections occurring at 14.4° 2θ and 15.3° 2θ, respectively. In OLC/TiO2, tetragonal anatase TiO2 (JCPDS 78-2486) is formed, as clearly indicated by the presence of the (101) reflection at 25.2° 2θ. The three multilayer systems, OLC/VTiO6:2, OLC/VTiO4:2, and OLC/VTiO2:2, all exhibit diffraction patterns that strongly resemble the monoclinic C2/m crystal lattice, similar to that of VO2(M) vanadia, while no additional peaks are found that might indicate the presence of an anatase-type crystalline phase, as observed for OLC/TiO2. This leads to the conclusion that in multilayer samples, the titania species is present in another configuration. The diffraction peaks detected for multilayer samples are found to be shifted towards lower diffraction angles, 2θ, with respect to peaks arising from the pure VO2(M) phase, as exemplified for the (110) reflection in Fig. 3B. Hence, the crystal structures of the newly formed multilayer phases are isomorphs of the monoclinic vanadia system. This could be explained by (1) the formation of a solid solution of vanadia and titania, V1−xTixO2, or (2) by the occurrence of crystal lattice strain in VO2(M), induced by epitaxially grown TiO2(B)-interlayers that also are isomorph to VO2(M) (JCPDS 74-1940, orange line in Fig. 3B).48 The observation of one sharp peak suggests the formation of a homogenous solid solution, since for two separate, epitaxially grown phases, two peaks in close proximity or a broad diffraction signal would be expected. The calculated lattice parameters of the multilayer phases are shown in Table 2. The atomically controlled insertion of two titania layers between arrays of six, four, or two vanadia layers via ALD led to an enlargement of the VO2(M) unit cell in all three crystallographic directions, leading to an expansion of the cell volume from 273 Å3 to 279 Å3, 280 Å3, and 281 Å3 for the samples OLC/VTiO6:2, OLC/VTiO4:2, and OLC/VTiO2:2, respectively. The expansion can be explained by the larger ionic radius of titanium (r = 0.60 Å) for Ti4+ in 6-fold coordination as opposed to r = 0.58 Å for V4+.49
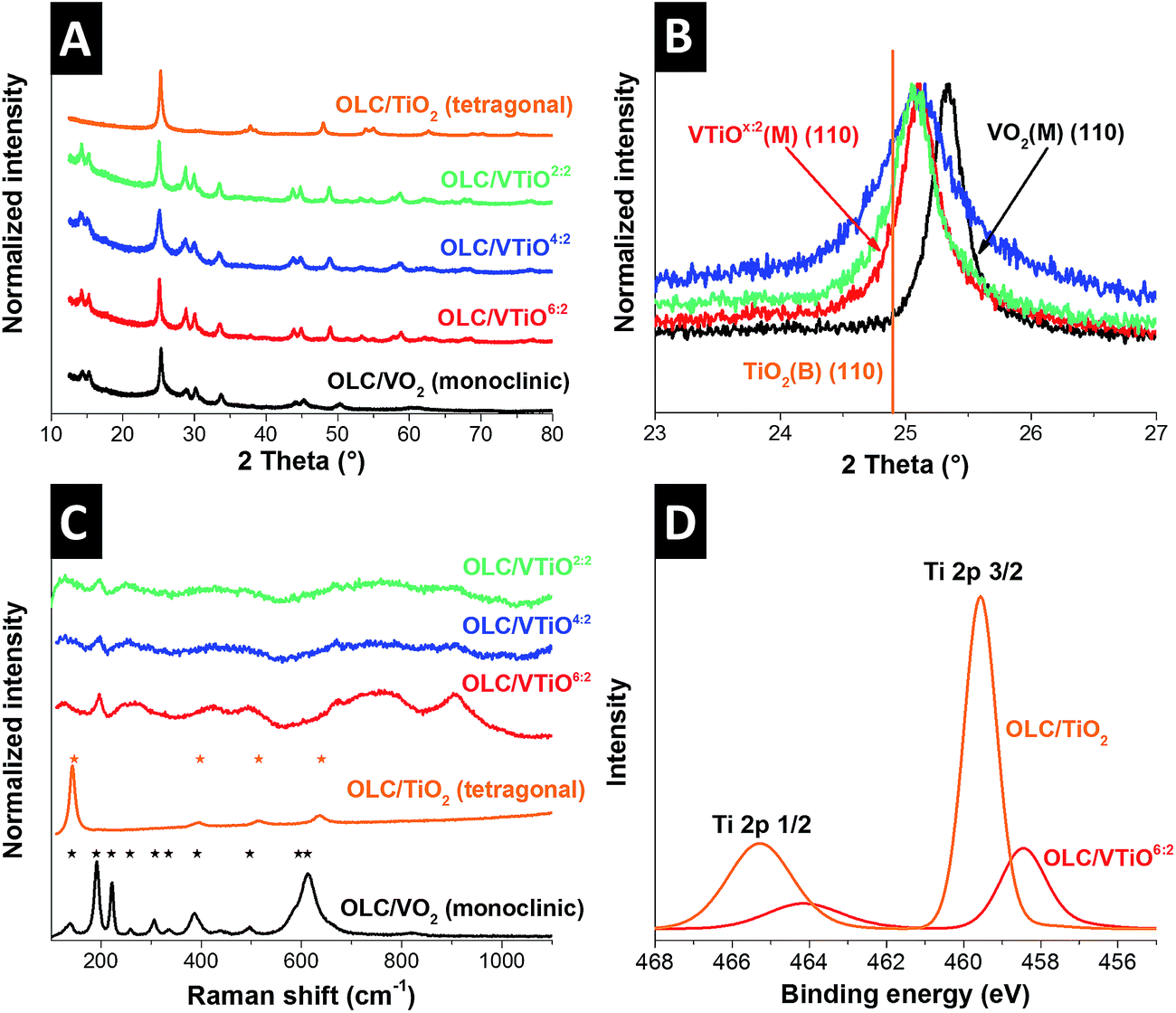 | ||
| Fig. 3 (A) Survey of X-ray diffractograms of all fabricated hybrid electrodes, (B) detailed view of the (110) signal of OLC/VO2 and all multilayer hybrid samples, including the (110) reflection position of TiO2(B), (C) Raman spectra of all hybrid samples, including ideal peak positions of VO2(M) and anatase, and (D) high resolution X-ray photoelectron spectra of the Ti 2p signal for hybrid samples OLC/TiO2 and OLC/VTiO6:2. | ||
| a (Å) | b (Å) | c (Å) | β (°) | Volume (Å3) | Type | |
|---|---|---|---|---|---|---|
| VTiO6:2 | 12.12 | 3.72 | 6.45 | 106.6 | 279.2 | Monoclinic |
| VTiO4:2 | 12.13 | 3.72 | 6.46 | 105.9 | 280.8 | Monoclinic |
| VTiO2:2 | 12.14 | 3.73 | 6.48 | 106.6 | 281.1 | Monoclinic |
| VO2 | 12.06 | 3.69 | 6.42 | 107.0 | 273.0 | Monoclinic |
| TiO2 | 3.79 | 3.79 | 9.51 | 90.0 | 136.7 | Tetragonal |
Raman spectroscopy was performed to further analyze the metal oxide structure (Fig. 3C). The findings of the XRD experiments for OLC/VO2 and OLC/TiO2 were confirmed: the observed Raman signals are characteristic of monoclinic vanadia and tetragonal titania.50,51 The Raman spectra of the multilayer samples are in stark contrast to the respective pure phases. A distinct peak at 195 cm−1, which decreases in intensity for increasing Ti-content, can be related to VO2(M). Furthermore, the broad shoulder centered at 910 cm−1 that also decreases with increasing Ti-content has been described for mixtures of vanadia with both alumina and titania.52,53 The signal is a result of polyvanadate chains bonded to the foreign metal.53 The broad feature around 250 cm−1 that becomes sharper for increasing Ti-content has also been described in the literature. It was attributed to a disordered rutile structure54 and second-order signals of octahedrally coordinated Ti in SrTiO3 compounds.51 Thus, the emergence of the fundamentally changed Raman spectra for the multilayer systems is a further strong indicator of the formation of a solid solution with modified bond structures.
X-ray photoelectron spectroscopy (XPS) was performed on OLC/VO2, OLC/TiO2, and OLC/VTiO6:2 samples to evaluate the change in vanadium and titanium binding energy when forming in a multilayer system. Survey spectra are given in Fig. S2A;† high resolution measurements of V 2p and Ti 2p are given in Fig. S2B† and 3D, respectively. The high resolution spectra of V 2p and Ti 2p in the multilayer sample show a reduced intensity, which is explained by the reduced content of the respective materials. While the signals of V 2p are located around roughly the same binding energy (517.7 ± 0.2 eV) for OLC/VO2 and OLC/VTiO6:2, a significant shift of the Ti 2p peaks is observed when being present in the multilayer system. The binding energy of titanium is reduced by a magnitude of 1.2 eV (Ti 2p3/2 shifts from 459.6 eV in anatase OLC/TiO2 (ref. 55) to 458.4 eV in OLC/VTiO6:2), which matches the value of previously reported titania doped vanadia phases.20 This is in agreement with the XRD and Raman results, suggesting the formation of a solid solution of vanadia and titania, with a change in the binding structure of titanium in the hybrid samples.
3.2 Electrochemical performance
The electrochemical properties of the hybrid electrodes were evaluated using cyclic voltammetry (CV) at a scanning rate of 1 mV s−1 (Fig. 4A and B). The OLCs used as substrates typically exhibit capacitive charge storage via double-layer formation, as has been well established in the literature.56–58 This behavior is reflected by a very small, rectangular shaped background in the CVs, best visible for sample OLC/TiO2 in the range between 0 V and +1.0 V vs. carbon. The main charge storage mechanism of the hybrid electrodes, however, is lithium intercalation into the redox-active metal oxide layers, which is synergistically promoted by the conducting network provided by OLCs. In the cathodic scan of sample OLC/VO2, a first lithiation peak occurs at −0.3 V vs. carbon, followed by a second, more pronounced peak at −0.5 V vs. carbon (corresponding to 2.9 V and 2.7 V vs. Li/Li+, respectively), indicating a two-step lithiation of the material, in accordance with the literature,59 with a total intercalation of about 0.8–1.1 Li per VO2.40 The anodic scan also exhibits two peaks, completing the delithiation around −0.1 V vs. carbon. In contrast, OLC/TiO2 exhibits only one pair of redox-peaks, with the lithiation process starting at −1.2 V and ending at −1.6 V vs. carbon (i.e., +2.0 V to +1.6 V vs. Li/Li+), which corresponds to the insertion of 0.4–0.5 Li per TiO2, as reported in the literature.60,61 The delithiation during the anodic scan is completed at −0.6 V vs. carbon. The larger voltage gap between the reactions, compared to OLC/VO2, indicates slower lithium intercalation kinetics in the OLC/TiO2 system. | ||
| Fig. 4 (A) Cyclic voltammograms of single phase and (B) multilayer hybrid electrodes recorded at a scan rate of 1 mV s−1. (C) Discharge capacity of all hybrid electrodes, derived from galvanostatic discharge from +1.0 V to −2.0 V vs. carbon at rates between 0.05 A g−1 and 20 A g−1. (D) Voltage–capacity profiles for sample OLC/VTiO6:2 at rates between 0.05 A g−1 and 20 A g−1 (corresponding to 0.13C to 52C) and (E) coulombic efficiency of all multilayer hybrid samples, derived from galvanostatic cycling at different rates. (F) Cycling stability test from galvanostatic cycling between +1.0 V and −2.0 V (−1.8 V) vs. carbon at 1 A g−1. | ||
The CV curves of the multilayer hybrid electrodes (Fig. 4B) exhibit a greater number of redox peaks, as compared to the single phase systems. In the cathodic scan of sample OLC/VTiO6:2, lithiation can already be detected starting at −0.2 V vs. carbon, going through at least three steps up to −1.2 V vs. carbon. At −1.3 V vs. carbon, a further, separated peak emerges, indicating a fourth step of lithium intercalation. The anodic scan shows little shift for all corresponding peaks, indicating fast lithium diffusion kinetics in OLC/VTiO6:2. With an increased titania content in the samples OLC/VTiO4:2 and OLC/VTiO2:2, two significant observations are made in comparison to OLC/VTiO6:2: (1) the area of the first set of peaks decreases, while the single, separated peak at a more negative potential increases in size and (2) the voltage shift between related cathodic and anodic peaks increases. This leads to the conclusions that (1) the cathodic peaks between −0.2 V and −1.2 V vs. carbon can be associated with a multistep reaction of Li with the vanadia species in the solid solution, while the peak at the more negative potential originates from the reaction between Li and the titania species. Also, (2) the intercalation kinetics drop with increasing titania content in the solid solution, in accordance with the observations made in pristine OLC/TiO2.
The capacity and power handling of the hybrid electrodes were evaluated using galvanostatic cycling with potential limitation (GCPL), shown in Fig. 4C. In the battery community, it is customary to normalize performance metrics to the mass of the redox-active material in the electrode. However, since both metal oxide and OLCs contribute to the electrochemical performance of the presented hybrid electrodes, it is appropriate to normalize to the entire electrode mass.62 For better comparability with literature values, the data for the best performing sample (OLC/VTiO6:2) will also be reported with respect to the metal oxide content.
The OLC/VO2 electrode exhibited a maximum specific discharge capacity of 198 mA h g−1, with a high capacity retention of 51 mA h g−1 at an ultrahigh discharge rate of 20 A g−1 or 101C, while the OLC/TiO2 electrode performed considerably worse with a maximum of 166 mA h g−1 and a retention of 20 mA h g−1 at 20 A g−1 or 120C. The multilayer hybrid electrodes exhibited a strongly improved maximum specific capacity of above 330 mA h g−1, with OLC/VTiO6:2 reaching the highest value of 382 mA h g−1 (554 mA h g−1 per metal oxide), thereby outperforming state-of-the-art metal oxide hybrid systems (ESI, Table S1†). This is a consequence of enhanced lithium intercalation capacity that can be related to the larger unit cell volume of VTiO, compared to VO2(M) and tetragonal TiO2. In addition, the number of possible lithiation reactions is increased due to the presence of both vanadia and titania species. The lithiation follows this reaction path, with x defining the composition of the multilayer system and y the number of lithium ions intercalated:
| VxTi1−xO2 + yLi+ + ye− ↔ LiyVxTi1−xO2 | (3) |
When evaluating the power handling ability of the multilayer hybrid electrodes, the trend of decreased kinetic properties with higher titania contents seen in the CVs is confirmed. The samples OLC/VTiO4:2 and OLC/VTiO2:2 dramatically decline in performance at high rates above 2.5 A g−1 (about 10C), while OLC/VTiO6:2 exhibits an extraordinary power handling, with a capacity retention of 82 mA h g−1 at 20 A g−1 or 52C (equivalent to 120 mA h g−1 normalized to the metal oxide mass), even reaching power values of novel hybrid supercapacitor materials (60 mA h g−1 at 17 A g−1 or 100C).63
The electrochemical performance of OLC/VTiO6:2 is further outlined by voltage–capacity profiles at various cycling rates (Fig. 4D). Two plateaus can be observed in the cathodic scan, corresponding to lithiation reactions with the vanadia and titania species of the material. When increasing the rate, the second plateau associated with lithiation of titania becomes smaller, thereby confirming that titania is the kinetically limiting part. The charge efficiency of all samples is determined to be well above 80% at very low rates of 0.05 A g−1, and reaches 100% at medium–high rates above 0.25 A g−1 (Fig. 4E). The charge efficiency below 100% at low rates is explained by the charge contribution of irreversible side reactions, which commonly occur with high surface area materials offering a high reaction interface between the electrode and electrolyte. The high degrees of lithiation reached at low specific currents are also in part not fully reversible at the same discharging rate, leading to irreversibly intercalated Li, further contributing to a charge efficiency below 100%.
The longevity of the hybrid electrodes was evaluated using GCPL at a rate of 1 A g−1 (Fig. 4F). For discharging to −2.0 V vs. carbon, after 200 cycles, a decline in discharge capacity was detected to 65% and 50% of the initial value for OLC/VO2 and OLC/VTiO6:2, respectively. This is characteristic of vanadia-containing electrodes that struggle with vanadium dissolution at low voltages.39,43 Decreasing the voltage window was found as a facile way to resolve this critical issue: by only discharging to a voltage of −1.8 V vs. carbon (i.e., 1.4 V vs. Li/Li+), the OLC/VTiO6:2 hybrid system showed a remarkably stable performance over 3000 cycles. After an initial drop to 83% after 200 cycles, stable cycling with minor capacity fading was observed. Although the maximum specific capacity was slightly reduced to 315 mA h g−1 (457 mA h g−1 per metal oxide), reducing the maximum voltage it is proposed as an effective strategy to significantly enhance the longevity of VO2-containing electrodes.
In order to demonstrate a device level application of the hybrid electrodes, charge-balanced full-cells composed of OLC/VTiO6:2 (anode) and activated carbon (cathode) were investigated. The CVs of the full-cells show electrolyte stability up to at least 3.2 V cell voltage (Fig. 5A). The CV shape clearly demonstrates two smeared out peaks between about 0 V and 1.8 V, and 2.3 V to 3.2 V, respectively, which correspond to the two regimes of lithium intercalation observed in the half-cell setup. This is confirmed by the two plateau-like regions in the voltage-profile (Fig. 5B). During galvanostatic cycling, the anode potential was monitored by a carbon reference and kept well above −1.8 V vs. carbon to stay within the stable potential range determined in half-cell experiments (blue line, Fig. 5B). The specific energy and power of full-cells are commonly displayed in a Ragone plot (Fig. 5C). The specific energy reaches a maximum value of 110 W h kg−1 for charging (76 W h kg−1 for discharging) with a maximum specific power of 6 kW kg−1 for charging (2.2 kW kg−1 for discharging). This outstanding device level performance is among the highest reported in the literature for hybrid supercapacitor full-cells and underlines the potential of the herein presented vanadia/titania multilayer system.
 | ||
| Fig. 5 Electrochemical benchmarking of hybrid supercapacitor full cells composed of OLC/VTiO6:2 (anode) and activated carbon (cathode). (A) Cyclic voltammograms to different maximum cell voltages between 3.0 V and 3.5 V. (B) Voltage profile of a galvanostatic cycle to 3.2 V at a rate of 0.05 A g−1. The blue curves show the potential evolution at the cathode and anode, monitored against a carbon reference. (C) Ragone-plot displaying specific energy and power, derived from galvanostatic cycling to 3.2 V at different rates. Inset: energy efficiency and coulombic efficiency at different cycling rates. | ||
4. Conclusions
Vanadia–titania/carbon onion hybrid electrodes were synthesized by atomic layer deposition with different stacking orders of the respective metal oxides. By employing this novel approach and repetitively stacking atomically controlled vanadia and titania layers with high conformity, a solid solution with an expanded vanadia unit cell was fabricated, leading to a highly improved specific capacity of 382 mA h g−1 (554 mA h g−1 normalized to metal oxide mass) in comparison to 198 mA h g−1 for pristine VO2. In synergy with the highly conductive network of carbon onions, an impressive capacity retention of 82 mA h g−1 (120 mA h g−1 per metal oxide) when discharging at a rate of 20 A g−1 or 52C was attained. By benchmarking for cycling stability, discharging to a reduced potential of −1.8 V vs. carbon was found as a solution to obtain longevity over 3000 cycles. Device level performance of up to 110 W h kg−1 in asymmetrical full-cells underlines the future potential of the presented vanadia/titania multilayer system.Acknowledgements
We acknowledge funding from the German Federal Ministry for research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. This work is part of the CREATe-Network Project, Horizon 2020 of the European Commission (RISE Project No. 644013). We thank Prof. Arzt (INM) for his continuing support and Juhan Lee (INM) for discussions.References
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- B. E. Conway, Electrochemical supercapacitors: scientific fundamentals and technological applications, Springer Science & Business Media, 2013 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
- H. Shi, Electrochim. Acta, 1996, 41, 1633–1639 CrossRef CAS.
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810–833 CrossRef CAS.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337–1344 CrossRef CAS PubMed.
- M. Zeiger, N. Jäckel, V. N. Mochalin and V. Presser, J. Mater. Chem. A, 2016, 4, 3172–3196 CAS.
- E. Bushueva, P. Galkin, A. Okotrub, L. Bulusheva, N. Gavrilov, V. Kuznetsov and S. Moiseekov, Phys. Status Solidi B, 2008, 245, 2296–2299 CrossRef CAS.
- E. Frackowiak, K. Metenier, V. Bertagna and F. Beguin, Appl. Phys. Lett., 2000, 77, 2421–2423 CrossRef CAS.
- T. M. Higgins, D. McAteer, J. C. M. Coelho, B. M. Sanchez, Z. Gholamvand, G. Moriarty, N. McEvoy, N. C. Berner, G. S. Duesberg and V. Nicolosi, ACS Nano, 2014, 8, 9567–9579 CrossRef CAS PubMed.
- J. S. Atchison, M. Zeiger, A. Tolosa, L. M. Funke, N. Jäckel and V. Presser, RSC Adv., 2015, 5, 35683–35692 RSC.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- T. A. Centeno, O. Sereda and F. Stoeckli, Phys. Chem. Chem. Phys., 2011, 13, 12403–12406 RSC.
- N. Jäckel, M. Rodner, A. Schreiber, J. Jeongwook, M. Zeiger, M. Aslan, D. Weingarth and V. Presser, J. Power Sources, 2016, 326, 660–671 CrossRef.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- K. Makgopa, P. M. Ejikeme, C. J. Jafta, K. Raju, M. Zeiger, V. Presser and K. I. Ozoemena, J. Mater. Chem. A, 2015, 3, 3480–3490 CAS.
- D. M. Anjos, J. K. McDonough, E. Perre, G. M. Brown, S. H. Overbury, Y. Gogotsi and V. Presser, Nano Energy, 2013, 2, 702–712 CrossRef CAS.
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS.
- P. H. Jampani, O. Velikokhatnyi, K. Kadakia, D. H. Hong, S. S. Damle, J. A. Poston, A. Manivannan and P. N. Kumta, J. Mater. Chem. A, 2015, 3, 8413–8432 CAS.
- S. Boukhalfa, K. Evanoff and G. Yushin, Energy Environ. Sci., 2012, 5, 6872–6879 CAS.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- M. V. K. Azhagan, M. V. Vaishampayan and M. V. Shelke, J. Mater. Chem. A, 2014, 2, 2152–2159 CAS.
- X. Wang and G. Yushin, Energy Environ. Sci., 2015, 8, 1889–1904 CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. Grey, B. Dunn and P. Simon, Nature Energy, 2016, 1, 16070 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS.
- M. Zeiger, N. Jäckel, M. Aslan, D. Weingarth and V. Presser, Carbon, 2015, 84, 584–598 CrossRef CAS.
- J. Liu, J. Essner and J. Li, Chem. Mater., 2010, 22, 5022–5030 CrossRef CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2012, 7, 174–182 CrossRef PubMed.
- M. Zeiger, S. Fleischmann, B. Krüner, A. Tolosa, S. Bechtel, M. Baltes, A. Schreiber, R. Moroni, S. Vierrath, S. Thiele and V. Presser, RSC Adv., 2016, 6, 107163–107179 RSC.
- K. E. Gregorczyk, A. C. Kozen, X. Chen, M. A. Schroeder, M. Noked, A. Cao, L. Hu and G. W. Rubloff, ACS Nano, 2014, 9, 464–473 CrossRef PubMed.
- M. D. Levi, M. R. Lukatskaya, S. Sigalov, M. Beidaghi, N. Shpigel, L. Daikhin, D. Aurbach, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1400815 CrossRef.
- X. Chen, H. Zhu, Y.-C. Chen, Y. Shang, A. Cao, L. Hu and G. W. Rubloff, ACS Nano, 2012, 6, 7948–7955 CrossRef CAS PubMed.
- M. Zeiger, T. Ariyanto, B. Krüner, N. J. Peter, S. Fleischmann, B. J. M. Etzold and V. Presser, J. Mater. Chem. A, 2016, 4, 18899–18909 CAS.
- I. Kovalenko, D. G. Bucknall and G. Yushin, Adv. Funct. Mater., 2010, 20, 3979–3986 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science Magazine, 2014, 343, 1210–1211 CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Skaarup, Electrochim. Acta, 1993, 38, 1215–1220 CrossRef CAS.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526–2552 RSC.
- D. Munoz-Rojas and E. Baudrin, Solid State Ionics, 2007, 178, 1268–1273 CrossRef CAS.
- L. Mai, Q. Wei, Q. An, X. Tian, Y. Zhao, X. Xu, L. Xu, L. Chang and Q. Zhang, Adv. Mater., 2013, 25, 2969–2973 CrossRef CAS PubMed.
- C. Tsang and A. Manthiram, J. Electrochem. Soc., 1997, 144, 520–524 CrossRef CAS.
- E. Baudrin, G. Sudant, D. Larcher, B. Dunn and J.-M. Tarascon, Chem. Mater., 2006, 18, 4369–4374 CrossRef CAS.
- K. Takahashi, Y. Wang, K. Lee and G. Cao, Appl. Phys. A, 2006, 82, 27–31 CrossRef CAS.
- S. Park, C. W. Lee, J.-C. Kim, H. J. Song, H.-W. Shim, S. Lee and D.-W. Kim, ACS Energy Lett., 2016, 1, 216–224 CrossRef CAS.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- R. Yoshida, Y. Suzuki and S. Yoshikawa, J. Solid State Chem., 2005, 178, 2179–2185 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. Petrov, V. Yakovlev and J. Squier, Appl. Phys. Lett., 2002, 81, 1023–1025 CrossRef CAS.
- U. Balachandran and N. Eror, J. Solid State Chem., 1982, 42, 276–282 CrossRef CAS.
- N.-Y. Topsøe, M. Anstrom and J. Dumesic, Catal. Lett., 2001, 76, 11–20 CrossRef.
- G. T. Went, S. T. Oyama and A. T. Bell, J. Phys. Chem., 1990, 94, 4240–4246 CrossRef CAS.
- Y. Hara and M. Nicol, Phys. Status Solidi B, 1979, 94, 317–322 CrossRef CAS.
- I. Chashechnikova, V. Vorotyntsev, V. Borovik, G. Golodets, I. Plyuto and A. Shpak, Theor. Exp. Chem., 1993, 28, 176–178 CrossRef.
- W. Gu, N. Peters and G. Yushin, Carbon, 2013, 53, 292–301 CrossRef CAS.
- M. Zeiger, N. Jäckel, D. Weingarth and V. Presser, Carbon, 2015, 94, 507–517 CrossRef CAS.
- J. K. McDonough, A. I. Frolov, V. Presser, J. Niu, C. H. Miller, T. Ubieto, M. V. Fedorov and Y. Gogotsi, Carbon, 2012, 50, 3298–3309 CrossRef CAS.
- D. Chao, C. Zhu, X. Xia, J. Liu, X. Zhang, J. Wang, P. Liang, J. Lin, H. Zhang and Z. X. Shen, Nano Lett., 2014, 15, 565–573 CrossRef PubMed.
- S. Huang, L. Kavan, I. Exnar and M. Grätzel, J. Electrochem. Soc., 1995, 142, L142–L144 CrossRef CAS.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H. M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef CAS.
- F. Gschwind, G. Rodriguez-Garcia, D. Sandbeck, A. Gross, M. Weil, M. Fichtner and N. Hörmann, J. Fluorine Chem., 2016, 182, 76–90 CrossRef CAS.
- K. Naoi, K. Kisu, E. Iwama, S. Nakashima, Y. Sakai, Y. Orikasa, P. Leone, N. Dupré, T. Brousse, P. Rozier, W. Naoi and P. Simon, Energy Environ. Sci., 2016, 9, 2143–2151 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta09890h |
| This journal is © The Royal Society of Chemistry 2017 |