
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBipolar nitrogen-doped graphene frameworks as high-performance cathodes for lithium ion batteries†
Yanshan
Huang
ab,
Dongqing
Wu
 *a,
Arezoo
Dianat
c,
Manferd
Bobeth
c,
Tao
Huang
a,
Yiyong
Mai
a,
Fan
Zhang
*a,
Gianaurelio
Cuniberti
c and
Xinliang
Feng
ad
*a,
Arezoo
Dianat
c,
Manferd
Bobeth
c,
Tao
Huang
a,
Yiyong
Mai
a,
Fan
Zhang
*a,
Gianaurelio
Cuniberti
c and
Xinliang
Feng
ad
aSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: wudongqing@sjtu.edu.cn; fan-zhang@sjtu.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China
cInstitute for Materials Science and Max Bergmann Center of Biomaterials, Technische Universität Dresden, 01062, Dresden, Germany
dFachrichtung Chemie und Lebensmittelchemie, Technische Universität Dresden, 01062, Dresden, Germany
First published on 1st December 2016
Abstract
Hierarchically porous nitrogen-doped graphene frameworks (N-GFs) are fabricated through the ice-templating of GO with polyethylenimine and the thermal treatment of the resultant hybrids. As cathode materials in lithium ion batteries (LIBs), the obtained N-GFs exhibit an outstanding specific capacity of 379 mA h g−1 at 0.5 A g−1 for 2500 cycles. Even at an ultrahigh current density of 5 A g−1, the N-GFs maintain a capacity of 94 mA h g−1, superior to that of most reported LIB cathode materials. The experimental results and quantum mechanics calculations suggest that pyridinic-like N and pyridinic N-oxide in graphene are responsible for the excellent cathodic performance of the bipolar N-GFs by providing fast surface faradaic reactions with both p- and n-doped states.
Introduction
As a crucial factor in determining the electrochemical performance of lithium ion batteries (LIBs), cathode materials with high specific capacity and rate capability are essential for satisfying the increasing demand for miniaturized LIBs with improved energy density.1,2 The cathode materials in present commercial LIBs are mainly based on inorganic intercalation compounds such as lithium manganese oxide (LiMn2O4) and lithium iron phosphate (LiFePO4). In addition to their high costs, these inorganic LIB cathodes have disadvantages of limited mineral resources, low theoretical capacities, and complicated manufacturing processes. As an alternative to inorganic intercalation materials, organic LIB cathodes including n-type (such as organo-disulfide and conjugated carbonyl compounds), p-type (conjugated amine and conjugated thioether compounds), and bipolar organics (conjugated hydrocarbon and nitroxyl radical) have recently received considerable attention because of their relatively low cost, easy processability and the molecular-level adjustable electrochemical properties.3–11 Compared to n- and p-type organic electrodes, the neutralize state of bipolar organic electrodes can be continuously oxidized by anions (such as ClO4−, PF6−, BF4− or N(CF3SO2)2− in the electrolyte) with high redox potential and reduced by cations (Li+) with low redox potential, respectively.12–14 Such a bipolar redox reaction with a linear transition from each doped state in the energy storing process can result in a remarkably high specific capacity. However, the major problems associated with organic cathode materials are their low electrical conductivity and high solubility in electrolytes, which hinder their utilization in high rate operation and long-cycle process of LIBs.Profiting from its excellent conductivity and high specific surface area, graphene has been extensively applied as a matrix to improve the conductivity of inorganic or organic cathode materials, thereby boosting their electrochemical performances.15–17 However, the structural defects of graphene in such hybrid electrodes inevitably retard Li diffusion during intercalation and the capacity of such hybrids is still almost lower than 200 mA h g−1.18–21 Several recent publications demonstrated that graphene oxide (GO) alone can serve as the cathode materials in LIBs and its Li storage properties are highly sensitive to the oxygen-functional groups.22,23 Nevertheless, the presence of oxygenated groups in the plane or at the edge of graphene also reduces the electrical conductivity, thus influencing the charge and ion transport behavior. Therefore, the tradeoff between the capacity and the conductivity must be addressed to enhance the effectiveness of graphene as a LIB cathode. By contrast, nitrogen (N)-doped graphene can both boost the conductivity of graphene and improve the wettability of graphene with an electrolyte. Previous studies have focused only on applications of N-doped graphene as LIB anodes,24,25 supercapacitors26 and electrocatalysts.27,28 In this study, for the first time, we demonstrate that N-doped graphene can function as a LIB cathode with excellent capacity and cycle stability. Hierarchically porous N-doped graphene frameworks (N-GFs) were fabricated through the ice-templating of GO with polyethylenimine (PEI) and the following thermal treatment of the resulting composites. With a N doping content of ∼5 at% and in the presence of pyridinic nitrogen and the pyridinic-oxide nitrogen, the resulting bipolar type N-GFs exhibit an outstanding specific capacity of 379 mA h g−1 at 0.5 A g−1 for 2500 cycles. Even at an ultrahigh current density of 5 A g−1, the N-GFs maintain a capacity of 94 mA h g−1, superior to that of most reported organic and inorganic LIB cathode materials (Table S1†).11,29–34 The strong affinity of lithium ions to active nitrogen species in the hybrids is verified by quantum mechanics calculations. All the results suggest that pyridinic-like nitrogen and pyridinic N-oxide in graphene provide fast surface faradaic reactions with both p- and n-doped states, which are responsible for the excellent cathodic performance of the N-GFs.
Experimental section
Preparation of N-GFs
Graphene oxide (GO) was synthesized from natural graphite flakes by a modified Hummers method. PEI (5 mg) was dissolved in the suspension of GO (100 mL, 1 mg mL−1). The mixture was stirred for 5 h. After that, the as-prepared suspension was washed and centrifuged with water 3 times. The resulting mixture was then placed in a 1 mL insulin syringe, which was then immersed in liquid nitrogen. The unidirectional frozen cryogels were then freeze-dried for 48 h, and removed from the syringe as intact monoliths. Finally, the as-prepared samples were thermally treated at 300, 450, and 600 °C in N2 for 2 h, respectively. And with the different thermal treatment temperatures, the samples were denoted as N-GF-300, N-GF-450, and N-GF-600, respectively. Instead of PEI, aqueous ammonia (NH3) and chitosan were also utilized as the nitrogen source to prepare N-GFs, and the resulting hybrids were denoted as N-GF-450-A (from NH3) and N-GF-450-C (from chitosan), respectively. The weight ratios of nitrogen sources and GO were both 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. The fabrication processes of N-GF-450-A and N-GF-450-C were similar to that of N-GF-450. As a control experiment, GF-450 without N-doping was also synthesized by the following process. The suspension of 100 mg, 5 mg mL−1 GO was placed in a vial and then immersed in liquid nitrogen. The unidirectional frozen cryogels were then freeze-dried for 48 h. Finally, the GF was thermally treated at 450 °C in N2 for 2 h.
:20. The fabrication processes of N-GF-450-A and N-GF-450-C were similar to that of N-GF-450. As a control experiment, GF-450 without N-doping was also synthesized by the following process. The suspension of 100 mg, 5 mg mL−1 GO was placed in a vial and then immersed in liquid nitrogen. The unidirectional frozen cryogels were then freeze-dried for 48 h. Finally, the GF was thermally treated at 450 °C in N2 for 2 h.
Characterization
Scanning electron microscopy (SEM) measurements were performed on an FEI Sirion-200 field emission scanning electron microscope. Transition electron microscopy (TEM) studies were conducted on a JEOL-2100 electron microscope at an operating voltage of 200 kV. The samples were dispersed in water and the suspension was dropped onto a copper grid covered with a carbon film. The X-ray diffraction (XRD) analysis was performed on a Rigaku D/Max 2500 X-ray diffractometer with Cu Kα radiation (λ = 1.54 Å) at a generator voltage of 40 kV and a generator current of 50 mA with a scanning speed of 5° min−1 from 10 to 80°. Nitrogen adsorption/desorption isotherms at 77 K were determined by using a Micromeritics ASAP 2010. X-ray photoelectron spectroscopy (XPS) experiments were carried out on an AXIS Ultra DLD system from Kratos with Al Kα radiation as the X-ray source for radiation. Elemental analysis was applied for the determination of C/N on a Vario EL Cube. Fourier transform infrared (FTIR) spectra were recorded on a NEXUS 670 spectrometer. Raman spectra were recorded on a SENTERRA with excitation from the 532 nm line of an Ar-ion laser with a power of about 5 mW.Electrochemical measurements
The cathode electrodes were prepared by mixing the hybrids, carbon black (Super-P), and poly(vinyl difluoride) (PVDF) with a weight ratio of 80:10:10 and pasting the mixture on aluminum foil with a thickness of 100 μm. Pure lithium foil was used as the counter-electrode. The electrolyte consisting of a solution of 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 by volume) was obtained from Ube Industries Ltd. The cells were assembled in an argon-filled glovebox with the concentrations of moisture and oxygen below 1 ppm. The electrochemical performance was tested at various rates in the voltage range of 1.50–4.50 V. The active material loading in these electrodes is about 0.6 mg cm−2.
Computational details
DFT calculations were performed with the Gaussian 09 program. All geometry optimizations were carried out at the RB3LYP level of density functional theory with the 6-31G(d) basis set. To compute the electronic charge density, a mixed basis-set approach was used here, where the Kohn–Sham orbitals are expanded into linear combinations of contracted Gaussian type orbitals (GTO) and complemented by a plane-wave basis set. In particular, for geometry optimization, we applied a DZVP (double zeta for valence electrons plus polarization functions) basis set complemented with a plane-wave basis with energy cut-off at 350 Ry. In all calculations, we employed the BLYP exchange–correlation functional and its corresponding norm-conserving pseudo-potential GTH (Goedecker, Teter and Hutter). Dispersion corrections were included through the standard D3 Grimme parameterization. Afterwards, all energies were computed by single point calculations with the B3LYP exchange–correlation functional. The convergence criteria for both geometry and energy calculations were set to 1 × 10−7 Hartree for the SCF energy and 9 × 10−4 Hartree per Å, respectively.Results and discussion
Scheme 1 illustrates the overall procedure for the fabrication of N-GFs. PEI is first added to the aqueous suspension of GO, and the rich amine groups on PEI enable it to anchor to the hydroxyl and carboxyl groups on GO through hydrogen bonding, forming PEI–GO.35 When the dispersion of PEI–GO is plunged into liquid nitrogen, the water in the suspension rapidly solidifies into ice crystals, which expel the PEI–GO nanosheets to form a three-dimensional (3D) framework along the solidification front. After the freeze-drying process, a macroscopic free-standing monolith (PEI-GOM) with a continuous and interconnected network architecture is formed. Finally, the thermal treatment of PEI–GO in a nitrogen atmosphere converts to N-doped graphene frameworks (N-GFs). During the process, PEI does not only serve as a cross-linker for binding GO sheets into a highly interconnected monolith in the ice-templating step. It also provides a nitrogen source for the N-doping in the graphene frameworks.36 The resulting N-GFs are denoted as N-GF-300, N-GF-450, and N-GF-600 according to the thermal treatment temperature at 300, 450, and 600 °C, respectively. In addition to PEI, NH3 and chitosan are also selected as nitrogen sources for the fabrication of N-GFs (ESI†). The resulting samples are denoted as N-GF-450-A (from NH3) and N-GF-450-C (from chitosan).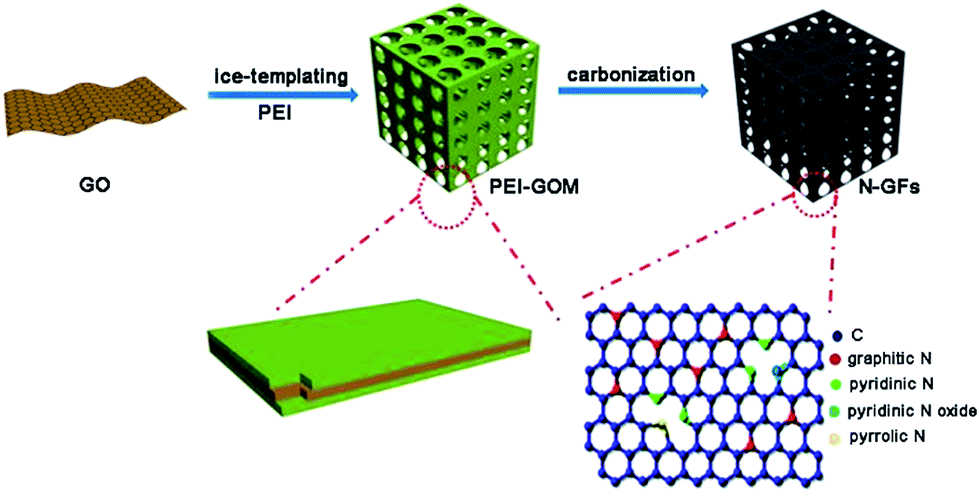 | ||
| Scheme 1 Schematic illustration of the synthesis of N-GFs. | ||
Cross-sectional scanning electron microscopy (SEM) images of the as-prepared N-GFs (Fig. 1a, b and S1†) indicate that all of the samples have highly interconnected 3D macroporous structures with diameters ranging from 5 to 20 μm. Moreover, alternative dark and light domains with different contrast levels are evident on the N-GF surfaces through transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM), which are attributable to the defects of the N-doped graphene framework (Fig. 1c and d). Furthermore, elemental mapping images of N-GF-450 show that C, N, and O atoms are homogeneously distributed in the N-GF (Fig. 1e–h). In addition, nitrogen physisorption measurements were conducted to gain insight into the porosities of N-GFs (Fig. S2a†). All samples exhibit type IV adsorption branches associated with a well-defined capillary condensation step. The Brunauer–Emmett–Teller (BET) surface areas of N-GF-300, N-GF-450, and N-GF-600 are 77, 111, and 121 m2 g−1, respectively. Furthermore, the existence of a high number of mesopores and micropores in N-GF-450 and N-GF-600 was verified according to the corresponding pore size distribution based on density functional theory (DFT, Fig. S2b†). Compared to N-GF-300, the considerably enhanced surface areas of N-GF-450 and N-GF-600 might be attributable to the micro- and mesopores generated through the conversion of PEI to N-doped carbon in the graphene frameworks.
 | ||
| Fig. 1 Structural characterization of N-GF-450: (a and b) SEM, (c) TEM, (d) HR-TEM and (e–h) elemental mapping images (by SEM): C (f), N (g) and O (h). | ||
According to the X-ray photoelectron spectroscopy (XPS) results, the N contents of N-GF-300, N-GF-450, and N-GF-600 are calculated to be 4.16, 5.14, and 4.31 at%, respectively, which are in agreement with the elemental analysis results (Table S1†). Moreover, the high resolution N1s and C1s and O1s spectra of the samples are shown in Fig. S3.† The N1s spectra of N-GFs can be fitted to four peaks at approximately 398.9, 400.2, 401.0, and 402.6 eV, corresponding to the pyridinic nitrogen (N1), pyrrole nitrogen (N2), graphitic nitrogen (N3), and pyridinic N-oxide (N4), respectively (Fig. S3†).37 As summarized in Fig. S3c,† the amounts of different N atoms in the N-GFs change with the thermal treatment temperature. The N4 in N-GF-300 and N-GF-450 is attributed to N1 coupled with the remaining O atoms on the graphene surface after thermal reduction at 300 or 450 °C. However, the amount of O atoms in N-GF-600 is considerably reduced and not sufficient to form N4. The C1s peak is dominated by a C–C bond at 248.8 eV. A small amount of carbonyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 290 eV) can also be detected from the XPS data for the O1s (531.5 eV). This type of the functional group on the graphene surface is well known to increase the capacity of the engineered graphene for its fast reaction with lithium.38
O, 290 eV) can also be detected from the XPS data for the O1s (531.5 eV). This type of the functional group on the graphene surface is well known to increase the capacity of the engineered graphene for its fast reaction with lithium.38
Cyclic voltammetry measurement (CV) was conducted to investigate the fundamental electrochemical behavior of the N-GFs ranging from 1.5 to 4.5 V versus Li/Li+ by using 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1:1) as the electrolyte at room temperature (Fig. 2a–c and S6†). Clearly, N-GFs exhibit the considerably improved electrochemical reactions of association and disassociation with Li+ compared with un-doped graphene.39 Furthermore, the electrochemical reaction of N-GF-450 is quite more active than those of N-GF-300 and N-GF-600. According to the aforementioned composition analysis, the doped N and O atoms are responsible for the electrochemical reaction. Among three samples, N-GF-600 with the least of the total of N and O contents shows the lowest electrochemical activity. With the most O content, the N-GF-300 does not show much higher gravimetric current and capacitance than N-GF-600. However, for N-GF-450, with the highest N contents, both the gravimetric current and capacitance are improved. The results indicate that the capacity of the sample is influenced by heteroatoms (mostly by N atoms) in their graphene framework. In particular, N1 and N4 on the N-GFs provide greater contribution than those of the other N species. To evaluate the reversible association and disassociation of Li+ in N-GFs, the CV of N-GF-450 at a slower scan rate of 0.1 mV s−1 is shown in Fig. 2b. In the initial cycle, three couples of redox peaks are observed at about 1.7/3.3, 3.8/4.1 and 4.2/4.3 V, resulting from the association and disassociation of Li+ with the heteroatoms (N and O) on the electrode materials. In the second cycle, the two cathodic peaks at 1.7 and 3.8 V shift to the higher potential at 1.8 and 3.9 V, respectively, hence reducing hysteresis between the anodic and cathodic peaks. This result indicates the good reversibility of the electrode.40 The redox reactions happening below and above the open-circuit voltage of 3 V suggest that the electrode has reversible p-doped (an oxidized state above 3 V vs. Li/Li+) and n-doped (a reduced state below 3 V vs. Li/Li+) redox activity. On the basis of the previously proposed lithium storage mechanism for N-rich organic electrodes,3,41 energy storage by N-GFs occurs through the reversible redox reaction accompanied by the association and disassociation of Li+ or electrolyte anions (PF6−); this mechanism can be defined as reaction (1) and (2):41–43
| [(N4)x+(PF6−)x] + xLi+ + xe− → (N1) + xLi+(PF6−) | (1) |
| (N1) + yLi+ + ye− → [(N1)y−(Li+)y] | (2) |
| (CO) + Li+ + e− → C–OLi | (3) |
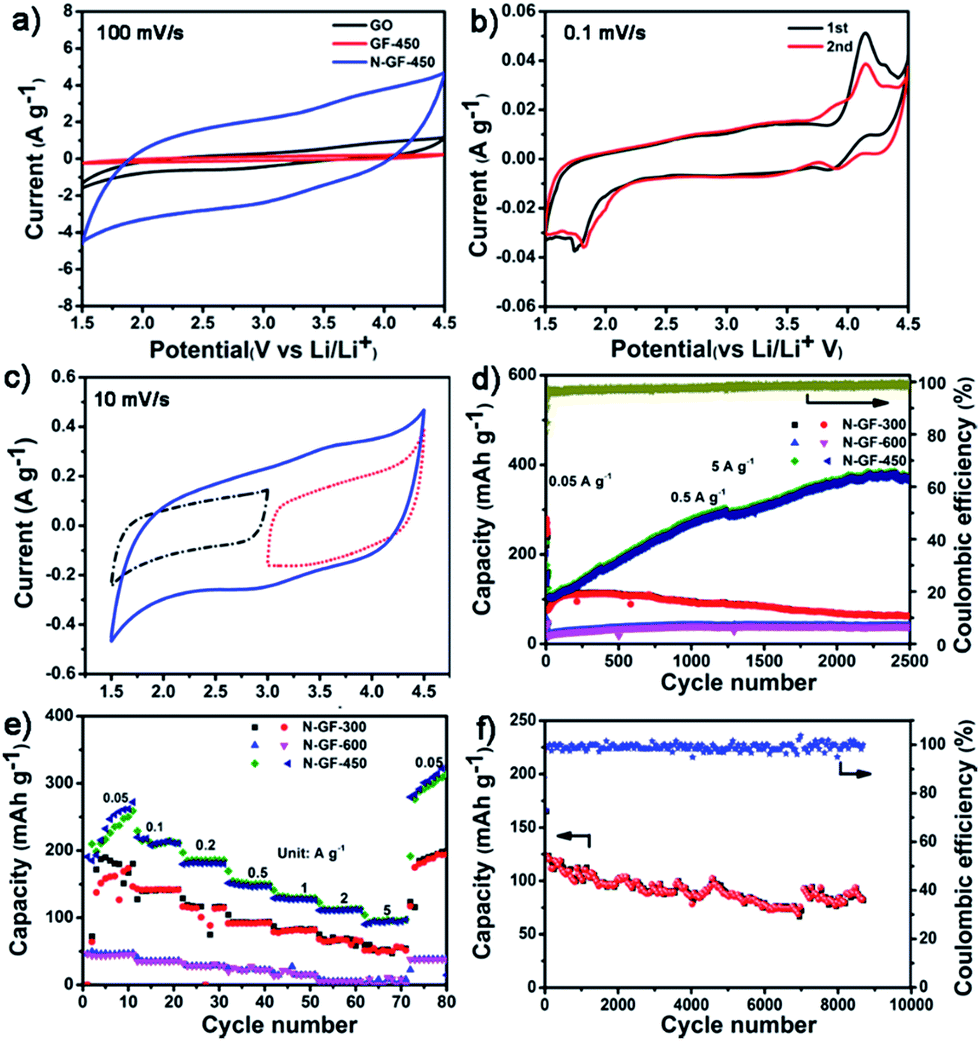 | ||
| Fig. 2 CV curves of (a) N-GF-450, GF-450, GO, N-GF-450-A and N-GF-C at the scan rate of 100 mV s−1, (b) CV curves of N-GF-450 at the scan rate of 0.1 mV s−1, (c) potential-dependent CV curves of N-GF-450 at 1.5–4.5 V, above and below the open-circuit voltage (3 V vs. Li/Li+), (d) cycling performance of the N-GFs at a current rate of 0.5 A g−1, (e) rate performance of the N-GFs at various current densities in the range of 0.05–5 A g−1 in the voltage range of 1.5–4.5 V, and (f) cycle performance of N-GF-450 up to 9000 cycles with a current density of 5.0 A g−1. | ||
The PF6− electrochemically doped in N-GF-450 at a full charge state (4.5 V, p-doped region) can be detected via FTIR spectra (Fig. S7a†). The coordination interaction between Li and N lone-pair electrons (Li–N bonding) is confirmed through XPS after full discharge to 1.5 V (Fig. S7b†),41 which indicates that the N1 and N4 atoms in the N-GFs are responsible for bipolar faradaic reactions to accommodate Li ions, and thus hold potential for the LIB cathode materials. Certainly, the CO also provides the capacity for lithium storage by faradaic reactions with lithium ions (3), similar to previously reported composites of carbon nanotubes.38,44,45 Thus a large working potential window (1.5–4.5 V versus Li/Li+) was achieved, which can thereby increase the energy density by yielding a high specific capacity, as shown in the typical profiles of voltage versus specific capacity at a current density of 0.5 A g−1 (Fig. S6a†). Furthermore, the CV profiles of N-GF-450 show a gradual deviation from the ideal rectangular shape with increased scan rate from 10 to 100 mV s−1 (Fig. 2a, S6b† and 2b), indicating the existence of both the redox reaction and double layer capacitance in the sample. In addition, the gravimetric current and the capacity increased considerably in the voltage window of 1.5–4.5 V vs. Li (blue solid curve), compared with a restricted voltage window of 1.5–3.0 V vs. Li (black dashed dotted curve) or 3.0–4.5 V vs. Li (red dotted curve) (Fig. 2c). The results indicate that the electrode cycling over the full voltage range yields much higher gravimetric currents from the faradaic reactions than double-layer capacitance.38,44
The galvanostatic discharge–charge measurements were performed on half cells at a current density of 0.5 A g−1. The capacity of N-GF-450 gradually increases from 163 to 370 mA h g−1 with almost 100% coulombic efficiency after 2500 cycles (Fig. 2d). Nevertheless, the capacities of N-GF-300 and N-GF-600 under the same conditions decrease to 60 and 40 mA h g−1, respectively. Notably, the capacity of the N-GF-450-based cathode remains at 250 mA h g−1 at a current density of 1 A g−1 even after 6500 cycles (Fig. S8†). The dramatic increase in the capacity of N-GF-450 could be attributed to the following reasons: the reversible formation and decomposition of an organic polymeric/gel-like film at the interface of the electrode materials, which provides the interfacial storage sites for excess Li+ through the pseudocapacitance-type behavior.46–48 And the full usage of the N atoms doped in the internal graphene frameworks with reactions described in eqn (1)–(3). With its extremely long cycle life and high charge–discharge rates, the superior performance of N-GF-450 undoubtedly goes far beyond that of the previously reported cathode materials (Table S1†).11,31–34,49–52 In addition, N-GF-450 exhibits excellent rate capability, delivering capacities of 259, 213, 147, 111, and 96 mA h g−1 at current densities of 0.05, 0.1, 0.5, 2, and 5 A g−1 respectively (Fig. 2e). When the current density is restored to 0.05 A g−1, N-GF-450 still achieves a high capacity of 313 mA h g−1. Moreover, N-GF-450 maintains a revisable capacity of 81 mA h g−1 for up to 9000 cycles at a remarkably high current density of 5 A g−1 (Fig. 2f). By contrast, the capacities of N-GF-300 and N-GF-600 at 5 A g−1 drastically decay to 55 and 8 mA h g−1, respectively.
Among the N-GFs in this work, the considerably pronounced electrochemical performance of N-GF-450 can be attributed to the combination of the high surface area with hierarchically porous architectures and the more N1 and N4 atoms in the graphene framework.53–56 In particular, the N-binding configuration has a remarkable effect on the Li+ storage behavior of the hybrids. According to the CV results and bipolar redox mechanism analysis, the N1 and N4 atoms in the N-GFs provide major active sites for accommodating Li ions.39 Additionally, the N4 atom has strong affinity for the PF6− anion in the electrolyte (Fig. S7a†), which can couple with Li+ when the electrode is in the oxidized state (p-doped). The presence of N atoms in the graphene frameworks can effectively reduce the charge-transfer resistance within the electrode, as verified according to the electrochemical impedance spectroscopy spectra of the samples (Fig. S9†). According to the equivalent circuit (inset Fig. S9†),39 N-GF-450 has lower contact resistance (Rf = 4.0 Ω) and charge-transfer resistance (Rct = 429.9 Ω) than other samples (Table S2†). By contrast, N-GF-600 without N4 species exhibits the highest charge-transfer resistance among the three N-GFs, which explains its low electrochemical activity as the LIB cathode.
The redox properties of N1, N2 and N3 species were estimated using density functional theory (DFT) calculation (Fig. S10†).5 The lowest-unoccupied molecular orbital (LUMO) energy level of the N1 (−1.742 eV) is slower than those of N2 (−1.611 eV) and N3 (1.646 eV). The net energy change of N1 (0.62 eV) is the highest compared to those of N2 and N3 (−1.37 and 0.14 eV, respectively), leading to the highest activity for association of Li atoms. The deeper insight into the association of Li ions with the different species of N atoms in the N-GFs is further supported through quantum mechanics and calculations. For our calculations, the association of Li ions with N-doped graphene was investigated within the framework of DFT by implementing the standard implementation in the CP2K software package.57,58 An N-doped graphene sheet with a total nitrogen doping concentration of 5 at%, of which N1, N2, and N3 atoms constituted approximately 57.4%, 35%, and 7.6%, respectively, was used in the simulation (Fig. 3a). As a first step, the interaction of Li atoms with N-doped graphene at different adsorption positions was investigated. The lithium adsorption energy was defined as:
| Eads = ELi@gr − Egr − ELi(gas phase) | (4) |
 | ||
| Fig. 3 (a) Adsorption energies of Li atoms at different positions on the N-doped graphene. Carbon, nitrogen, hydrogen, and lithium atoms are represented by the colors cyan, blue, white, and pink, respectively. (b) Calculated average voltages as a function of the Li concentration. The inset shows the top and side views of the atomic structure of Li on N-doped graphene for a Li concentration of 9 at%. | ||
The Li adsorption energies and the corresponding adsorption positions are illustrated in Fig. 3b. The most stable adsorption position is that of the Li atom between the N1 and N2 atoms, followed by the hollow sites of benzene and pyridinic rings. The Li atom on top of the N3 atom has the lowest adsorption energy. To evaluate the electrochemical activity of the Li/N-doped-graphene system, the change in the partial charge of the Li ions was calculated through Bader charge analysis. For Li atoms adsorbed between the N1 and N2 atoms, a Li partial charge of approximately +0.44e was used as an example.
Battery voltages or potential differences between the bulk lithium metal as the anode and N-doped graphene with different Li concentrations as the cathode were calculated. The voltage between the two electrodes was derived from the difference in the chemical potential of Li in the anode and cathode. At zero temperature, the average voltage V was obtained from the total energy through the formula:59
 | (5) |
Conclusions
Herein, 3D macroporous GFs coupled with N atoms were successfully developed as the cathodes for rechargeable LIBs with high capacity and a long cycle life. And their energy storage principle was further investigated by experiment and calculation, which implies that the excellent cathodic performance of the hybrids is mainly due to pyridinic N and pyridinic N-oxide in graphene. More importantly, the effect of nitrogen doping demonstrated in our study will inspire the design and development of various heteroatom doped carbon-based cathode materials for LIBs in future.Acknowledgements
This work was financially supported by the 973 Program of China (2013CBA01602 and 2014CB239701), Natural Science Foundation of China (21320102006, 61235007, 61575121, 21572132 and 21372155), Professor of Special Appointment at Shanghai Institutions of Higher Learning (Eastern Scholar), China Postdoctoral Science Foundation ( KLH1615152), and MPI-SJTU Partner Group Project for Polymer Chemistry of Graphene Nanoribbons. We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for some Measurements and the Center of Information Services and High Performance Computing (ZIH) at TU Dresden for computational resources.Notes and references
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS PubMed.
- K. Higashiguchi, K. Yasui and H. Kikuchi, J. Am. Chem. Soc., 2008, 130, 6326–6327 CrossRef CAS PubMed.
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J.-I. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS PubMed.
- A. Vu and A. Stein, Chem. Mater., 2011, 23, 3237–3245 CrossRef CAS.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- T. Song, J. Xia, J.-H. Lee, D. H. Lee, M.-S. Kwon, J.-M. Choi, J. Wu, S. K. Doo, H. Chang, W. I. Park, D. S. Zang, H. Kim, Y. Huang, K.-C. Hwang, J. A. Rogers and U. Paik, Nano Lett., 2010, 10, 1710–1716 CrossRef CAS PubMed.
- C. Luo, R. Huang, R. Kevorkyants, M. Pavanello, H. He and C. Wang, Nano Lett., 2014, 14, 1596–1602 CrossRef CAS PubMed.
- H. Wu, Q. Meng, Q. Yang, M. Zhang, K. Lu and Z. Wei, Adv. Mater., 2015, 27, 6504–6510 CrossRef CAS PubMed.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef.
- P. J. Nigrey, D. MacInnes, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Electrochem. Soc., 1981, 128, 1651–1654 CrossRef CAS.
- K. Sakaushi, E. Hosono, G. Nickerl, H. Zhou, S. Kaskel and J. Eckert, J. Power Sources, 2014, 245, 553–556 CrossRef CAS.
- P. Xiong, B. Liu, V. Teran, Y. Zhao, L. Peng, X. Wang and G. Yu, ACS Nano, 2014, 8, 8610–8616 CrossRef CAS PubMed.
- M. Srivastava, J. Singh, T. Kuila, R. K. Layek, N. H. Kim and J. H. Lee, Nanoscale, 2015, 7, 4820–4868 RSC.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Manivannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS PubMed.
- Y. Meng, H. Wu, Y. Zhang and Z. Wei, J. Mater. Chem. A, 2014, 2, 10842–10846 CAS.
- H. Kim, K.-Y. Park, J. Hong and K. Kang, Sci. Rep., 2014, 4, 5278 CAS.
- J. Yang, J. Wang, Y. Tang, D. Wang, X. Li, Y. Hu, R. Li, G. Liang, T.-K. Sham and X. Sun, Energy Environ. Sci., 2013, 6, 1521–1528 CAS.
- H. Kim, H.-D. Lim, S.-W. Kim, J. Hong, D.-H. Seo, D.-C. Kim, S. Jeon, S. Park and K. Kang, Sci. Rep., 2013, 3, 1506 Search PubMed.
- W. Ai, Z. Du, Z. Fan, J. Jiang, Y. Wang, H. Zhang, L. Xie, W. Huang and T. Yu, Carbon, 2014, 76, 148–154 CrossRef CAS.
- Y. Xie, L. Fang, H. Cheng, C. Hu, H. Zhao, J. Xu, J. Fang, X. Lu and J. Zhang, J. Mater. Chem. A, 2016, 4, 15612–15620 CAS.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef CAS PubMed.
- Y. Meng, K. Wang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2011, 134, 15–18 CrossRef PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed.
- F. Xu, X. Chen, Z. Tang, D. Wu, R. Fu and D. Jiang, Chem. Commun., 2014, 50, 4788–4790 RSC.
- S. Han, J. Wang, S. Li, D. Wu and X. Feng, J. Mater. Chem. A, 2014, 2, 6174–6179 CAS.
- Y. Bai, K. Jiang, S. Sun, Q. Wu, X. Lu and N. Wan, Electrochim. Acta, 2014, 134, 347–354 CrossRef CAS.
- C. Li, X. Mu, P. A. van Aken and J. Maier, Adv. Energy Mater., 2013, 3, 113–119 CrossRef CAS.
- M. Lee, J. Hong, H. Kim, H.-D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26, 2558–2565 CrossRef CAS PubMed.
- Y. Huang, D. Wu, J. Wang, S. Han, L. Lv, F. Zhang and X. Feng, Small, 2014, 10, 2226–2232 CrossRef CAS PubMed.
- A. G. Zestos, C. B. Jacobs, E. Trikantzopoulos, A. E. Ross and B. J. Venton, Anal. Chem., 2014, 86, 8568–8575 CrossRef CAS PubMed.
- C. Hu, H. Chen, Y. Xie, L. Fang, J. Fang, J. Xu, H. Zhao and J. Zhang, J. Mater. Chem. A, 2016, 4, 18284–18288 CAS.
- H. R. Byon, B. M. Gallant, S. W. Lee and Y. Shao-Horn, Adv. Funct. Mater., 2013, 23, 1037–1045 CrossRef CAS.
- K. Sakaushi, G. Nickerl, F. M. Wisser, D. Nishio-Hamane, E. Hosono, H. Zhou, S. Kaskel and J. Eckert, Angew. Chem., Int. Ed., 2012, 51, 7850–7854 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812–1816 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef PubMed.
- A. L. M. Reddy, S. Nagarajan, P. Chumyim, S. R. Gowda, P. Pradhan, S. R. Jadhav, M. Dubey, G. John and P. M. Ajayan, Sci. Rep., 2012, 2, 960 Search PubMed.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B.-S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252–5256 CAS.
- J.-Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 3096–3101 CrossRef CAS PubMed.
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed.
- K. Zhang, C. Guo, Q. Zhao, Z. Niu and J. Chen, Adv. Sci., 2015 DOI:10.1002/advs.201500018.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015 DOI:10.1002/advs.201500124.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- Y. Mao, H. Duan, B. Xu, L. Zhang, Y. Hu, C. Zhao, Z. Wang, L. Chen and Y. Yang, Energy Environ. Sci., 2012, 5, 7950–7955 CAS.
- H. Zhu, X. Wang, X. Liu and X. Yang, Adv. Mater., 2012, 24, 6524–6529 CrossRef CAS PubMed.
- J. V. Vondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. V. Vondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta09161j |
| This journal is © The Royal Society of Chemistry 2017 |