DOI:
10.1039/C6TA07997K
(Paper)
J. Mater. Chem. A, 2016,
4, 17809-17818
WO3 nanosponge photoanodes with high applied bias photon-to-current efficiency for solar hydrogen and peroxydisulfate production†
Received
15th September 2016
, Accepted 27th October 2016
First published on 7th November 2016
Abstract
We prepared WO3 nanosponge photoanodes by nanoparticle/solution hybrid dispersion–deposition. The WO3 nanosponge photoanodes showed a large photocurrent (3.04 mA cm−2) in 1.0 M H2SO4 electrolyte under 1 Sun illumination. This high photocurrent arose from the robust inter-nanoparticle connections and preservation of the nanopores in the photoanode, achieved by the addition of tungsten phenoxide and the very fine nanoparticles obtained by wet milling. The absorbed photon-to-current efficiency spectra showed a high maximum of 95.4% at 410 nm. The Faraday efficiency of S2O82− and H2 evolution was almost 100%, and the applied bias photon-to-current efficiency for S2O82− and H2 was calculated to be 2.45%, which is the highest reported value. Obtaining this kind of high-value-added product efficiently at photoanodes in photoelectrochemical systems is important for the use of sunlight in sustainable industrial chemistry.
Introduction
Solar hydrogen production has great potential as a sustainable green energy source.1–6 In particular, the use of semiconductor photoelectrodes for solar water splitting has been studied extensively,7–11 because photoelectrochemical (PEC) systems have the following major advantages for water splitting: (1) easing the energy level restrictions in semiconductor photoelectrodes by using an external bias voltage; and (2) allowing charge separation and straightforward product collection at separate reaction sites for oxidation (anode) and reduction (cathode). There have been numerous reports on water splitting in PEC systems,12–19 and TiO2 and WO3 photoelectrodes have been widely used because of their high chemical stability. Although pure TiO2 shows high activity under only ultraviolet (UV) light due to its wide band gap at 3.2 eV,20–22 black TiO2, to which hydrogen and anions (S, N, and I), or oxygen deficiencies have been introduced, has achieved an enhanced photoresponse, even under sunlight.23–25 Black TiO2 photoelectrode systems showed an increase in the solar-to-hydrogen (STH) efficiency of 1.5–2.0%.
Solar hydrogen production from water is an important goal for energy production; however, the current STH efficiency is still not commercially feasible. The STH efficiency should stably exceed 10–20%.26,27 Thus, we have been searching for other applications of photoelectrodes driven by sunlight. Solar hydrogen production in PEC systems is a cathode reaction, and oxygen gas is obtained at the photoanodes. Oxygen gas has a market value less than several tens of percent that of hydrogen gas, and few studies have focused on the other products besides oxygen at photoanodes. However, Mi et al. reported that some products, such as peroxydisulfate (S2O82−) and H2O2, can be obtained at WO3 photoanodes in aqueous electrolytes.28 Fuku et al. obtained Ce4+, IO4−, and H2O2 in the WO3 photoanode system in addition to S2O82−.29,30 These reagents have a much higher market value than oxygen. In this work, we focused on the availability of these high-value-added anode products obtained by PEC solar hydrogen production. For example, S2O82− is important as an industrial bleaching and oxidising reagent, and it generally has a market price more than one hundred times that of oxygen. Therefore, peroxydisulfate evolution at the photoanode in addition to hydrogen gas evolution at the cathode is a possible application of sunlight. The peroxydisulfate ions were evolved by an anion-derived hole transfer process in accordance with the following equation reported by Mi et al.:28
| | | 2HSO4− → S2O82− + 2H+ + 2e−, S2O82−/HSO4− = +2.12 V vs. RHE | (1) |
The present highest applied bias photon-to-current efficiency for S2O82− and H2 (ABPES2O82−) is 2.2% in WO3 photoanodes.29 WO3 photoanodes have a narrower band gap (2.6–2.8 eV) than TiO2 and are highly stable under acidic conditions. In addition, they have a deep valence band potential at +3.0 eV; thus, WO3 photoanodes are suitable for obtaining various oxidants under sunlight. However, the reported value of the photocurrent in the WO3 photoanodes (single junction) for water splitting15,31,32 generally remains 1.0–2.5 mA cm−2 under simulated sunlight (AM1.5G), which is far below the theoretical limit33 of the photocurrent of other semiconductor photoanodes. Therefore, the fabrication process for WO3 photoanodes must be improved to realize the potential of the material. We refocused on effective nanostructures for maximizing PEC properties in the WO3. The excited electron/hole transporting property is one of the most important key factors for enhancing PEC reactions. While there are many reports on a fabrication process for inter-particle necking to enhance transporting properties in TiO2 photoelectrodes,34,35 it has been rarely studied in the WO3. In this study, we have developed a new WO3 nanoparticle/solution (NPS) hybrid dispersion–deposition process to make rigid inter-particle connections in the WO3 nanostructure. The obtained WO3 nanosponge photoanodes prepared by using this fabrication process showed very high photocurrent and a maximum ABPES2O82− of 2.45%.
Experimental procedure
Preparation and characterisation of WO3 nanosponge layers
A WO3 nanoparticle dispersion was prepared by wet-ball milling of WO3 nanocrystal powders (EM Japan) in isopropanol (IPA) with a tilted rotation planetary ball mill (Planet M, Nagao System) using zirconia balls at 650 rpm with or without a toluene solution of tungsten phenoxide (Gelest) as a source of W ions in the dispersion ink. The dispersion containing WO3 nanocrystals and W ion solution is called WO3 NPS hybrid dispersion ink. Polyethylene glycol (PEG; molecular weight: 300, Wako Pure Chemical) was added to the WO3 dispersion ink in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. The WO3 dispersion ink with PEG was spin-coated at 2000 rpm for 10 s onto F-doped SnO2 (FTO) glass substrates. The films were preheated at 500 °C for 5 min. The coating and preheating were repeated several times to increase the film thickness, and then the preheated films were fired at 550 °C for 30 min in air. The preparation conditions and film thickness of representative photoanodes are shown in Table 1. The phase purity and crystallinity of the WO3 thin films on the FTO substrates were determined by X-ray diffraction (XRD; SmartLab, Rigaku). The distribution of particle size (d) for the WO3 dispersion was checked with a particle size analyser (ELSZ-1000, Otsuka Electronics). The surface morphology and cross-sectional structure were determined by field emission scanning electron microscopy (FESEM; SU9000, Hitachi). The optical absorbance spectra and light harvesting efficiency (LHE) of the WO3 films were evaluated by using a UV-visible spectrometer (UV-3150, Shimadzu) equipped with an integrating sphere (MPC-3100, Shimadzu). X-ray photoemission spectroscopy (XPS) was carried out with an Al Kα source.
:1 volume ratio. The WO3 dispersion ink with PEG was spin-coated at 2000 rpm for 10 s onto F-doped SnO2 (FTO) glass substrates. The films were preheated at 500 °C for 5 min. The coating and preheating were repeated several times to increase the film thickness, and then the preheated films were fired at 550 °C for 30 min in air. The preparation conditions and film thickness of representative photoanodes are shown in Table 1. The phase purity and crystallinity of the WO3 thin films on the FTO substrates were determined by X-ray diffraction (XRD; SmartLab, Rigaku). The distribution of particle size (d) for the WO3 dispersion was checked with a particle size analyser (ELSZ-1000, Otsuka Electronics). The surface morphology and cross-sectional structure were determined by field emission scanning electron microscopy (FESEM; SU9000, Hitachi). The optical absorbance spectra and light harvesting efficiency (LHE) of the WO3 films were evaluated by using a UV-visible spectrometer (UV-3150, Shimadzu) equipped with an integrating sphere (MPC-3100, Shimadzu). X-ray photoemission spectroscopy (XPS) was carried out with an Al Kα source.
Table 1 Preparation conditions and thickness of representative photoanodes
| Sample |
Thickness/μm |
Dispersion |
WO3 nanoparticle/wt% |
W phenoxide/mM |
Milling time/min |
| W1 |
7.3(4) |
D1 |
10 |
0.0 |
15 |
| W2 |
7.3(2) |
D2 |
23 |
0.0 |
360 |
| W3 |
7.4(5) |
D3 |
23 |
5.0 |
360 |
PEC performance evaluation
Linear sweep voltammetry for solar water splitting photocurrent measurements was performed under 100 mW cm−2 AM 1.5G simulated sunlight (1 Sun) from a 150 W Xe lamp (XES-40S2-CE, San-Ei Electric), in 1.0 M H2SO4 (pH = 0.0) at 0.4–2.0 V vs. a reversible hydrogen electrode (VRHE) in a three-electrode electrochemical cell equipped with a quartz glass window. The 1 Sun simulated sunlight was calibrated by using a spectroradiometer (Fig. S1;† SOMA Optics). An Ag/AgCl reference electrode in saturated KCl solution and a Pt wire counter electrode were used, and the scan rate was 5 mV s−1. The measured potential vs. Ag/AgCl (VAg/AgCl) in the three-electrode system was converted to VRHE according to VRHE = VAg/AgCl + 0.059pH + V0Ag/AgCl, where V0Ag/AgCl = 0.1976 at 25 °C. Mott–Schottky plots were obtained by using a potentiostat (Autolab PGSTAT204, Metrohm) equipped with an impedance analyser (Autolab FRA32M, Metrohm) in 1 M H2SO4. The excitation voltage and frequency were 10 mV and 1 kHz, respectively. Intensity modulated photocurrent spectroscopy (IMPS) was performed with a potentiostat (PGSTAT204, Metrohm), an impedance analyser (FRA32M, Metrohm), and a light-emitting diode (LED) driver kit (Metrohm) that drove a 365 nm power UV LED (NCSU033B, Hamamatsu Photonics). The LED intensity was modulated by 10% in the range of 100 kHz–0.1 Hz. Incident photon-to-current conversion efficiency (IPCE) was evaluated by using the three-electrode setup with the equation IPCE = 1240J/λI, where λ is the incident light wavelength (nm), and I is the intensity of the light source at each wavelength (mW cm−2). The absorbed photon-to-current efficiency (APCE) was calculated by APCE = IPCE/LHE. The bias voltage was 1.50VRHE. The monochromatic light was generated by a Xe lamp with a monochromator (SPG-120S-REV, Shimadzu). The ABPES2O82− of the WO3 photoanodes was measured by using a two-electrode configuration cell with a Pt wire counter electrode and an ion-exchange membrane (Nafion). ABPES2O82− values were estimated by ABPES2O82− = J(2.12 − ECE)η(S2O82−)/IAM1.5, where J is the photocurrent density (mA cm−2), ECE is the applied bias voltage vs. the counter electrode, η(S2O82−) is the Faraday efficiency of S2O82−production, and IAM1.5 is the irradiance of AM 1.5 simulated sunlight at 100 mW cm−2. The production of S2O82− was evaluated by colorimetry by using UV-VIS spectroscopy at 310 nm after the reaction with FeSO4 solution (ESI†). The evolved H2 gas was detected by using a thermal conductivity detector gas chromatograph (GC-8AIT, Shimadzu).
Results and discussion
Preparation and structural/optical properties of WO3 nanosponge photoanodes
Fig. 1 shows the WO3 nanoparticle morphology of the original and wet-ball milled samples (360 min). The original WO3 nanoparticle diameter (d) was 20–200 nm (Fig. 1a), and included big nanoparticles with d ≈ 100–200 nm and small nanoparticles with d ≈ 20–50 nm. The original nanoparticles were wet-ball milled into smaller particles with a diameter of 5–150 nm (Fig. 1b), and the bimodal size distribution remained, with big nanoparticles with d ≈ 100–150 nm and very small granular nanoparticles with d ≈ 5–20 nm.
 |
| | Fig. 1 FESEM images of nanoparticle morphology of WO3 nanoparticles (a) before and (b) after wet-ball milling for 360 min. | |
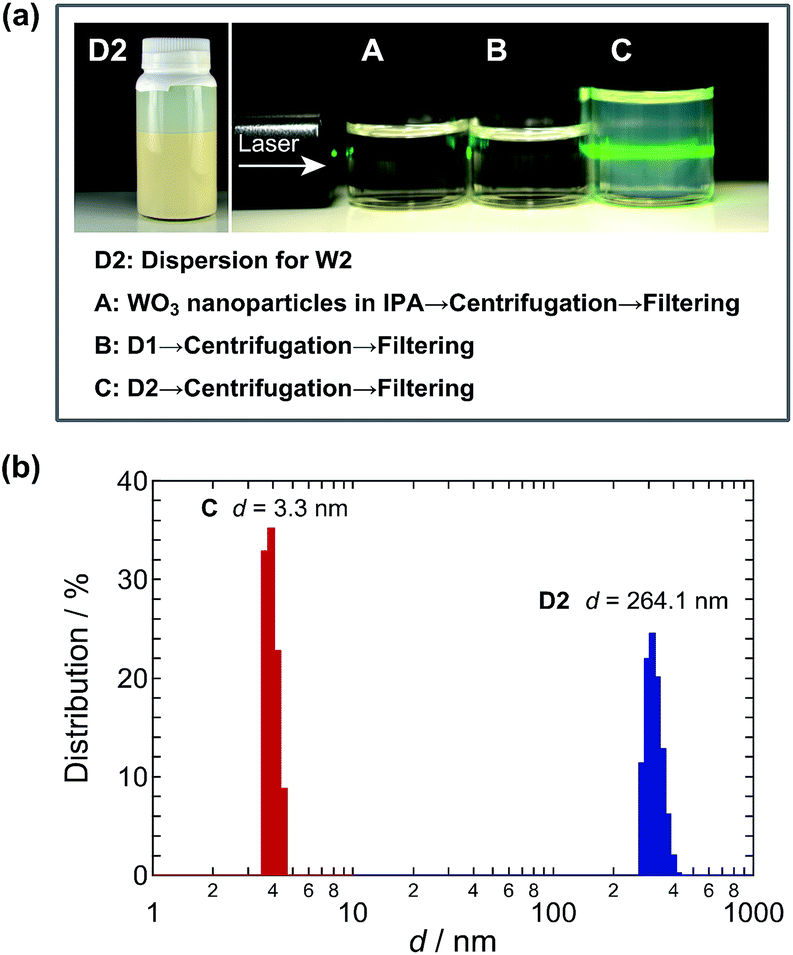
Fig. 2a shows photographs of the WO3 nanoparticle dispersions. Dispersion D2, used to fabricate photoanode W2, was beige, and the dispersed nanoparticle size was evaluated by using a particle size analyzer to be mainly 264.1 nm in the range of 250–400 nm (Fig. 2b). This value indicates that the nanoparticles aggregated and were detected as secondary particles. We examined the effect of milling by preparing dispersions A, B, and C. Dispersion A was obtained by centrifuging and filtering (100 nm pore size) the original WO3 nanoparticle dispersion in IPA (WO3 nanoparticles were stirred vigorously in IPA). Dispersion B was obtained by centrifuging and filtering (100 nm pore size) dispersion D1, used to fabricate photoanode W1. Dispersion C was obtained by centrifuging and filtering (100 nm pore size) dispersion D2, used to fabricate photoanode W2. Dispersions A and B were completely transparent; however, a light suspension was visible in dispersion C. Under green laser irradiation, this colloidal suspension exhibited Tyndall scattering (Fig. 2a) that originated from very fine, highly monodisperse WO3 nanoparticles with d = 3.3 nm, which corresponds to 4–5 unit cells of WO3 (Fig. 2b). This means that wet-ball milling for 360 min produced very fine nanoparticles in the dispersion, whereas milling for 15 min did not. In the D3 dispersion (the WO3 NPS hybrid dispersion) for the W3, the solvated W ions from tungsten phenoxide were added to the WO3 nanoparticle dispersion for D2 that contained very fine nanoparticles as seen in dispersion C.
 |
| | Fig. 2 (a) Photographs of the (O) WO3 nanoparticle dispersion used for photoanode W2 and centrifuged/filtered dispersions A, B and C. The original sources of A, B and C were as follows: (A) original WO3 nanoparticle dispersion in IPA (WO3 nanoparticles were stirred vigorously in IPA), (B) dispersion W1 and (C) dispersion W2. The filtering of the centrifuged supernatant liquid was carried out by using 100 nm pore filters. (b) Particle diameter (d) distribution for the dispersion O and C. | |
The crystallinity and structure of photoanodes W1, W2, and W3, and the original WO3 nanoparticles were analyzed by XRD (Fig. 3). The XRD pattern of the original WO3 nanoparticles was from a pure γ-phase of WO3 with a monoclinic P21/c space group. The diffraction peaks were fitted without a specific crystal orientation by the Rietveld method by using RIETAN-FP,36 and the unit cell parameters were refined as a = 7.3204(5) Å, b = 7.5301(5) Å, c = 7.6917(5) Å, and β = 90.67(1)°, which were almost consistent with the reported values (JCPDS 20-1324 (ref. 37)). Photoanodes W1, W2, and W3 crystallised at 550 °C in the pure γ-phase of WO3. Photoanode W3 showed preferential (001)-orientation growth, whereas photoanodes W1 and W2 exhibited almost no specific orientation.
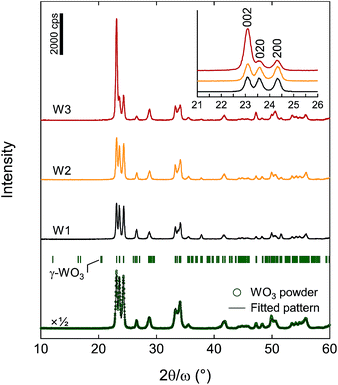 |
| | Fig. 3 XRD patterns of photoanodes W1, W2, and W3, and the original WO3 nanoparticles. The diffraction pattern of the WO3 nanoparticles was fitted by the Rietveld method. The vertical marks indicate the positions calculated for the Bragg reflections for γ-WO3. | |
Fig. 4 shows the FESEM for photoanodes W1, W2, and W3 at the WO3 film surface and the interface between the WO3 and FTO substrate. All the photoanodes had a granular morphology and 5–100 nm nanopores between the nanoparticles. Faceted-grown plate-like nanoparticles grew on the top surface of W3 (Fig. 4e), although the plate-like particles did not have a specific orientation in the films, as observed in the cross-sectional view (Fig. 4f). In contrast, no faceted-grown nanoparticles were observed in photoanodes W1 and W2, and they were more spherical (Fig. 4a–d). The preferential (001)-orientation growth observed in the XRD pattern of photoanode W3 originated from these faceted-grown (001)-face up plate-like nanoparticles on the surface. Therefore, there was an additional driving force for the crystal growth in photoanode W3. The W ions introduced by adding tungsten phenoxide to the WO3 NPS hybrid dispersion ink spread effectively on the nanoparticles during firing, and acted as an accelerator for crystal surface reconstruction, leading to the faceted growth. Another effect of tungsten phenoxide addition appeared at the interface between the WO3 film and FTO glass substrate. In photoanode W3, very fine WO3 nanoparticles covered the FTO glass surface, which was due to crystal nucleation from tungsten phenoxide solution. However, the interfaces had hollows with a size of approximately 100 nm in places in photoanodes W1 and W2. The high coverage of the top surface of the FTO bottom electrode facilitated effective excited electron transfer from the WO3 layer.
 |
| | Fig. 4 FESEM images of the top surface of the WO3 film and interface at the FTO glass substrate of the (a and b) W1, (c and d) W2 and (e and f) W3 films. | |
The cross-sectional FESEM image of the whole of photoanode W3, including the FTO bottom electrode, is shown in Fig. 5a. The WO3 film thickness was 7.4 μm with large numbers of nanopores (Fig. 4f and 5b), and the morphology was like a nanosponge. Microscopically the nanosponge consisted of large plate-like nanoparticles with an average size of 117 nm, and small granular nanoparticles with an average size of 23 nm (Fig. 5b). The large plate-like nanoparticles were bound by the small nanoparticles, and each particle had strong connections with distinct crystal grain boundaries. Thus, photoanode W3 exhibited the good crystallinity of WO3 and finely connected nanoparticle networks with large numbers of nanopores. These features are desirable for excited electron/hole transfer.
 |
| | Fig. 5 Cross-sectional view of FESEM for photoanode W3. (a) Broad overview and (b) enlarged view of WO3 nanoparticles. The white arrows indicate the grain boundaries of WO3 nanoparticles. | |
Fig. 6 shows Tauc plots for photoanodes W1, W2, and W3, and original WO3 nanoparticles that were evaluated from the transparency spectra obtained by using an integrating sphere. The band gap energy (Eg) of the source nanoparticles of 2.61 eV was narrow compared with the literature value of WO3 of 2.8 eV, indicating that there were defects in the crystal lattice of the nanoparticles. Photoanode W1 was fabricated from nanoparticle dispersion D1 that was subjected to short wet-ball milling (15 min), and firing at 550 °C recovered these defects to some extent. However, the finely crushed nanoparticles obtained by long wet-ball milling (360 min) that were used to fabricate photoanodes W2 and W3 retained slight surface defects owing to the large surface area of the very small diameter particles. Moreover, photoanode W3 contained small nanoparticles that originated from the pyrolysis and crystallisation of tungsten phenoxide. This increased the amount of very small nanoparticles, leading to a slight red shift in the absorption edge. The surface oxygen deficiency varied slightly between photoanode W1 and photoanodes W2 and W3. The reduced W5+ state was not detected by XPS by strong W6+ signals (Fig. S2†).38 This means that there should be fewer oxygen defects than in photoanodes treated in reducing atmospheres.39
 |
| | Fig. 6 Tauc plots for (a) WO3 nanoparticles, (b) W1, (c) W2, and (d) W3. | |
Fig. 7a shows the absorbance spectra of photoanodes W1, W2, and W3 collected with and without the integrating sphere. The optical band edges of each photoanode corresponded to the results of the Tauc plots (Fig. 6). However, a big difference was observed between the absorbance spectra of photoanode W1 and photoanodes W2 and W3 measured with and without the integrating sphere at wavelengths longer than 500 nm. In photoanode W1, a large pseudo-absorption was observed that arose from the incident light diffusion of the WO3 layer above 500 nm. However, the absorption above 500 nm in photoanodes W2 and W3 was much smaller than that of photoanode W1 owing to the small light diffusion of the WO3 layers. Photoanode W1 was light yellow compared with photoanodes W2 and W3 due to light diffusion (Fig. 7b). The incident white light (AM1.5G) was changed to red-orange by the relatively large transmission above 500 nm in photoanodes W2 and W3. The small light diffusion indicates that there were robust interconnections at the grain boundaries of the nanoparticles. Although the addition of tungsten phenoxide would affect the inter-nanoparticle bonding, the very small nanoparticles produced by the long wet-milling time of WO3 nanoparticles (Fig. 2) would also play an important role by lowering the crystal growth temperature. The very small particles can reduce the melting temperature, as described by40
| |  | (2) |
where Δ
T is the melting point depression,
γls is the solid–liquid interface energy,
Tm is the bulk melting point,
ρs is the density of the solid, Δ
Hf is the bulk heat of fusion, and
r is the particle radius. Hence, Δ
T of the small nanoparticles (
d ≈ 3.3 nm) was 30 to 45 times higher than that of large nanoparticles (
d ≈ 100–150 nm) according to this equation. It is unclear whether the small nanoparticles actually melted at 550 °C. However, the reactivity for liquid–solid or solid–solid sintering is increased by very small nanoparticles between the large nanoparticles in addition to the new nucleation and crystal growth by tungsten ions around the nanoparticles derived from the tungsten phenoxide solution (
Fig. 8). Thus, we prepared WO
3 nanosponge layers with sufficient inter-nanoparticle bonding and many nanopores.
 |
| | Fig. 7 (a) Absorption spectra of photoanodes W1, W2, and W3 collected with and without the integrating sphere. (b) Photographic images of W1, W2, and W3 photoanodes (top panel), and transmitted white light (AM1.5G) from the photoanodes through the photomask (bottom panel, TLS: transmitted simulated sunlight). | |
 |
| | Fig. 8 Schematic illustration of crystal growth of W3 by NPS hybrid dispersion ink. | |
PEC properties of WO3 nanosponge photoanodes for hydrogen and peroxydisulfate evolution under simulated sunlight
To evaluate the preparation conditions for optimising the PEC properties, we first examined the photocurrent density (J) of the ca. 2 μm-thick WO3 nanosponge photoanodes in aqueous 1 M H2SO4 solution under 1 Sun simulated sunlight from the front of the photoanodes at 1.5VRHE. Fig. 9 shows the dependence of J on the concentration of tungsten phenoxide in the WO3 NPS hybrid dispersions (cw), the wet-milling time (tmill), and the concentration of WO3 nanoparticles in the dispersions (cp). cw reached a maximum at 5 mM, and then J decreased with increasing concentration. This indicates that adding tungsten phenoxide improved the crystal quality of WO3 nanosponges (Fig. 4f and 5). Excess tungsten phenoxide addition could reduce the surface area by filling the nanopores. We measured tmill up to 360 min, and a long tmill increased J because of the production of very fine nanoparticles that improved the inter-nanoparticle bonding. cp affected the coating process. A high particle concentration was unsuitable for obtaining a homogeneous coating, and the optimum cp was 10–25%. Thus, we concluded that the optimum conditions for a WO3 nanosponge with high J were cw = 5 mM, tmill = 360 min, and cp = 23 wt% (dispersion D3 in Table 1).
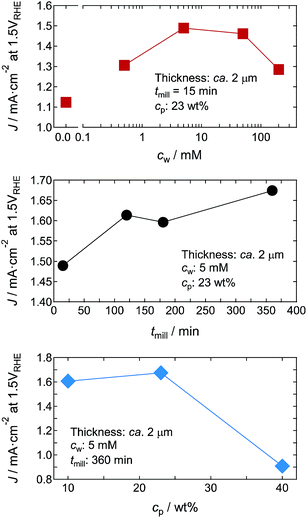 |
| | Fig. 9
c
w, tmill, and cp dependences of J for WO3 nanosponge photoanodes ca. 2 μm thick in aqueous 1 M H2SO4 solution under 1 Sun simulated sunlight from the front of the photoanodes at 1.5VRHE. | |
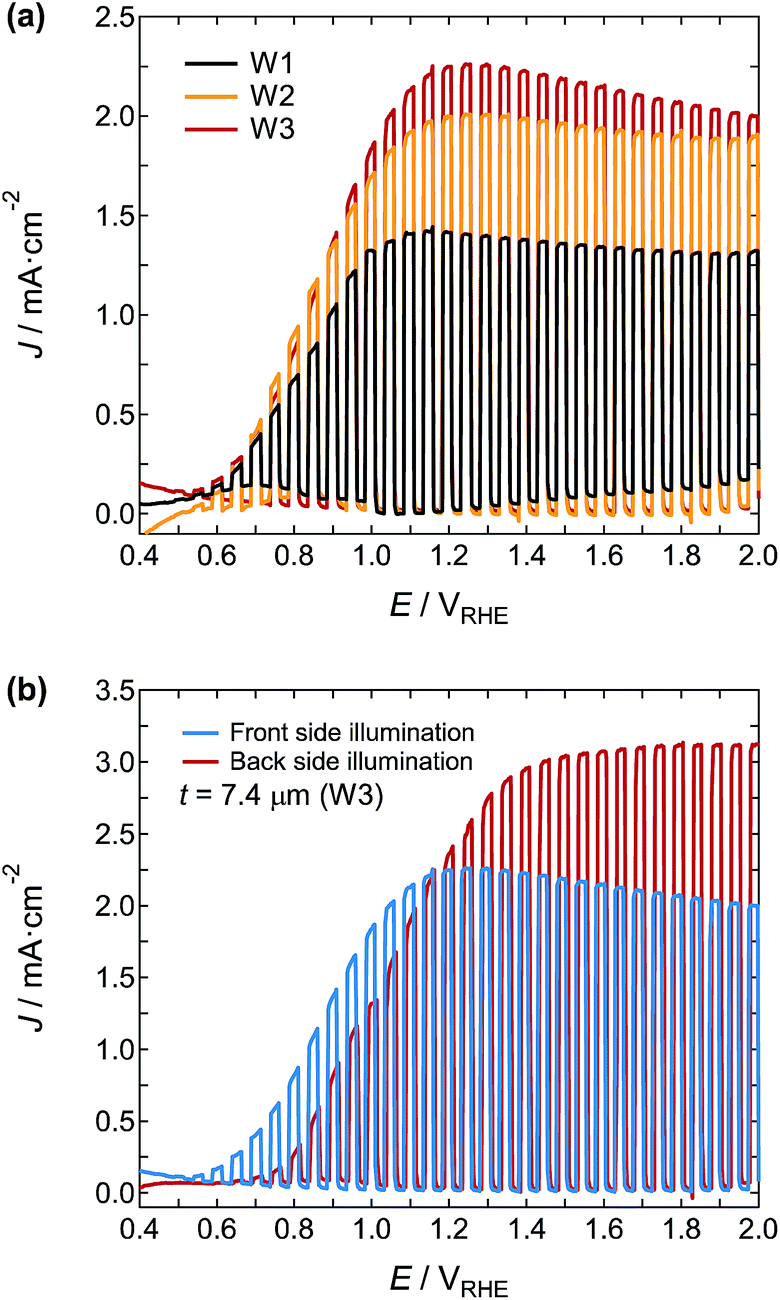
Fig. 10 shows the film thickness dependence of J under 1 Sun simulated sunlight from the front and back of the photoanodes at 1.5VRHE for the WO3 nanosponge photoanodes W1 and W3 prepared with dispersions D1 and D3, respectively (Table 1). For photoanode W1 prepared with dispersion D1, J measured under back illumination had a maximum (Jmax) of 1.58 mA cm−2 at 4.8 μm (Fig. 10a). Under front illumination, J was saturated up to 6–8 μm, and Jmax was 1.22 mA cm−2 at 5.8 μm. For WO3 nanosponge photoanode W3 prepared by using dispersion D3, which was obtained by a long wet-milling time with tungsten phenoxide, J was markedly enhanced. Under front illumination, J was saturated up to 7 μm, and Jmax was 2.28 mA cm−2 (average of 10 samples) at 7.4 μm (87% higher than Jmax for photoanode W1). The J under the front illumination did not decrease below 15 μm; however, it will start decreasing at thicker thickness because of the limit of excitation penetration depth and long transporting distance of excited electrons. Under back illumination, J reached a maximum at 6–8 μm, and a Jmax of 3.01 mA cm−2 was high (average of 10 samples) at 7.4 μm (91% higher than Jmax for W1). Back illumination produces a high photocurrent because the photoexcited electrons can be transferred smoothly to the FTO bottom electrode.41,42 The thickness dependence of J should be determined mainly by a competition between the light absorption and excited electron/hole transfer. The photoresponse of WO3 nanosponges prepared by the D3 NPS hybrid dispersion showed a slight anodic shift with the increasing film thickness, and the thick WO3 photoanode (14.4 μm) exhibited a gradual increase of the J–V curve (Fig. S3†). This indicated that too high photocarrier density near the bottom electrode due to the large film thickness could cause sluggish transport of excited electrons, resulting in the appearance of Jmax in the thickness dependence of J. Nevertheless, Jmax in the photoanode W3 prepared from dispersion D3 appeared thicker than photoanode W1 prepared from dispersion D1, suggesting that the WO3 layer prepared from dispersion D3 effectively collected photoexcited electrons further from the bottom electrode. This arose from the good inter-particle connections that allowed photocarrier transportation.
 |
| | Fig. 10 Thickness dependence of J at 1.5VRHE in 1 M H2SO4 electrolyte for the WO3 films prepared from nanoparticle dispersions (a) D1 and (b) D3. The front and back of the WO3 photoanodes were illuminated with incident light. | |
The J–V curves under chopped simulated sunlight at 1 Sun from the front of photoanodes W1, W2, and W3 were plotted (Fig. 11a). The onset potential of each photoanode was very similar at around 0.49VRHE, and the J–V curves were saturated at around 1.1–1.2VRHE. The J of W3 at 1.2VRHE was 2.25 mA cm−2, which was 60% and 13% higher than that of W1 and W2, respectively. Fig. 11b shows the J–V curves of the W3 under excitation from the front and back illumination. Although the onset potential under back illumination showed an anodic shift to 0.63VRHE, J increased markedly to 3.04 mA cm−2 at 1.50VRHE. This value is very high for a single junction photoanode, and reaches approximately half the theoretical limit of J under AM 1.5G illumination for the stoichiometric WO3 with a band gap of 2.8 eV. The reported photocurrent densities of WO3 photoanodes by using H2SO4 electrolyte are summarized in Table 2.
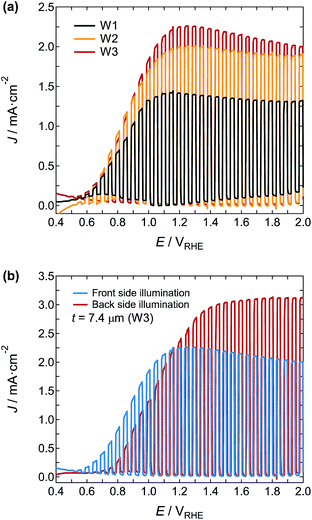 |
| | Fig. 11 (a) J–V curves under chopped simulated sunlight at 1 Sun in 1 M H2SO4 electrolyte from the front of the W1, W2, and W3 photoanodes. (b) LSV curves of the W3 photoanode illuminated with excitation light from the front and back. | |
Table 2 Preparation conditions, nanostructure and PEC properties of WO3 photoanodes in H2SO4 electrolyte
| Preparation method |
Nanostructure morphologya |
Conc.b |
Light source |
J/mA cm−2 |
Ref. |
|
d and t represent the particle size and film thickness, respectively.
Concentration of H2SO4 electrolyte.
AM 1.5G: 100 mW cm−2.
|
| Chemical vapor deposition |
Nanowire |
0.5M |
AM1.5Gc |
1.2 (1.2 V vs. SCE) |
43
|
| Hydrothermal |
Spherical nanoparticles (d = 60 nm) |
0.5M |
AM1.5Gc |
0.6 (1.2 V vs. NHE) |
44
|
| Hydrothermal |
Nanobar building blocks (d = 60 nm) |
0.5M |
AM1.5Gc |
1.87 (1.6 V vs. Ag/AgCl) |
45
|
| Pulsed laser deposition |
Nanotree |
1M |
AM1.5Gc |
1.85 (0.8 V vs. RHE) |
46
|
| Plasma-assisted deposition |
Nanofilament (d = 100–200 nm) |
3M |
AM1.5Gc |
1.0 (1.7 V vs. RHE) |
47
|
| Electrodeposition |
Thin film (t = 300 nm) |
1M |
AM1.5Gc |
1.3 (1.4 V vs. NHE) |
28
|
| Chemical solution deposition |
Spherical nanoparticles (d = 50–100 nm) |
1M |
AM1.5Gc |
2.6 (1.5 V vs. RHE) |
29
|
| NPS hybrid dispersion deposition |
Plate-like nanoparticles (d = 20–120 nm) |
1M |
AM1.5Gc |
3.04 (1.5 V vs. RHE) |
This work |
| Chemical solution deposition |
Droplet-like particles (d = 200–300 nm) |
1M |
86 mW cm−2 |
1.4 (1.2 V vs. SCE) |
48
|
| Chemical solution deposition |
Nanoparticles (d = 40 nm) |
1M |
140 mW cm−2 |
1.6 (1.4 V vs. SCE) |
42
|
| Anodization |
Worm-like structure |
1M |
150 mW cm−2 |
3.5 (1.5 V vs. SCE) |
49
|
| Anodization |
Nanoporous (d = 70–90 nm) |
0.5M |
100 mW cm−2 |
3.45 (1.6 V vs. Ag/AgCl) |
50
|
The photocurrent was maintained at ca. 85% after 60 min, and it was recovered to the original photocurrent value by washing the photoanode (Fig. S4†). It could be mainly due to accumulation of the products in the inner photoanode nanostructure. The nanostructure of W3, e.g., faceted nanocrystals and pore size, was unchanged before/after the reaction under 1 Sun illumination at 1.5VRHE for 120 min (Fig. S5†); therefore, the obtained photoanodes would have sufficient stability for the use of this PEC reaction.
We performed Mott–Schottky and IMPS analyses of the effect of the physical properties of the photoanodes on the PEC performance. The Mott–Schottky plots for photoanodes W1, W2, and W3 exhibited n-type characteristics with a positive slope (Fig. 12), indicating that electrons were the main carrier, as in n-type semiconductors. The curves were fitted by
| | | 1/C2 = (2e0ε0εNd)(E − EFB − kT/e0) | (3) |
where
C is the space charge capacitance,
e0 is the electron charge,
ε0 is the vacuum permittivity,
ε is the dielectric constant of WO
3 (= 20),
38Nd is the donor density,
k is the Boltzmann constant,
T is the temperature, and
E and
EFB are the applied and flat band potentials, respectively. The
Nd of photoanodes W2 and W3 was 2.44 × 10
21 and 3.06 × 10
21 cm
−3, respectively (
Table 3). These values were 30–40 times higher than that of photoanode W1. The high carrier density observed in photoanodes W2 and W3 could be due to the partial oxygen deficiency
51 at the WO
3 nanoparticle surface, as indirectly observed in the red shift of the optical absorption edges for photoanodes W2 and W3 (
Fig. 6 and
7).
 |
| | Fig. 12 Mott–Schottky plots of photoanodes W1, W2, and W3 at a frequency of 1.0 kHz in 1 M H2SO4 electrolyte. | |
Table 3 Calculated Nd, EFB, and τe for photoanodes W1, W2, and W3
| Sample |
N
d/cm−3 |
E
FB/VAg/AgCl |
τ
e (front)/ms |
τ
e (back)/ms |
| W1 |
7.32 × 1019 |
0.48 |
7.11 |
0.63 |
| W2 |
2.44 × 1021 |
0.57 |
7.98 |
1.79 |
| W3 |
3.06 × 1021 |
0.67 |
8.95 |
5.65 |
The electron transport in the photoanodes was evaluated by IMPS. Fig. 13a and b show the IMPS spectra of photoanodes W1, W2, and W3 under front and back illumination with UV LEDs at 365 nm. The photoexcited electron transport time (τe) is evaluated by an imaginary minimum (fmin) of the semicircles and is expressed as
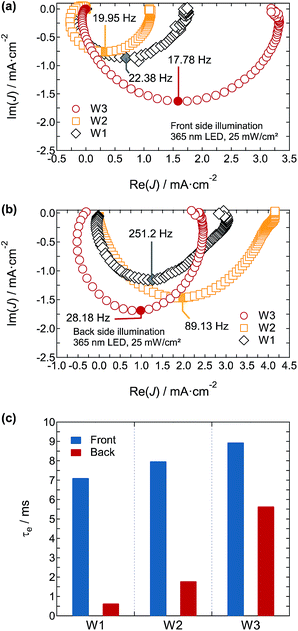 |
| | Fig. 13 IMPS responses of photoanodes W1, W2, and W3 biased at 1.5VRHE in 1 M H2SO4 electrolyte under (a) front and (b) back illumination with the UV LED (365 nm). The LED was driven as a modulated light source with a DC intensity of 25 mW cm−2 and superimposed 10% AC intensity. (c) Photoexcited electron transport time τe for photoanodes W1, W2, and W3 under front and back illumination. | |
The calculated τe values of photoanodes W1, W2, and W3 under front illumination were 7.11, 7.98, and 8.95 ms, respectively (Fig. 13c and Table 3). Back illumination made a large difference in τe of 0.63, 1.79, and 5.65 ms for photoanodes W1, W2, and W3, respectively (Fig. 13c and Table 3). Because of the short wavelength LED (365 nm) illumination, the penetration depth of excitation light was shallow, and the effect on transport time to the bottom electrode was maximised. The much longer transport time in photoanode W3 than in photoanodes W1 and W2 reflected the ease of charge transport from the regions further from the bottom electrode, suggesting that the good inter-nanoparticle connections played an important role in increasing carrier transport. Based on the Mott–Schottky and IMPS analyses, photoanode W3 had a high carrier density and much better carrier transport, which affect the increase in photocurrent.
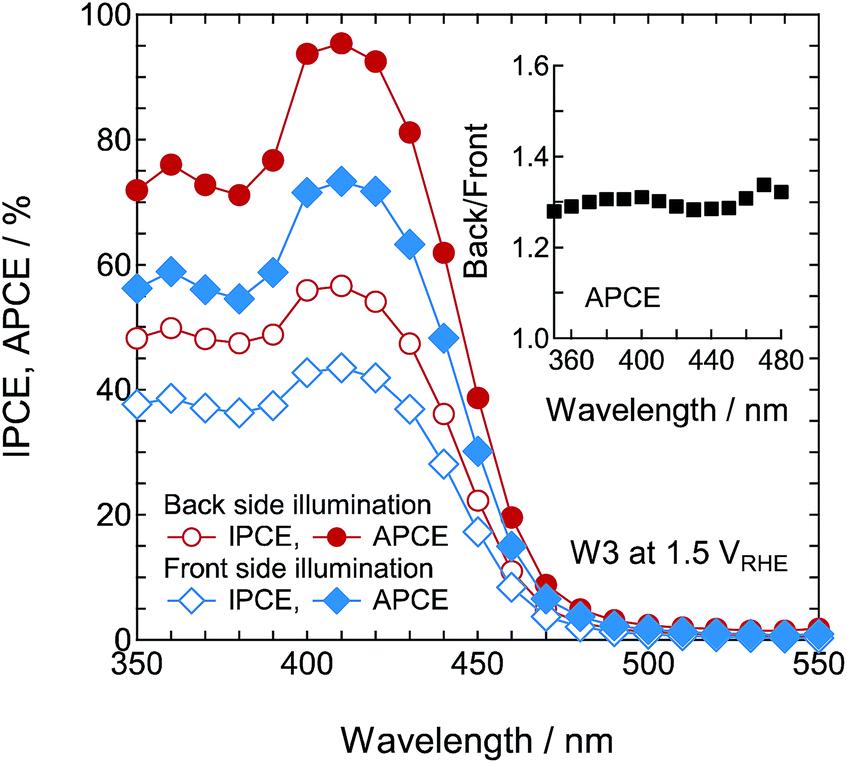
To evaluate the wavelength dependence of the PEC reaction in photoanode W3 in 1 M H2SO4 electrolyte, we measured the IPCE and APCE spectra (Fig. 14). As the wavelength of light decreased, the back illumination APCE spectra increased below 480 nm, and reached a high maximum of about 95.4% at 410 nm. This means that almost all the absorbed light was used for the PEC reaction around this wavelength. The APCE values were about 30% higher for back illumination than for front illumination, and it was almost independent of the wavelength. The IPCE values for back and front illumination were about 40% less than the APCE values. This arose from the light reflection of the WO3 nanosponge photoanodes (Fig. S6†), and was linked to the decrease in J from the theoretical limit in photoanode W3. Therefore, the light harvesting properties in this system will be important for decreasing the gap between the current value and the theoretical limit.
 |
| | Fig. 14 IPCE and APCE spectra of photoanodes W1, W2, and W3 as a function of the excitation wavelength at 1.50VRHE under front and back illumination. The inset shows the relative APCE under back illumination compared with front illumination. | |
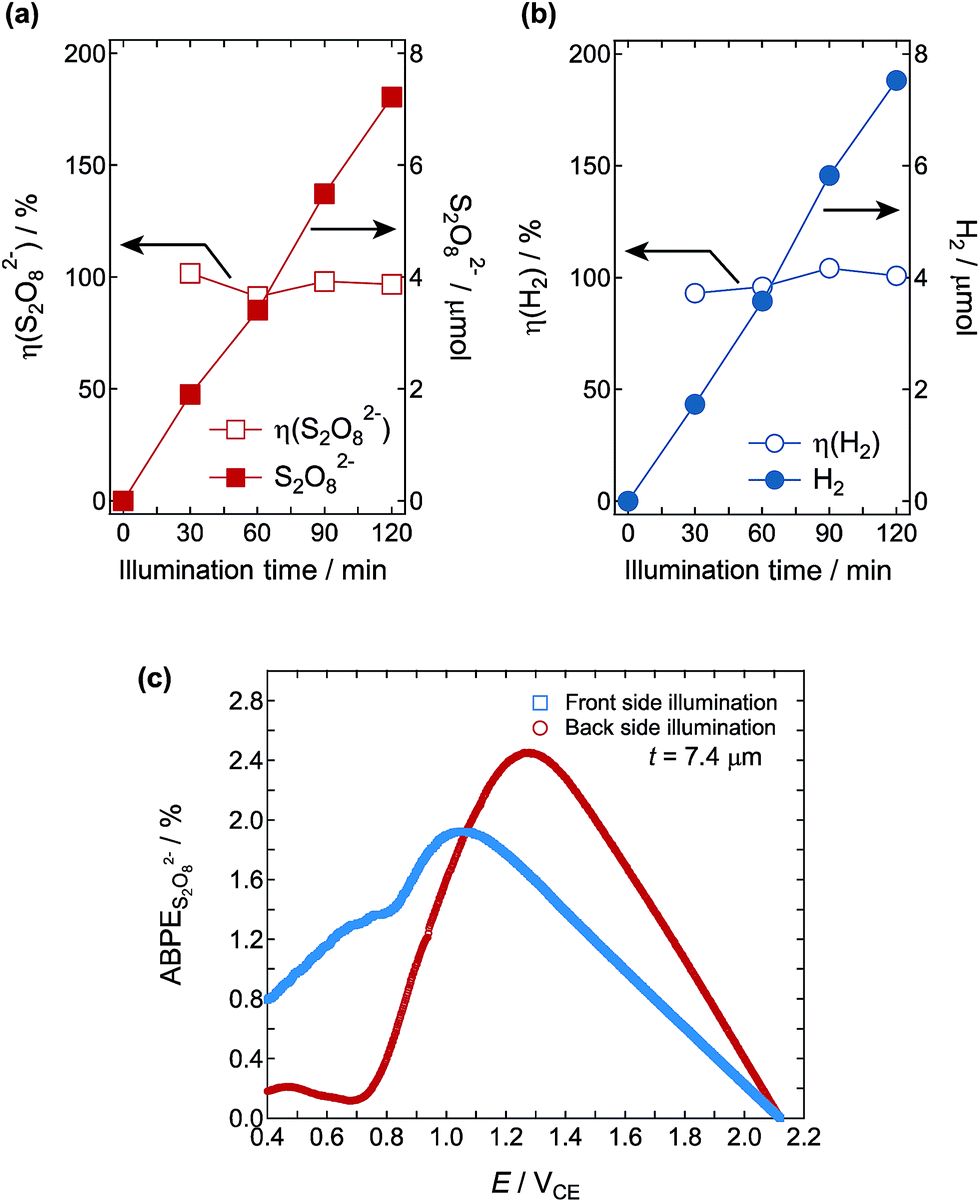
We confirmed the generation efficiency of S2O82− at the W3 photoanode and H2 gas at the Pt cathode in 1 M H2SO4 electrolyte. The simulated sunlight was illuminated from the back of the photoanode. The Faraday efficiency of η(S2O82−) and η(H2) was calculated by η(S2O82−/H2) = (generated amount of S2O82−(H2)/theoretical amount of S2O82−(H2)) × 100. Both S2O82− and H2 were stably generated under excitation light illumination, and stoichiometric evolution of S2O82− and H2 was confirmed as Faraday efficiencies of almost 100% (Fig. 15a and b). These results were consistent with previous reports.28,29
 |
| | Fig. 15 Time dependence of Faraday efficiency and generated amounts of (a) S2O82− (anode) and (b) H2 (cathode). The reaction was carried out under 1 Sun simulated sunlight illumination from the back of the W3 photoanode with a steady photocurrent at 0.2 mA. (c) ABPES2O82− calculated for the evolution of S2O82− and H2 in 1.0 M H2SO4 solution under simulated sunlight illumination from the front and back of photoanode W3. | |
Finally, we measured the J–V curves with a two-electrode setup in 1.0 M H2SO4 electrolyte under 1 Sun illumination from the front and back of photoanode W3. Fig. 15c shows the calculated ABPES2O82−, and the maximum values for the back illumination of 2.45% at 1.26VCE and 1.92% at 1.03VCE, respectively, were remarkably high. The 2.45% ABPES2O82− is the highest efficiency reported for the production of S2O82− and H2.
Conclusions
We prepared WO3 nanosponge photoanodes by NPS hybrid dispersion–deposition, and the WO3 nanosponge photoanodes showed a large photocurrent (3.04 mA cm−2) at 1.50VRHE in 1.0 M H2SO4 electrolyte under 1 Sun back illumination of the photoanodes. This high photocurrent was obtained mainly because of the robust inter-nanoparticle connections and the preservation of the nanopores. The connections were produced by adding tungsten phenoxide and using very fine nanoparticles that were obtained by wet-milling. The APCE spectra showed a high maximum of about 95.4% at 410 nm. The PEC reaction of this system was confirmed as a stoichiometric reaction for S2O82− and H2 evolution, and ABPES2O82− was calculated to be 2.45% at 1.26VCE for the back illumination, which is the highest reported value. Obtaining this kind of high-value-added product at the photoanodes in PEC systems will be crucial for the sustainable industrial chemistry using sunlight.
Acknowledgements
The authors thank Dr K. Fuku and Dr Y. Miseki for valuable discussion. This research was (partly) supported by the International Joint Research Program for Innovative Energy Technology of the Ministry of Economy, Trade and Industry (METI).
Notes and references
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- T. Takata, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Catal. Today, 1998, 44, 17 CrossRef CAS.
- A. Steinfeld, Sol. Energy, 2005, 78, 603 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- T. Nakajima, T. Kitamura and T. Tsuchiya, Appl. Catal., B, 2011, 108–109, 47 CrossRef CAS.
- B. D. Alexander, P. J. Kulesza, I. Rutkowska, R. Solarska and J. Augustynski, J. Mater. Chem., 2008, 18, 2298 RSC.
- J. Nowotny, T. Bak, M. K. Nowotny and L. R. Sheppard, Int. J. Hydrogen Energy, 2007, 32, 2609 CrossRef CAS.
- C. X. Kronawitter, L. Vayssieres, S. Shen, L. Guo, D. A. Wheeler, J. Z. Zhang, B. R. Antoun and S. S. Mao, Energy Environ. Sci., 2011, 4, 3889 CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321 RSC.
- R. van de Krol, Y. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- T. Hisatomi, F. L. Formal, M. Cornuz, J. Brillet, N. Tétreault, K. Sivula and M. Grätzel, Energy Environ. Sci., 2011, 4, 2512 CAS.
- X. Liu, F. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7894 RSC.
- R. Saito, Y. Miseki and K. Sayama, Chem. Commun., 2012, 48, 3833 RSC.
- B. A. Pinaud, P. C. K. Vesborg and T. F. Jaramillo, J. Phys. Chem. C, 2012, 116, 15918 CAS.
- T. Nakajima, T. Nakamura, K. Shinoda and T. Tsuchiya, J. Mater. Chem. A, 2014, 2, 6762 CAS.
- T. Nakajima, T. Nakamura and T. Tsuchiya, Phys. Chem. Chem. Phys., 2014, 16, 26901 RSC.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- C. Yang, Z. Wang, T. Lin, H. Yin, X. Lü, D. Wan, T. Xu, C. Zheng, J. Lin, F. Huang, X. Xie and M. Jiang, J. Am. Chem. Soc., 2013, 135, 17831 CrossRef CAS PubMed.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007 CAS.
- R. Sathre, C. D. Scown, W. R. Morrow III, J. C. Stevens, I. D. Sharp, J. W. Ager III, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3264 CAS.
- J. Nowotny, T. Bak, M. K. Nowotny and L. R. Sheppard, Int. J. Hydrogen Energy, 2007, 32, 2609 CrossRef CAS.
- Q. Mi, A. Zhanaidarova, B. S. Brunschwig, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 5694 CAS.
- K. Fuku, N. Wang, Y. Miseki, T. Funaki and K. Sayama, ChemSusChem, 2015, 8, 1593 CrossRef CAS PubMed.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406 RSC.
- T. Zhu, M. N. Chong and E. S. Chan, ChemSusChem, 2014, 7, 2974 CrossRef CAS PubMed.
- C. A. Bignozzi, S. Caramori, V. Cristino, R. Argazzi, L. Meda and A. Tacca, Chem. Soc. Rev., 2013, 42, 2228 RSC.
- A. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Scwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 33 Search PubMed.
- D. Zhang, T. Yoshida, K. Furuta and H. Minoura, J. Photochem. Photobiol., A, 2004, 164, 159 CrossRef CAS.
- S. Kambe, S. Nakade, Y. Wada, T. Kitamura and S. Yanagida, J. Mater. Chem., 2002, 12, 723 RSC.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 CrossRef CAS.
- B. O. Loopstra and H. M. Rietveld, J. Solid State Chem., 1991, 91, 286 CrossRef.
- J. Yan, T. Wang, G. Wu, W. Dai, N. Guan, L. Li and J. Gong, Adv. Mater., 2015, 27, 1580 CrossRef CAS PubMed.
- T. Singh, R. Müller, J. Singh and S. Mathur, Appl. Surf. Sci., 2015, 347, 448 CrossRef CAS.
-
R. M. German, Liquid Phase Sintering, Plenum Publishing Corporation, New York, 1985 Search PubMed.
- F. Amano, D. Li and B. Ohtani, J. Electrochem. Soc., 2011, 158, K42 CrossRef CAS.
- L. Meda, G. Tozzola, A. Tacca, G. L. Marra, S. Caramori, V. Cristino and C. A. Bignozzi, Sol. Energy Mater. Sol. Cells, 2010, 94, 788 CrossRef CAS.
- V. Chakrapani, J. Thangala and M. K. Sunkara, Int. J. Hydrogen Energy, 2009, 34, 9050 CrossRef CAS.
- S. J. Hong, H. Jun, P. H. Borse and J. S. Lee, Int. J. Hydrogen Energy, 2009, 34, 3234 CrossRef CAS.
- S. K. Biswas and J.-O. Baeg, Int. J. Hydrogen Energy, 2013, 38, 3177 CrossRef CAS.
- S. Shin, H. S. Han, J. S. Kim, I. J. Park, M. H. Lee, K. S. Hong and I. S. Cho, J. Mater. Chem. A, 2015, 3, 12920 CAS.
- M. de Respinis, G. D. Temmerman, I. Tanyeli, M. C. M. van de Sanden, R. P. Doerner, M. J. Baldwin and R. van de Krol, ACS Appl. Mater. Interfaces, 2013, 5, 7621 CAS.
- B. Yang, Y. Zhang, E. Drabarek, P. R. F. Barnes and V. Luca, Chem. Mater., 2007, 19, 5664 CrossRef CAS.
- V. Cristino, S. Caramori, R. Argazzi, L. Meda, G. L. Marra and C. A. Bignozzi, Langmuir, 2011, 27, 7276 CrossRef CAS PubMed.
- W. Z. Li, J. Li, X. Wang, S. Luo, J. Xiao and Q. Y. Chen, Electrochim. Acta, 2010, 56, 620 CrossRef CAS.
- S. K. Deb, Phys. Rev. B: Solid State, 1977, 16, 1020 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta07997k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence