
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow α-Fe2O3 nanofibres for solar water oxidation: improving the photoelectrochemical performance by formation of α-Fe2O3/ITO-composite photoanodes†
M.
Einert
a,
R.
Ostermann
a,
T.
Weller
a,
S.
Zellmer
b,
G.
Garnweitner
b,
B. M.
Smarsly
a and
R.
Marschall
*a
aInstitute of Physical Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: roland.marschall@phys.chemie.uni-giessen.de
bInstitute for Particle Technology and Laboratory for Emerging Nanometrology (LENA), Technische Universität Braunschweig, Volkmaroder Str. 5, 38104 Braunschweig, Germany
First published on 28th October 2016
Abstract
We demonstrate the synthesis and photoelectrochemical performance of high-aspect ratio dense and hollow α-Fe2O3 nanofibres, and the formation of core–shell-like α-Fe2O3/indium-tin oxide (ITO) nanocomposites utilised as a photoanode for solar water splitting. α-Fe2O3 nanofibres were prepared via a single-nozzle electrospinning technique using iron chloride (FeCl3) and poly(vinylpyrrolidone) (PVP) as precursors, followed by calcination. A new synthetic formation mechanism has been proposed taking into account the significance of three control parameters: (i) the iron precursor, (ii) the role of a co-solvent and (iii) the influence of the humidity on the tube evolution of α-Fe2O3 nanotubes. Hollow α-Fe2O3 fibres showed enhanced photocurrents and incident photon-to-current efficiency (IPCE) values compared to dense fibres, which are ascribed to the superior surface area of hollow fibres offering a good accessibility for the electrolyte and thus leading to improved mass transport. The photoelectrochemical properties of the α-Fe2O3 nanofibres could be further enhanced by the combination with highly crystalline, uniform ITO nanocrystals (Ø 10 nm), thus forming a core–shell-like α-Fe2O3/ITO fibre nanocomposite. The doubled photocurrent of the α-Fe2O3/ITO nanocomposite can most likely be attributed to the fast interfacial charge carrier exchange between the highly conductive ITO nanoparticles and α-Fe2O3, thus inhibiting the recombination of the electron–hole pairs in the semiconductor by spatial separation.
Introduction
One of the major challenging tasks of our generation is to tackle the substitution of fossil resources in order to reduce the ongoing rise of CO2 emission, considering alternative and renewable energy sources. Semiconductor-assisted solar water splitting is such a sustainable process in which hydrogen is simply produced by a chemical reduction reaction. Owing to its high gravimetric energy density, “clean hydrogen” is a promising candidate for the storage of chemical energy1,2 and further stands out by emitting no greenhouse gases using it e.g. in fuel cells for automobiles,3 as its reaction product is solely pure water. Since Fujishima and Honda first developed photoelectrochemical water splitting under UV-irradiation in 1972,4 enormous efforts have been focused on improving solar-to-hydrogen efficiency of various n-type metal oxide semiconductors such as TiO2,5–7 WO3,8–10 α-Fe2O3,11–15 ZnO,16,17 and BiVO4.18,19 Among these photoanodes, pristine α-Fe2O3 with its optical band gap of around 2.0 eV (λ ≈ 620 nm)20,21 fulfils the requirements for efficient visible-light absorption. Furthermore, α-Fe2O3 is characterised by its high chemical stability in aqueous and alkaline media as well as stable performance under anodic polarisation.12 The low-cost of α-Fe2O3 and its non-toxicity are advantageous properties compared to other n-type semiconductors. When utilising α-Fe2O3 as a photoanode in a photoelectrochemical cell (PEC), the valence band (VB) maximum of 2.5 eV vs. normal hydrogen electrode (NHE) enables water oxidation (E0Ox = 1.23 eV vs. NHE), whereas the conduction band (CB) level is positioned below the hydrogen redox potential which requires an external bias for hydrogen production (at the photocathode).22 The unique properties of α-Fe2O3 enable a theoretical solar-to-hydrogen conversion efficiency of 16.8%.23 However, the typical PEC performance of α-Fe2O3 photoanodes is far below this theoretical value ranging between 1 and 5%.11,24,25 The reason can be found in the poor absorptivity (photon penetration depth ≈ 118 nm at λ = 550 nm) owing to its indirect band gap, the low mobility of majority charge carriers (electrons, 0.2 cm2 V−1 s−1), short minority carrier (holes) diffusion length LD of 2–4 nm (for comparison: TiO2 104 nm),26 and slow oxidation kinetics.12,14These limiting intrinsic properties – leading to fast recombination of photogenerated electron–hole pairs and hence low photoresponse – require a precise structural design of α-Fe2O3 on the nanoscale. While the large overpotential of α-Fe2O3 photoanodes for water oxidation,27 requiring a high external bias, can be reduced by surface modification and/or the usage of catalysts (Co2+,28 IrO2 (ref. 29)), the photocurrent density is tuneable through structure control. Discussing the optimum morphology of α-Fe2O3, the material has to be tailored with regard to its short hole diffusion length allowing the minority carriers to migrate from the point of excitation to the semiconductor liquid junction (SCLJ) before recombination occurs.30
Therefore, the necessity of an appropriate and facile synthesis approach supplying nanoscaled materials is obvious. Electrospinning is a straightforward technique to synthesise polymeric and composite fibres, emerging as a suitable method to prepare 1-dimensional nanostructured materials.31–34 Especially, the formation of tubular fibres has attracted great attention. The diffusion paths of migrating charge carriers are typically an order of magnitude lower (tens compared to hundreds of nanometre) in hollow nanowires influencing electron–hole lifetimes significantly. The advantage of tubular structured α-Fe2O3 fibres was demonstrated by Chaudahri et al. who revealed their superior cycling performance towards lithium storage.35 Besides hollow morphologies, recently electrospinning of α-Fe2O3 fibres from PVA/iron nitrate solutions has been carried out and the product utilised after calcination as a photoanode in a PEC.36 Further, electrospun α-Fe2O3 fibres were photocatalytically investigated in terms of degradation of methylene blue.37 Li et al.38 reported on the photoelectrochemical characterisation of electrospun La-doped α-Fe2O3 nanotubes observing increased conductivity and light absorption due to the effect of doping.
Among the known preparation mechanisms of hollow structures via electrospinning,39 the single-nozzle electrospinning is readily scalable and is also the most elegant strategy tailoring delicately the composition and the blend ratio of two immiscible polymers dissolved in a spinning solution.40,41 Phase separation is initiated when the solvent evaporates during electrospinning providing core/sheath fibres which are collected at the counter electrode. The single-nozzle technique was further applied for the preparation of ceramic fibres by simply adding a suitable metal precursor to the bi-polymer/solvent system.41 Interestingly, for the formation of hollow α-Fe2O3 nanofibres the usage of solely PVP as a spinning polymer has been reported.35,39,42 However, the influence of distinct conditions, i.e. necessity of low humidity during preparation and a suitable co-solvent, has not been discussed so far. Therefore, further research for the thorough understanding of the formation mechanism of hollow nanofibres by the single-nozzle electrospinning technique with reproducible results is highly indicated.
The formation of semiconductor composites43 is a further promising approach for the suppression of recombination reactions, as one of the main challenges in semiconductor-assisted solar water splitting is the sufficiently fast separation of electron–hole pairs (excitons) after excitation by sunlight with wavelengths equal to the band gap of the photocatalyst. In order to inhibit recombination by relaxation of the charge carriers from the CB to the VB and to enable oxidation and reduction reactions at the semiconductor's surface, the electrons have to be transferred within the range of a few picoseconds44,45 into the CB of an adjacent photocatalyst or into a conductive scaffold. Since α-Fe2O3 suffers from ultrafast recombination rates, excited electrons are capable of recombining with holes or trap states within merely 8 ps.45
Transparent conducting oxides (TCOs), such as FTO (F:SnO2),46 ATO (Sb:SnO2)47 or AZO (Al:ZnO),14 have been shown to function as the host scaffold for α-Fe2O3. Since TCOs combine high electronic conductivity with transparency in the visible spectrum of light, the formation of TCO/α-Fe2O3 composite photoanodes leads to enhanced charge carrier separation and hence to highly improved photocurrents up to 3.3 mA cm−2 at 1.23 eV versus NHE.47 Marschall48 gives a comprehensive review about the formation of semiconductor composites and their application in photocatalysis. In terms of formation of nanocomposites by single-nozzle electrospinning, for example, Au nanoparticle-embedded TiO2 composite nanofibres,49 or tin nanoparticles in multichannel carbon tubes50 have been reported for diverse applications, but never in the context of photoelectrochemical water splitting.
The goal of the present work is the investigation and comparison of the structural and physicochemical properties of dense and hollow α-Fe2O3 nanofibres and to ascertain how their photoelectrochemical performance is influenced by the structural modification. Since the photoelectrochemical characterisation of electrospun α-Fe2O3 nanofibres has rarely been studied until now and – to the best of our knowledge – not been carried out in terms of single-nozzle electrospinning of a nanoparticle based dispersion in coexistence of a molecular (ferric) spinning solution, this work paves the way towards the facile formation of photoactive composite nanostructures and a profound understanding of charge transfer processes within the bi-component semiconducting system. Utilised as a photoanode in a PEC, the α-Fe2O3/ITO nanocomposite fibres show an improved photoresponse (relative to pristine Fe2O3) which is attributed to the fast spatial charge carrier separation transferring electrons from the α-Fe2O3 phase to the highly accessible nanoparticular ITO moiety.
Experimental
Materials
All materials were of analytical grade and used as received: polyvinylpyrrolidone (PVP, MW = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1, Alfa Aesar), ferric chloride (97%, Sigma Aldrich), In(III) acetylacetonate (≥99.99%, trace metals basis, Aldrich), Sn(IV) tert-butoxide (≥99.99%, trace metals basis, Aldrich), ethylene diamine (≥99.5%, GC, Fluka), dimethylformamide (DMF, Sigma Aldrich) and ethanol (EtOH, VWR).
300000 g mol−1, Alfa Aesar), ferric chloride (97%, Sigma Aldrich), In(III) acetylacetonate (≥99.99%, trace metals basis, Aldrich), Sn(IV) tert-butoxide (≥99.99%, trace metals basis, Aldrich), ethylene diamine (≥99.5%, GC, Fluka), dimethylformamide (DMF, Sigma Aldrich) and ethanol (EtOH, VWR).
Preparation of ITO nanoparticle dispersion
ITO nanoparticles were prepared via non-aqueous sol–gel synthesis using indium(III) acetylacetonate and tin(IV) tert-butoxide as precursors in benzyl alcohol as a high boiling solvent as described earlier.51–53 After the nanoparticle synthesis two washing steps with chloroform were performed to remove adsorbed reaction residues. As we reported recently, ethylene diamine acts as a highly efficient stabiliser for ITO nanoparticles in ethanol.54 Hence, subsequently ethylene diamine as an additive (molar ratio of ITO to stabiliser 1:4) and ethanol (p.a.) as a solvent were added and stirred at room temperature to obtain a stable colloidal ITO nanoparticle dispersion.
Preparation of electrospinning solution
For all experiments, typically 2 g of spinning solution was prepared: for dense α-Fe2O3 fibres, 50 mg FeCl3 was dissolved in 250 mg DMF and subsequently 1040 mg EtOH and 660 mg of a solution of 15 wt% PVP in EtOH were added (5 wt% PVP/EtOH solution). For hollow α-Fe2O3 fibres, 50 mg FeCl3 was refluxed in 250 mg DMF at 125 °C for 15 min, before 740 mg EtOH and 960 mg 15 wt% PVP in EtOH were added, thus obtaining a 7 wt% PVP/EtOH solution. For the α-Fe2O3/ITO composite fibres, 50 mg of FeCl3 was dissolved in 250 mg DMF and afterwards successively 250 mg EtOH, 660 mg 15 wt% PVP in EtOH and 800 mg of 5 wt% ITO-NP in EtOH were added. All solutions were homogenised in a shaker.Electrospinning of nanofibres
Electrospinning was carried out with a setup consisting of a high-voltage supply (Scientific Instruments, TSI-HV), a syringe pump (Harvard Apparatus, 11 Plus MAI 70-2208), a syringe, a needle (inner diameter 4.73 mm) and aluminium foil covered circular counter electrode (diameter 10 cm) in a humidity-controlled chamber. Large amounts of fibre mats were prepared by utilisation of a rotating drum (diameter ca. 10 cm) serving as a collector electrode. Stable jet formation for all spinning systems was achieved by adjusting the solution flow rate to 0.3 mL h−1 and applying a voltage of 11 kV (−2 kV vs. 9 kV) at a tip-to-collector distance of 15 cm. The relative humidity – which was chosen as a control parameter in the electrospinning experiments – was set to 20% for the hollow and composite systems, while for the dense system 45% rel. humidity was adjusted by supplying a constant flow of dry air into the closed chamber at 25 °C. For the photoelectrochemical characterisation, the corresponding spinning solutions were electrospun on F:SnO2 (FTO) substrates which have been dip-coated with a constant withdrawal of 2 mm cm−1 in a 1 wt% PVP in EtOH solution (humidity chamber 30–40%) prior to the electrospinning procedure. The PVP coating is prepared to compensate shrinkage effects during thermal treatment, which would induce both micro- and macroscopic cracks throughout the fibrous electrode. All as-spun fibres (fibres as powder and on FTO) were annealed to 550 °C in air with a heating ramp of 5 °C min−1 and kept at this temperature for 2 h. For determination of the irradiated mass of the calcined fibres on the FTO-substrate, the samples were weighed with a microbalance from Mettler Toledo (Model AX26 Delta Range) and afterwards treated with an aqueous 1 M HCl solution for 1 h to dissolve the absorber. Finally, the samples were cleaned with ethanol, dried in air for 1 h, and subsequently weighed again.Characterisation
The nanofibres were investigated by scanning electron microscopy (SEM, Zeiss Merlin) at an acceleration voltage of 3 kV and a current of 90 pA. Before analysis, the samples were sputter-coated with platinum for 45 seconds. Elemental mapping was performed by EDX-analysis using an Inca Energy System from Oxford Instruments. Transmission electron microscopy (TEM) images and selected area electron diffraction (SAED) were accomplished on a Philips CM30 at 300 kV acceleration voltage. Wide-Angle X-ray scattering (WAXS) was carried out on an X'Pert PRO diffractometer from PANalytical Instruments (Cu Kα radiation, λ = 154.18 pm) utilising a θ – 2θ geometry. The data were recorded in a step scan mode from 10 to 90° with a step size of 0.03°, an acceleration voltage of 40 kV and an emission current of 40 mA. Rietveld refinement was carried out using FULLPROF 2.05 and a modified Thompson–Cox–Hastings Pseudo-Voigt profile function. The weighted profile R-factor (Rwp) and the goodness of fit (χ2) were taken for demonstrating the quality of the fit. Raman spectra were acquired on a SENTERRA dispersive Raman microscope from Bruker optics equipped with a Nd:YAG laser (λexc = 532 nm, P = 0.2–5 mW) and an objective from Olympus (MPLan N 100x). Nitrogen physisorption was performed at 77 K using the Autosorb-6 automated gas adsorption station from Quantachrome Corporation. Before analysis, the fibre mats were degassed in vacuum at 120 °C. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) models were employed for evaluation of the surface area and pore size distribution. Thermogravimetric analyses (TGA) were conducted with a Netzsch STA40PC thermoscale connected to a QMG421 quadrupole mass spectrometer (MS) from Balzers. A few milligrams of fibres were heated to 800 °C with a ramp of 5 °C min−1 in synthetic air. UV-visible light spectroscopy was employed by measuring the diffuse reflectance with a Perkin Elmer Lambda 750 UV-Vis-NIR spectrometer equipped with a Praying-Mantis mirror. BaSO4 was utilised as a standard reference. The optical band gap (Egap) was determined from the Tauc plots. All photoelectrochemical experiments were carried out in a three-electrode photoelectrochemical cell (PEC) filled with an aqueous 1 M NaOH electrolyte (pH = 13.6) using the electrospun nanofibres on FTO as the working electrode, a platinum wire as the counter electrode, and Ag/AgCl as the reference electrode (3 M NaCl). All electrodes were scanned from 700 mV to −600 mV versus Ag/AgCl utilising a Zahner Zennium potentiostat. Calculation of the potential of the RHE is given by VRHE = VAg/AgCl + 0.209 V + 0.059 V × pH. Photocurrent density with intermittent light irradiation was measured with a scan speed of 5 mV s−1 and a light period time of 10 s using a white light LED (average λ = 536 nm, P = 100 mW cm−2) illuminating an area of 1 cm2 from a distance of 10 cm. For incident photon-to-current efficiency (IPCE) measurements, a Zahner tunable light source system, model CIMPS TLS03, was employed exhibiting a LED array for monochromatic light excitation. The light source was operated with a frequency of 1 Hz at 430 mV (versus Ag/AgCl). Mott–Schottky plots were acquired in non-irradiated mode for frequencies between 10 Hz and 10 kHz with a step width of 50 mV s−1.Results & discussion
Electrospinning of nanofibres and their characterisation
For the preparation of α-Fe2O3 nanofibres via electrospinning it is essential to figure out a suitable composition ratio of the metal precursor and spinning polymer. PVP (poly(vinylpyrrolidone) with MW = 1300000 g mol−1) has been shown to be a versatile electrospinning polymer that allows producing nanofibres of various metal oxides,55 and was therefore selected in our attempt to produce α-Fe2O3 nanofibres. Together with iron(III) chloride as the metal oxide precursor stable electrospinning could be achieved. Although PVP and the precursor FeCl3·H2O are individually soluble in methanol or ethanol, precipitation occurs when both solutions are combined. It is known that PVP forms complexes with trivalent metal ions M3+,56 such as Fe3+, and we found that the resultant Fe–PVP complex is insoluble in alcohols. However, adding DMF as a co-solvent allows obtaining stable electrospinning solutions, as DMF has a similar structure to 2-pyrrolidone and prevents the formation of insoluble Fe–PVP complexes. In order to investigate the formation mechanism and the morphology of α-Fe2O3 nanofibres, nanotubes and their ITO α-Fe2O3 composites, SEM and TEM studies were carried out. For the preparation of dense and hollow α-Fe2O3 fibres, it is apparent that the concentration of PVP, the iron precursor and the humidity have a major impact on the structural development during electrospinning. As illustrated in Fig. 1A, dense α-Fe2O3 fibres were obtained by electrospinning of 5 wt% PVP concentrated DMF/EtOH solution in a chamber with a relative humidity of around 35%. In contrast, well-defined nanotubes were observed (Fig. 1B1–B3) when increasing the PVP content to 7 wt% and decreasing the humidity below 20% by supplying dry air to the spinning chamber. The results suggest that the tube formation is predominantly influenced by the presence of water. We verified this assumption by addition of 1 wt% of water to the initial spinning solution, resulting in dense fibres. Therefore, it is not surprising that compact fibres are found at high humidity in the case of PVP-based systems, as the water is absorbed in the hydrophilic PVP and leads to hydrolysis and condensation within the emerging fibres.
 | ||
| Fig. 1 Nanofibres prepared by annealing at 550 °C: (A1, A2) SEM images of electrospun, dense α-Fe2O3 fibres; (B1) bundle of electrospun α-Fe2O3 hollow fibres, (B2) high-resolution SEM image of a single α-Fe2O3 hollow fibre and (B3) the corresponding TEM image; (C) high-resolution SEM image of ITO nanoparticles; (D1) electrospun α-Fe2O3/ITO composite fibres as (D2) high-resolution SEM- and (D3) corresponding TEM picture with SAED as inlet evincing a highly crystalline phase; (D4) SEM-image of α-Fe2O3/ITO composite fibres showing the corresponding elemental mapping of (D5) iron, (D6) indium, (D7) tin and (D8) oxygen. | ||
The as-spun FeCl3/PVP fibres are between 200 and 400 nm in diameter possessing a smooth surface and uniform structure (Fig. S1, ESI†), whereas shrinkage of size occurs down to 70–100 nm after calcination at 550 °C. The reduction in diameter is mainly ascribed to the thermal degradation of PVP (see TGA). The inner diameter of the hollow fibres ranges between 200 nm and 400 nm with a wall thickness of approximately 40–60 nm after calcination. SEM images in Fig. 1 clearly indicate that the nanotubes are perfectly developed in contrast to fibres prepared with Fe(NO3)3·9H2O as the precursor showing less defined hollow structures that still contain some compact sections owing to the presence of water prematurely initiating hydrolysis and condensation reactions during the spinning process (Fig. S2, ESI†). In addition to the reported results from the literature, the empirically obtained data of this work strongly suggest that the hydrolysis and condensation reactions during electrospinning most likely have a significant impact on the tube formation process. Eid et al.42 reported the fabrication of α-Fe2O3 by electrospinning and subsequent removal of the PVP–polymer in air or H2/Ar, respectively. They used anhydrous iron acetate as the precursor, but did not specify the humidity during electrospinning. Cheng et al.39 prepared α-Fe2O3 fibres using Fe(NO3)3·9H2O proposing the formation of a gel layer on the surface of the fibres. However, the Laplace pressure of a tube with an inner diameter of 100 nm would be several atmospheres and gas would diffuse through a gel layer of PVP and hydroxides, as a nanometre layer of PVP does not act as a diffusion barrier.
Here we propose a different formation mechanism for hollow α-Fe2O3 fibres as follows: initially, Fe3+ ions and PVP are dissolved homogeneously in EtOH and DMF. During electrospinning the solvents quickly evaporate at the surface of the emerging fibre and a skin forms. However, with increasing concentration of Fe3+ and PVP near the surface of the fibre, a precipitate of Fe–PVP will form even before the fibre fully solidifies. Therefore, a core–shell/skin morphology can develop inside the fibres with a polymer-rich core, as the Fe3+–DMF complex migrates towards the surface. Upon calcination the iron-rich skin transforms into the solid FeOx shell, while the polymer-rich core acts as a template, resulting in hollow nanotubes after calcination. These observations and assumptions are summarised in Fig. 2 and taken together provide a reasonable explanation for the nanotube formation, but have to be confirmed with other material systems.
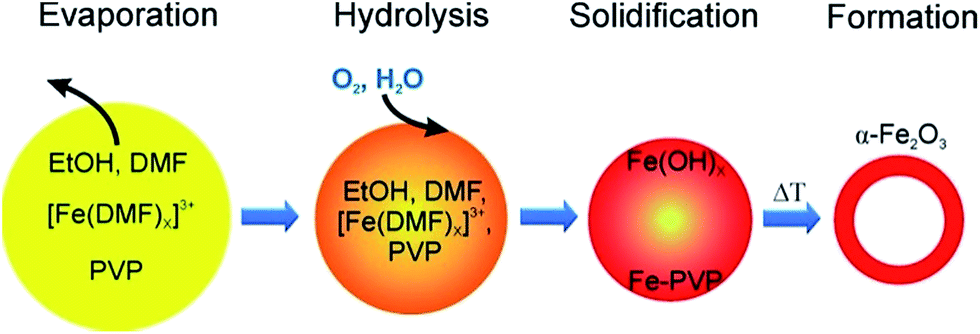 | ||
| Fig. 2 Proposed mechanism of α-Fe2O3 tube formation: a homogeneous electrospinning jet dries and a solid Fe3+–PVP complex precipitates to stabilise the skin that later transforms into iron hydroxide and upon calcination to iron oxide. | ||
Based on the obtained information about the formation mechanism of tube structuring, we successfully prepared – under nearly identical conditions – core/shell ITO/α-Fe2O3 composite fibres of about 150–300 nm in diameter (Fig. 1D1 and D2). The highly stable colloidal ITO nanoparticle dispersion (Fig. 1C) was added to the FeCl3/PVP/DMF/EtOH (2.5/7/10/80.5 by weight) solution, and electrospun at a humidity of approximately 20%. The TEM picture in Fig. 1D3 reveals two adjacent fibres with α-Fe2O3 wall thickness of ca. 15 nm. However, thicknesses up to 50 nm have been observed as well (compare Fig. 1D2) indicating a non-uniform shell structure throughout the composite fibres. This inhomogeneity can be ascribed to the agglomeration effect, as ITO nanoparticles possess – due to their size – the tendency to minimise their surface energies by agglomeration which occurs most likely during solvent evaporation. The ITO agglomerates break sporadically through the α-Fe2O3 fibre wall which is obvious on distinct spots on the fibre surface. Selected-area electron diffraction (SAED) on composite fibres confirms the presence of highly crystalline ITO nanoparticles distributed throughout the fibre visualised by the strong and separated diffraction signals in a reciprocal plane (Fig. 1D3).
Lateral elemental mapping of α-Fe2O3/ITO composite fibres was employed by EDX analysis which is shown in Fig. 1D4–D8 for iron, indium, tin and oxygen, respectively. The imaging of the detected elements further verifies the homogeneous distribution of the aforementioned elements within the fibre structure.
For the evaluation of the crystal structure and the degree of crystallinity, X-ray diffraction (XRD) analyses were applied to ITO nanoparticles (as-prepared and calcined at 550 °C), dense α-Fe2O3-, hollow α-Fe2O3- and composite ITO/α-Fe2O3-fibres as depicted in Fig. 3A. The XRD patterns of dense and hollow α-Fe2O3 fibres show the reflection signals of trigonal-crystallised α-Fe2O3 (JCPDS database card no. 003-0800), whereas the reflections of the as-prepared ITO nanoparticles are consistent with reference of indium oxide (In2O3, JCPDS database card no. 006-0416). The XRD pattern of the composite α-Fe2O3/ITO sample matches with the reference of both the α-Fe2O3 and indium oxide crystal phase. In order to evaluate the ratio of α-Fe2O3 to ITO in the composite fibres, Rietveld analyses were employed (using the FULLPROF software) refining the values of scale factors utilising the Hill and Howard approach.57 It was found that the composite contains 30 wt% ITO nanocrystals and 70 wt% α-Fe2O3 giving a slightly lower value for ITO compared to the theoretically determined value of 40:60 from the initial spinning solution. This might be due to small amounts of ITO being dissolved in the acidic FeCl3 solution. Interestingly, the indium oxide phase can be deconvoluted into a cubic and rhombohedral modification (ratio 70:30) giving rise to the assumption that a phase transformation occurred during the annealing process. The transformation might be explained by the presence of hematite acting as a catalyst agent, since the diffraction patterns of the bare ITO particles calcined at 550 °C (Fig. S3†) do not show any phase modifications besides the cubic structure.
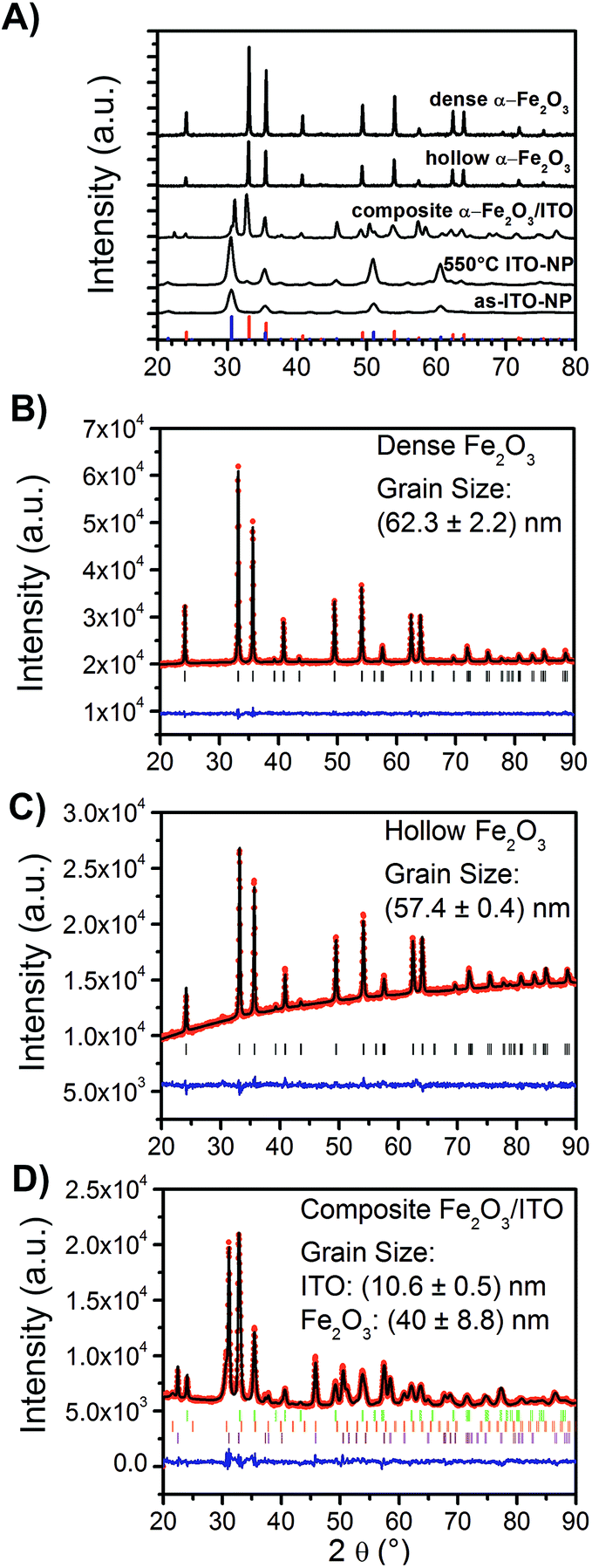 | ||
| Fig. 3 (A) XRD patterns of dense α-Fe2O3-, hollow α-Fe2O3- and composite α-Fe2O3/ITO fibres annealed at 550 °C, and of the as-prepared and 550 °C calcined ITO nanoparticles. The blue bars represent the reference card JCPDS no. 006-0416 for cubic In2O3, whereas the red bars correspond to the JCPDS no. 003-0800 for trigonal α-Fe2O3. Refined XRD patterns of (B) dense α-Fe2O3, (C) hollow α-Fe2O3, and (D) composite α-Fe2O3/ITO fibres (reference codes: 006-0416 and 022-0336 for In2O3, and 003-0800 for α-Fe2O3). | ||
As known from solid state physics, the crystalline structure of a material determines the electronic band structure, and hence the mobility of charge carriers required for the redox reactions at the surface of the photocatalyst. Therefore, the pronounced reflection signals of the α-Fe2O3 nanofibres suggest that these materials most likely meet the requirements for high photoactivity as, in general, the photoelectrochemical properties strongly correlate, among others, with the crystallinity and grain (crystallite) size of the photocatalyst.58
Thus, the grain sizes were evaluated by Rietveld refinement and were found to be 62.3 ± 2.2 nm (χ2 = 1.1 and Rwp = 7.6) and 57.4 ± 0.4 nm (χ2 = 1.4 and Rwp = 13.6) for the dense and hollow α-Fe2O3 nanofibres, respectively. The composite α-Fe2O3/ITO sample exhibits grain sizes of 10.6 ± 0.5 nm and 40 ± 8.8 nm (χ2 = 3.1 and Rwp = 11.2) for the ITO and α-Fe2O3 phase, respectively. The results support the SEM investigations, indicating that the particles are composed of single-crystals as confirmed by TEM and SAED measurements (Fig. 1). Further, it can be concluded that the α-Fe2O3 fibre walls in the composite are smaller relative to the pristine morphologies, which are reasonable values as confirmed by the SEM investigation. The bare ITO nanocrystals (Fig. S1D and C†) are 10 nm (χ2 = 11.9 and Rwp = 11.4), the same grain size (within the error of 0.4 nm) as in the composite fibre after annealing at 550 °C.
Nanostructures provide short diffusion pathways for charge carriers, which are a prerequisite for the inhibition of recombination processes. In this way, electrons and holes are capable of migrating from the semiconductor to the back contact (FTO substrate) of the electrode and to the electrolyte, respectively. For the photoelectrochemical experiments conducted, all samples were treated at 550 °C providing high crystallinity as well as small crystallite size. The results further clarify that the walls of the electrospun α-Fe2O3 fibres are composed of merely a few crystallites which might be an advantageous structure concerning photoelectrochemical activity.
For a further detailed analysis of the crystal structure and exclusion of impurities, the bare hematite and composite samples were further investigated by Raman spectroscopy as illustrated in Fig. 4. Hematite crystallises in the corundum structure (see XRD analysis, JCPDS no. 003-0800), and Raman spectroscopy analysis showed, according to its classification as the trigonal D63d space group symmetry, seven active Raman phonon modes (2A1g + 5Eg modes). The Raman spectra of dense and hollow hematite nanofibres (Fig. 4A) all possessed typical phonon modes appearing in particular at 242, 289, 406 and 608 cm−1 which can be assigned to vibrations with symmetry Eg, whereas the A1g mode is exhibited at 223 and 498 cm−1. We noted that the signal at 289 cm−1 can be deconvoluted into two Eg modes indicated by the appearance of a pronounced shoulder. The mode at 660 cm−1 which is not allowed for defect-free hematite, but was also observed for other sol–gel derived, nanostructured iron oxide materials,59 is most likely assigned to grain boundaries and disorder effects at the fibre surface. In nanomaterials the lack of long-range order is capable of leading to reduction of space symmetry and activation of extra modes.60,61 Interestingly, the intensity of the broad band at 660 cm−1 was highest for the composite system rather than for hematite (dotted line in Fig. 4A), which gives rise to the assumption that the composite fibres show a slightly higher degree of disorder of the surface atoms.
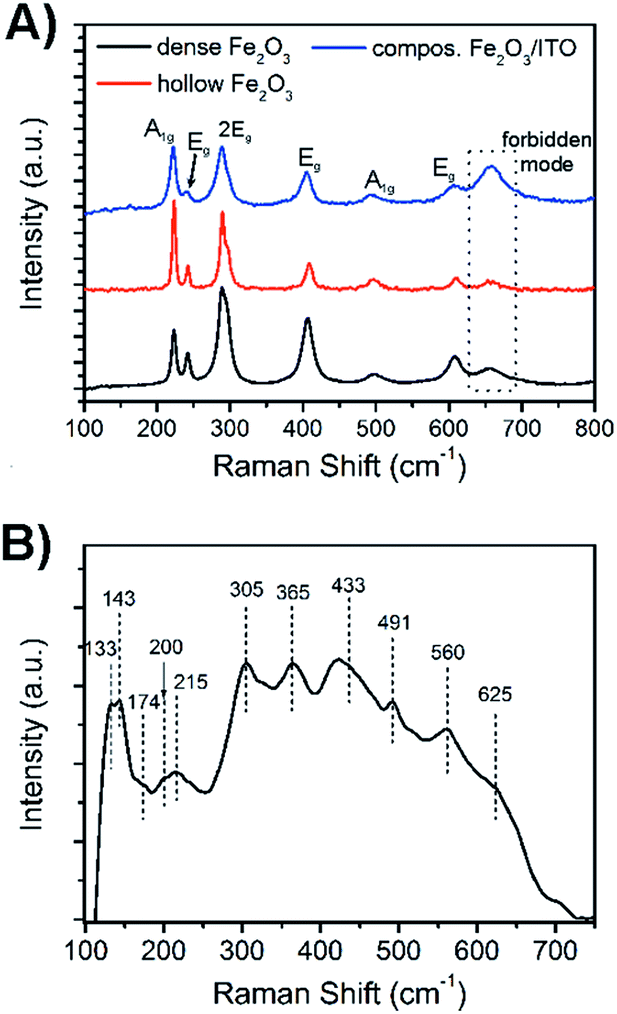 | ||
| Fig. 4 (A) Raman spectra of dense α-Fe2O3 fibres (black), hollow α-Fe2O3 (red) and composite α-Fe2O3/ITO fibres. (B) Raman spectrum of ITO nanoparticles calcined at 550 °C. The numbers in the spectra reveal the wavenumber the Raman mode corresponds to. | ||
This observation is reasonable taking into account that the ITO nanoparticles are distributed throughout the hematite fibre, which possibly disturb the fibre structure and induce stress and strain along the fibre surface. XRD analysis demonstrated that the ITO sample possessed the cubic bixbyite structure with space group Ia,3Th7 (JCPDS no. 071-2195). According to the group theory, 22 Raman-active phonon modes (4Ag + 4Eg + 14F2g) are expected theoretically for In2O3.62 However, it has been shown by Kranert et al. that the number of appearing Raman modes is dependent on the excitation wavelength of the employed laser.63 In general, the authors described that the usage of a laser in the visible spectral range (non-resonant excitation) leads to the reduction of the observed Raman modes for In2O3 ranging from 6 cm−1 to 17 cm−1, while a spectrum excited with λexc = 325 nm reveals almost all Raman vibrations. In our work, the Raman spectrum of the ITO nanoparticles annealed to 550 °C and excited with a laser wavelength of λexc = 532 nm exhibited 11 phonon modes, as presented in Fig. 4B. Among these, the A2g modes can be found at 133, 491 and 560 cm−1, while vibrations with F2g symmetry appear at 143, 202, 215, 305, 365, 433, 517, and 625 cm−1. The single Eg peak can be assigned to 177 cm−1. The evaluated Raman modes are in accordance with the literature.63,64 The broad Raman signals observed for the ITO nanoparticles and hematite fibres and the red shift among the hematite samples can both be attributed to the phonon confinement effect and are well known for other nanoscopic metal oxides.65–67
We note that for the composite fibres no ITO bands appeared in the Raman spectrum which might be explained by the superposition of both spectra and the less pronounced signal intensity of ITO modes. The hematite samples were excited with a laser intensity of 0.2 mW, whereas the ITO samples needed an excitation power of 2 mW to obtain a well-defined spectrum typical of ITO.64
Besides the described impact of the crystal structure and the nanoscopic morphology of the investigated materials on their photoelectrochemical properties, the precise determination of the specific surface area is of great importance and offers detailed information about the absolute area of the interface between the photoactive material and the electrolyte, which in turn determines the charge carrier exchange and thus the overall photoresponse. Therefore, the nanofibres were characterised by physisorption experiments using nitrogen as the adsorptive (T = 77 K). Fig. 5 shows the isotherms of the dense α-Fe2O3, hollow α-Fe2O3 and composite α-Fe2O3/ITO nanofibres annealed at 550 °C exhibiting specific surface areas of 3 m2 g−1, 8 m2 g−1, and 7 m2 g−1, respectively. The specific surface areas were determined based on the Brunauer–Emmett–Teller theory (BET).68 As ceramic nanofibres (prepared by the electrospinning method) generally do not possess any micro- and/or mesopores, the evaluated specific surface areas are most likely assigned to the inter-fibre space and are comparable to the reports of other α-Fe2O3 hollow fibres (12 m2 g−1)35,37 with respect to the average (outer) diameter of 300 nm.
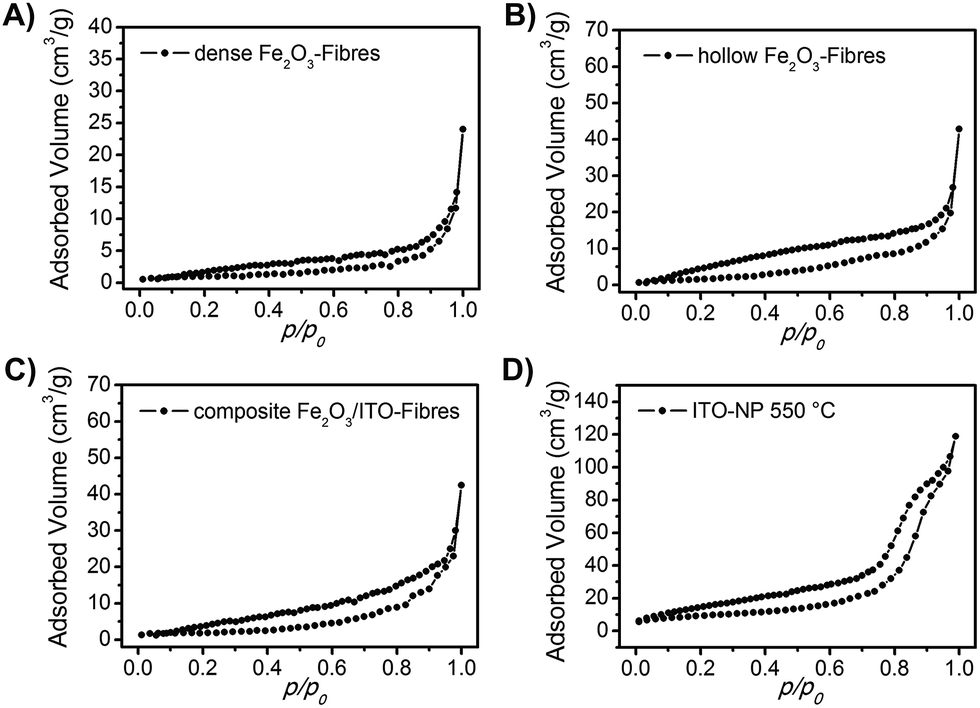 | ||
| Fig. 5 N2 physisorption isotherms of (A) dense α-Fe2O3 fibres, (B) hollow α-Fe2O3 fibres, (C) composite α-Fe2O3/ITO fibres and (D) ITO nanoparticles. All samples have been calcined at 550 °C. | ||
We note that the hollow fibres showed an almost three times higher surface area compared to the dense material which is reasonable as the tube structuring comes along with an increase of surface. Interestingly, the pristine ITO nanoparticles calcined at 550 °C (Fig. 5D) exhibited a distinct isotherm of type IV with a H1 hysteresis typical of interparticle pores69 with sizes in the range of the nanoparticles. The shape of the isotherm can be attributed to the capillary condensation of nitrogen in the interparticular voids69,70 created by the spatial arrangement of the spherical ITO-nanoparticles as presented by the pore size distribution analysis of Barrett–Joyner–Halenda (BJH, Fig. S4, ESI†) showing a strong increase of the cumulative pore volume predominantly between 5 and 10 nm. The specific surface area was determined to be 32 m2 g−1. The physisorption measurements support the SEM and TEM investigation concerning the structural composition of the composite fibres suggesting that the ITO nanoparticles are mainly distributed within the fibrous structure and hence are not accessible for the adsorption gas during the measurement.
Fig. 6A exhibits the mass change during the formation of as-spun dense (black) and hollow α-Fe2O3 (red) fibres and α-Fe2O3 (blue) composite fibres during thermogravimetric measurements in air. The residual mass of the three samples amounts to 13%, 6.5% and 19.5% indicating the highest mass loss for hollow fibres. This finding can be explained by the PVP content of the as-spun hollow and dense fibres, which is approximately 70% and 50%, respectively. As expected, the composite fibres show the lowest decline owing to the presence of ITO nanoparticles. The dried nanoparticles show a weight loss of around 6% due to 2–3 wt% of bound ethylene diamine utilised as the stabiliser and 3–4 wt% of benzyl alcohol remaining from the particle synthesis.54 The obtained residual masses are in agreement with theoretically determined values.
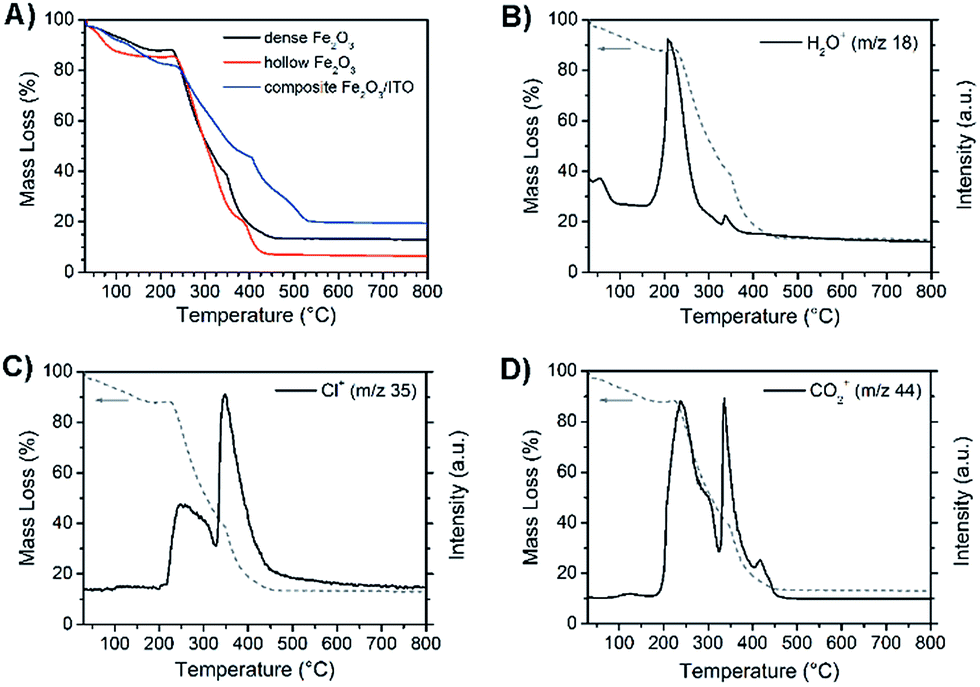 | ||
| Fig. 6 (A) Thermogravimetric analysis (TGA) of dense (black) and hollow α-Fe2O3 fibres (red) and composite α-Fe2O3/ITO fibres (blue) heated in air. Mass spectrometry of ionic fragments with (B) m/z = 18, (C) m/z = 35 and (D) m/z = 44 detected during pyrolysis of dense α-Fe2O3 fibres in air. | ||
All three samples reveal comparable mass loss transients, starting with a weak decline in mass due to desorption of water (m/z = 18) up to 100 °C as illustrated exemplarily by the MS analysis of dense α-Fe2O3 fibres (Fig. 6B). The main signal of m/z = 18 is observed at 200 °C and causes a weight loss of about 15%. The thermal degradation of the α-Fe2O3 nanofibres predominantly occurs between 220 and 460 °C. In this range the dense and hollow fibres lose 75% and 78% of their total mass, respectively. This observation goes in line with the detection of the ionic degradation products with m/z = 35 (Fig. 6C) and 44 (Fig. 6D), which can be assigned to fragmentation signals of Cl+ and CO2+. As depicted in Fig. 6C the strongest signals for the Cl+ fragments, which can be ascribed to the decomposition of FeCl3,71 were observed at around 250 °C and 350 °C, whereas CO2+ was mainly detected between 250 and 450 °C (Fig. 6D). The detection of CO2+ is in general an indication of the degradation of organic compounds in an oxygenic atmosphere72 and can particularly be attributed to the decomposition of PVP which most likely degrades in this temperature range as confirmed by the thermogravimetric analysis of pure PVP-fibres (Fig. S5, ESI†).
Compared to pure α-Fe2O3, the composite fibres show a weaker decrease in mass loss, which is ascribed to the presence of ITO nanocrystals exhibiting a weight loss of only 6 wt% as they are already fully crystallised and contain only a low amount of surface-bound organics. From the results it can be noted that the samples require a temperature of at least 500 °C before the mass change ceases and a metal oxide network is formed. When discussing the photoelectrochemical properties of α-Fe2O3, thorough investigation of its absorption is of fundamental interest as the absorption of light precedes the generation of electron–hole pairs. Hence, the dense, hollow and composite α-Fe2O3/ITO nanofibres were characterised by UV-Vis spectroscopy. All samples showed a strong absorption between 400 nm and 650 nm as indicated by the Kubelka–Munk function, F(R), in dependence of the wavelength (Fig. S6A, ESI†) which is typical of the absorption spectrum of α-Fe2O3.73 In Fig. 7A the presentation of the Tauc plots of dense, hollow and composite fibres showed that the band gap absorption, considering an indirect absorption process in α-Fe2O3,74 is between 2.0 and 2.1 eV.75 However, assuming a direct optical transition, which is also reported in the literature,76,77 a slight decrease of the band gap energies is observed (Fig. 7B). The evaluation of the intersection of the slope of the (direct) Tauc plot data with the baseline resulted in energies of 1.9 eV, 2.0 eV and 2.1 eV for dense, hollow and composite fibres. The slight deviations between indirect and direct transitions are in agreement with other α-Fe2O3 nanostructures.30,59 Interestingly, the Tauc plots of the composite α-Fe2O3/ITO fibres showed a steeper increase for energies above 3 eV most likely indicating the presence of ITO nanoparticles. For a profound understanding of the absorption properties of the composite fibres, the bare ITO nanoparticles were investigated by UV-Vis spectroscopy for comparison. As shown in Fig. 7D, the as-prepared ITO nanocrystals possess a direct optical band gap of around 3.8 eV decreasing to 3.6 eV after calcination at 550 °C which corresponds well with other reports.78,79 The results indicate that the band gap of the merely 7 nm-sized ITO nanoparticles decreases as a consequence of the temperature treatment which is probably attributed to the nano-confinement effect.80 As demonstrated by XRD analysis, the crystallite size increases up to 10 nm during annealing at 550 °C resulting in less confined charge carriers within the crystal structure which possibly lead to band-gap narrowing. When assuming an indirect optical transition, the same trend is conspicuous with a band gap of 3.3 eV for the pyrolysed and 3.5 eV for the as-prepared ITO particles (Fig. 7C). Consequently, the slope of the Tauc plot data between 3 and 4 eV for the α-Fe2O3/ITO composite fibres can be interpreted as the superposition of both the absorption spectra of α-Fe2O3 and ITO. The ITO particles revealed a pronounced absorption of wavelengths between 200 nm and 400 nm (Fig. S6B, ESI†).
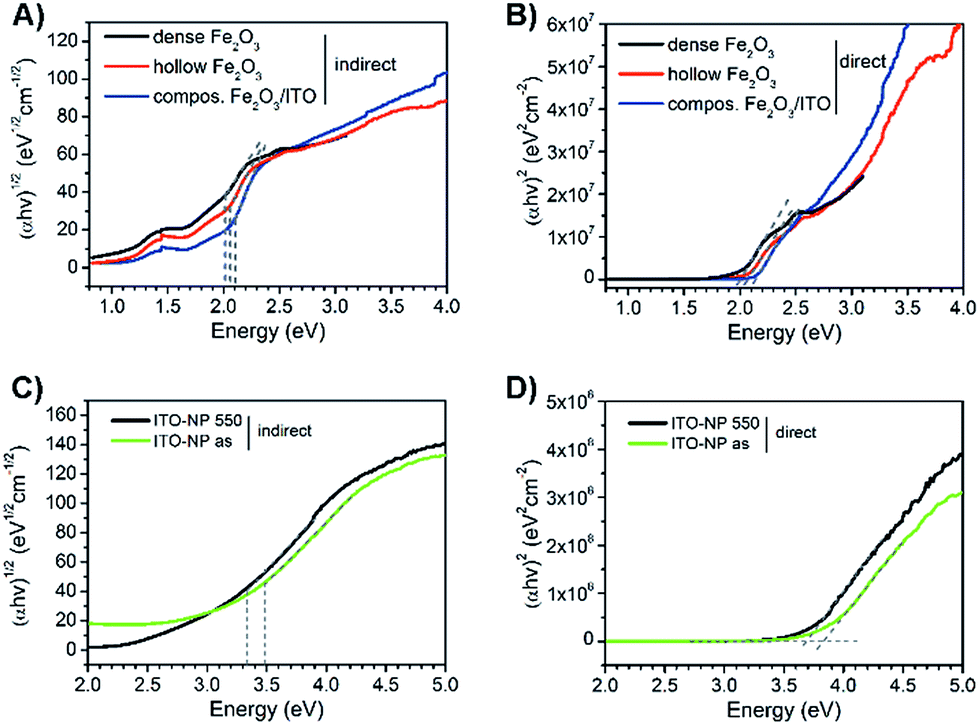 | ||
| Fig. 7 Tauc plots of dense (black), hollow (red) and composite α-Fe2O3/ITO nanofibres (blue) with evaluation of the optical band gap assuming (A) indirect and (B) direct absorption behaviour. Tauc plots of as-prepared (green) and 550 °C annealed ITO nanoparticles (black) for (C) indirect and (D) direct absorption characteristics. The dotted lines serve as guidance for the eyes for optical band gap determination. | ||
Photoelectrochemistry
To study the photoelectrochemical performance of dense and hollow α-Fe2O3 nanofibre photoanodes and to compare them with α-Fe2O3/ITO (ratio 2:3 by weight) composite fibre anodes, the photocurrent response under illumination (white light LED, emission spectrum shown in Fig. S8, ESI†) in a three-electrode setup was determined. Fig. 8A depicts the photocurrents under intermittent light irradiation of dense (black), hollow (red) and composite α-Fe2O3/ITO fibres (blue). At a potential of 0.4 V vs. Ag/AgCl in an aqueous 1 M NaOH (pH = 13.6) – corresponding to 1.4 V vs. reversible hydrogen electrode (RHE) – the dense, hollow α-Fe2O3 and composite α-Fe2O3/ITO photoanodes showed photocurrent densities of 2.1 μA cm−2, 3.4 μA cm−2 and 3.9 μA cm−2 (Fig. S7D†), respectively, with the highest value observed for the composite fibres. Since the photocurrent densities are relatively low compared to other nanostructured hematite anodes11 owing to the extremely small amount of active mass per square centimetre, which was determined to be 15–19 μg, the obtained photocurrents were normalised to the active mass and are shown as gravimetric currents in Fig. 8A. The mass activities of the dense, hollow, and composite photoanodes were 0.11 mA mg−1, 0.16 mA mg−1, and 0.25 mA mg−1, respectively. Thus, the superior performance of the composite relative to the dense system is even more pronounced, gravimetric photocurrent being more than doubled. The results suggest that the evaluation not only of the current densities, but also the normalisation to the active mass is more meaningful concerning the photoelectrochemical performance of such highly porous anode materials. The determined values are compared in Table 1. Furthermore, the performance of hollow nanofibres revealed a low signal-to-noise ratio, which can be explained by the high degree of porosity and thus the lower amount of active material compared to compact fibres.
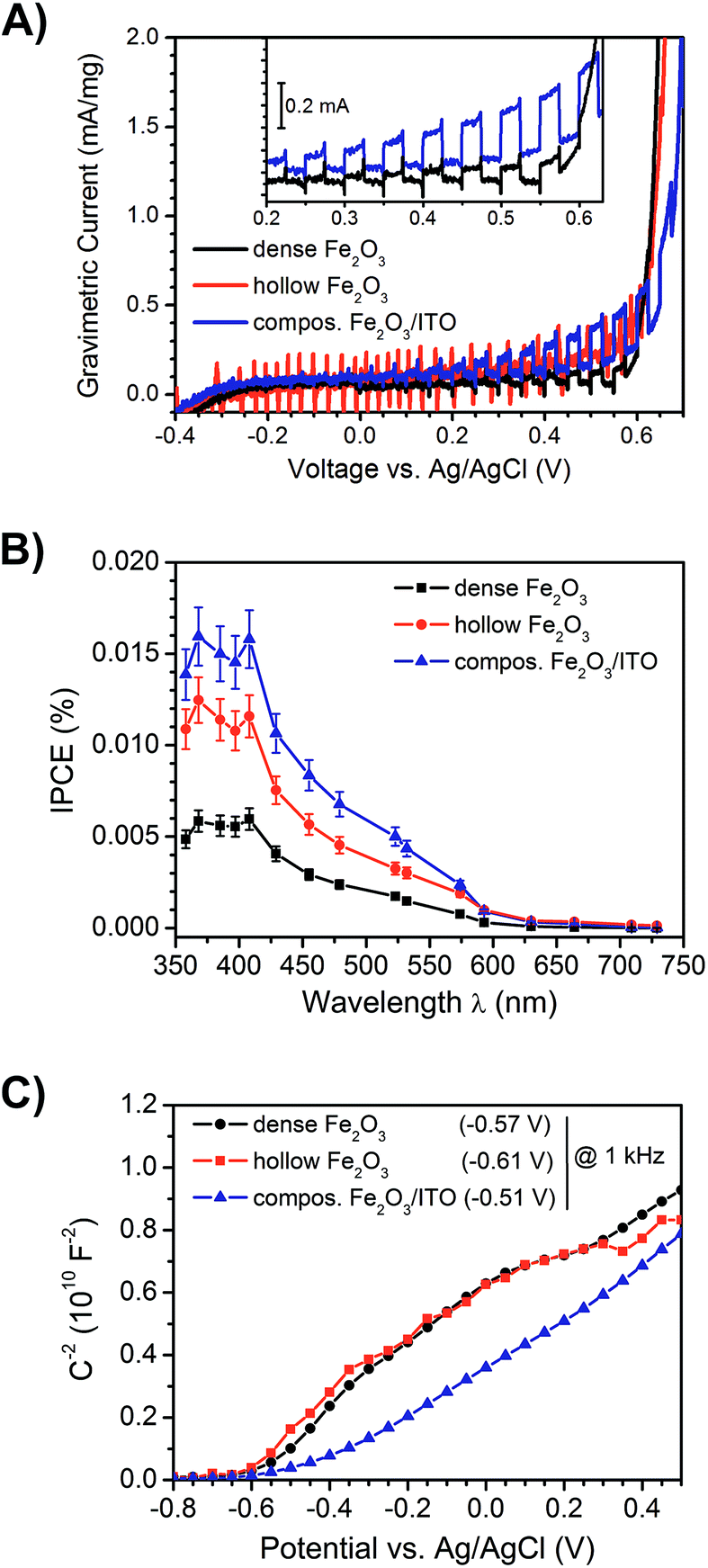 | ||
| Fig. 8 Photoelectrochemical characterisation of dense (black), hollow α-Fe2O3 (red) and composite α-Fe2O3/ITO fibres (blue) serving as anode materials in a three electrode cell vs. Ag/AgCl in 1 M NaOH (pH 13.6): (A) gravimetric photocurrents, (B) IPCE determined at a potential of 430 mV and (C) Mott–Schottky plot with evaluation of the flat band potential Vfb. The connecting lines between the data points serve as guidance for the eye. | ||
| Dense α-Fe2O3 | Hollow α-Fe2O3 | Composite α-Fe2O3/ITO | ITO nanoparticles | |
|---|---|---|---|---|
| Size/diameter, Ø (nm) | 70–100 | 200–400 | 150–300 | 10–12 |
| Crystallite size (nm) | 62 | 57 | 10.6 (ITO), 40 (Fe2O3) | 10 |
| BET surface area (m2 g−1) | 3 | 8 | 6.5 | 32 |
| Weight loss at 550 °C (%) | 86.5 | 93 | 80 | 94 |
| Band gap, Egap (eV) | 2.0 | 2.1 | 2.2 | 3.6 |
| Gravimetric photocurrents at 0.4 V (mA mg−1) | 0.11 | 0.16 | 0.25 | n.e. |
| Photocurrent density at 0.4 V (μA cm−2) | 2.1 | 3.4 | 3.9 | n.e. |
| IPCE at 368 nm (m%) | 15.9 | 12.5 | 5.8 | n.e. |
| Flat band potential, Vfb (V) | −0.57 | −0.61 | −0.51 | n.e. |
| Donor density, ND′ (cm3)−1 | 1.8 × 1020 | 1.9 × 1020 | 2.4 × 1020 | n.e. |
Interestingly, the α-Fe2O3/ITO composite exhibited the highest photocurrents and extremely weak anodic transients after switching on the light source for a time period of 10 seconds. In contrast, the photocurrent transient for pure (dense) α-Fe2O3 fibres was well pronounced and revealed long lasting decays to the steady-state value after illumination, which clearly indicated that the recombination of the photogenerated electron–hole pairs was substantially increased in the pure materials. These observations are reasonable with respect to the ultrashort diffusion length of minority charge carriers (which are only 2–4 nm) promoting recombination before the migration to the SCLJ can occur. However, it has to be mentioned that the detection of photocurrents for α-Fe2O3 structures prepared at calcination temperatures as low as 550 °C has rarely been reported, as electron transport processes are strongly limited by recombination in bulk or at grain boundaries.56,81 Therefore, the enhanced photocurrents of hollow fibres can be ascribed to the relatively short diffusion pathways of 30–60 nm compared to dense morphologies revealing dimensions between 70 and 100 nm (both illustrated by our SEM investigations). On the other hand, the presence of weak anodic and cathodic transients – which were observed for the composite system – is an indication of suppressed recombination of the photo-induced electron–hole pairs. Considering the timescale, the probability of recombination processes which occur within a few picoseconds45 increases with time, therefore fast charge carrier transfer and separation is essential to circumvent these processes. The recombination rate of the composite nanofibres seems to be significantly decreased, which was shown by improved photocurrents and a substantially decreased transient decay (Fig. 8A, inset). Therefore, the presence of ITO-nanoparticles, embedded throughout the α-Fe2O3 fibre string, presumably enabled fast electron transfer, and was additionally favoured by the large contact area between both phases. As ITO is a highly conductive transparent oxide (ρ ≈ 10−3 Ω cm for sol–gel derived thin films)82,83 it is most likely that electrons were transferred from the excitation sites of the poorly conductive α-Fe2O3 to the ITO particles inhibiting the recombination process and thus improving the photocurrent response of composite fibres, as illustrated in Fig. 9.
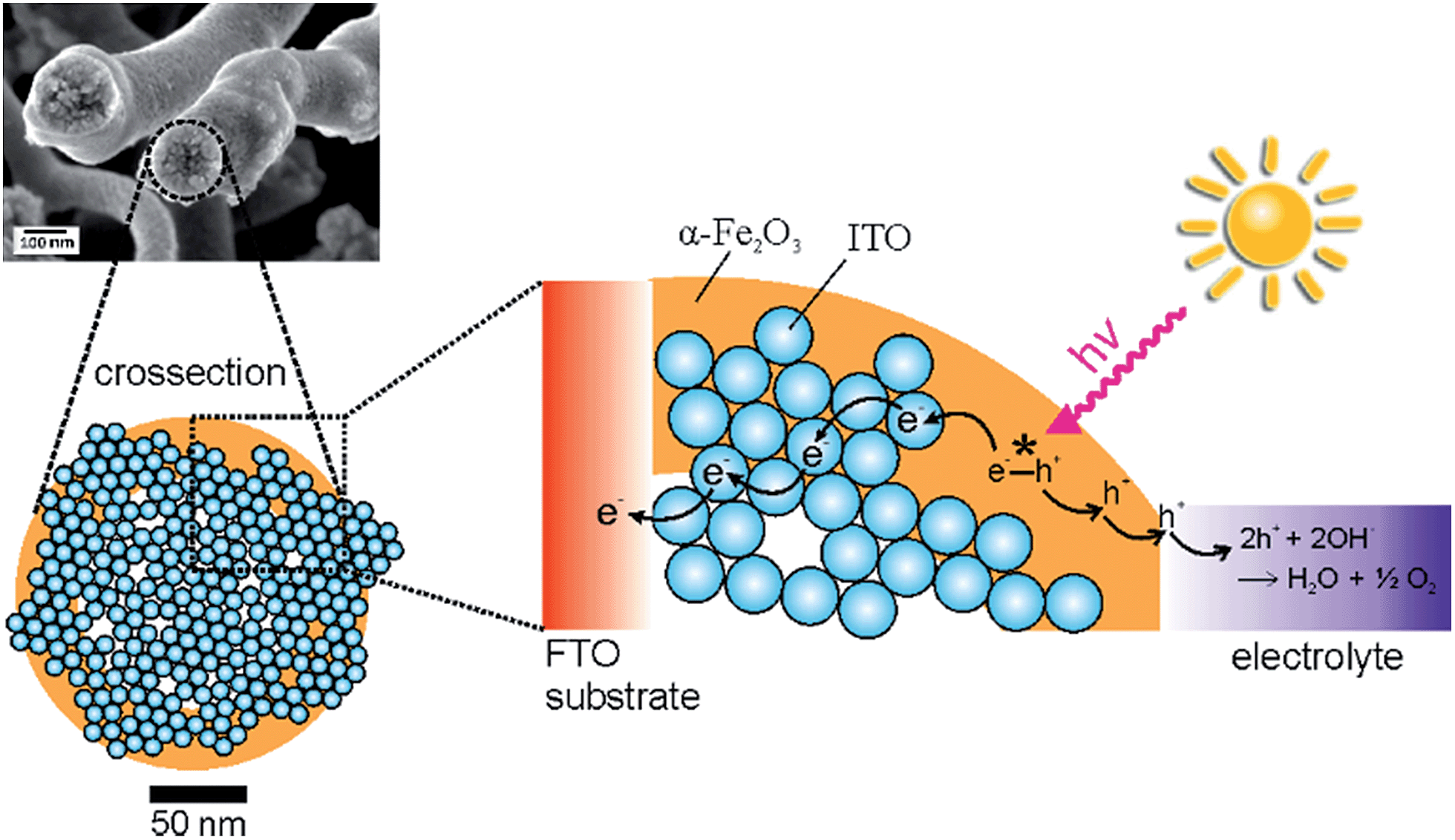 | ||
| Fig. 9 Schematic illustration of the charge transport process of photogenerated charge carriers in α-Fe2O3/ITO composite fibres, shown as a cross-section: electrons are transferred from hematite to the highly conductive, nanoparticular ITO agglomerates, while the holes migrate to the fibre's surface, which is in direct contact with the electrolyte thus efficiently promoting spatial charge carrier separation. | ||
The obtained photocurrents were further confirmed by evaluation of the IPCE at a potential of 430 mV vs. Ag/AgCl (detailed description of the experiment is given in the ESI†) as illustrated in Fig. 8B. All samples showed the highest efficiency between 350 nm and 420 nm followed by a steady decrease and finally almost no detection of IPCE signals above 600 nm, which is in good accordance with the determined band gap energies (2.0–2.1 eV, see Fig. 7). The highest IPCE value was found for the mixed ITO/α-Fe2O3 morphology, followed by the pure α-Fe2O3 nanotubes indicating again that in the composite morphology more electrons are transported to the back contact of the electrode.
The generally small photocurrents and IPCE values of the electrospun nanofibres investigated in this work can be predominantly ascribed to the minor amount of active material resulting from the (macro-)porous nature of the fibrous electrodes, and the absence of any co-catalyst and/or doping metals, usually enhancing the performance of hematite significantly.29,30
From Mott–Schottky measurements (Fig. 8C) the flat band potentials Vfb were determined by extrapolating the slope of the curves between −0.4 V and 0.2 V. The Vfb were found to be −0.57 V, −0.61 V, and −0.51 V vs. Ag/AgCl measured at 1 kHz for the dense, hollow and composite system, respectively. These values are in good agreement with the ones reported for α-Fe2O3 in the literature,84 but it has been shown that the accurate evaluation of the Vfb depends strongly on the applied frequency.85 However, for all samples the value of Vfb was constant for frequencies ranging from 10 Hz to 10 kHz (Fig. S7A, ESI†). Besides, the curves of pure and composite fibres differ from each other. Whilst the pure fibres possessed a convex trend, the composite exhibited a more concave curving upwards, which is most likely ascribed to the surface roughness effect.84,86 Such interpretation is reasonable, as the composite fibres exhibit a rougher surface due to ITO particle agglomeration. According to the Mott–Schottky equation,
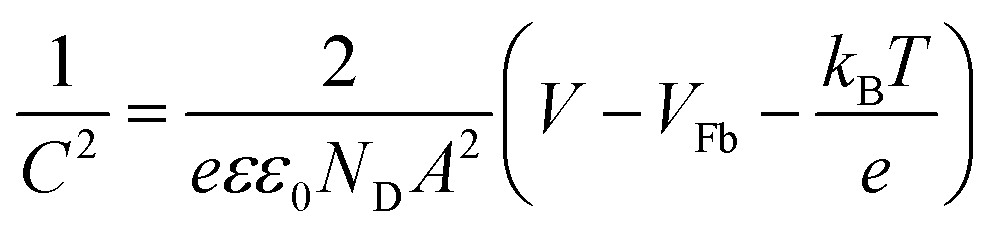
Conclusions
In this work, we have performed single-nozzle electrospinning of FeCl3 and PVP in an EtOH/DMF mixture. The obtained as-spun fibres were successfully transformed into dense and hollow α-Fe2O3 nanofibres with diameters ranging from 70 nm to 100 nm, and wall thicknesses of 10 nm to 60 nm after annealing at 550 °C. Concerning the formation mechanism of α-Fe2O3 hollow fibres, we have found that the humidity in the spinning chamber and the water content in the precursor solution exert a major impact on the morphological evolution of the α-Fe2O3 system during electrospinning. By simply adding a dispersion of stabilised ITO nanoparticles in ethanol to the initial ferric solution, a stable colloidal ITO/FeCl3 solution was obtained, which was utilised to produce highly accessible, α-Fe2O3/ITO nanocomposite fibres by electrospinning. The dense, hollow and composite nanofibres were characterised by means of SEM, TEM, XRD and Raman spectroscopy revealing trigonal α-Fe2O3 regarding the characterisation of α-Fe2O3/ITO composites – a homogeneous distribution of nanoparticular ITO throughout the hematite fibres revealing a core/shell-like morphology. Photoelectrochemical characterisation was performed through photocurrent density, IPCE and Mott–Schottky measurements, clearly indicating that the tube structuring of α-Fe2O3 fibres led to a superior photoresponse when investigated in aqueous alkaline media as the photoanode in a photoelectrochemical cell. A doubling of the photocurrent (relative to pristine α-Fe2O3) could be achieved through the design of α-Fe2O3/ITO nanocomposites. The results suggest that the presence of ITO nanocrystals serving as a conducting component promotes the electron transport and thus spatial charge carrier separation, leading to suppressed electron–hole recombination within the composite material.Acknowledgements
The authors would like to thank Christoph Seitz for fruitful discussion and support during characterisation of the ITO nanoparticles, Rüdiger Ellinghaus for physisorption measurements, Christian Suchomski for his support in Raman investigations, and Hubert Wörner for accomplishment of TGA experiments (all Justus-Liebig-University Giessen). R. M. gratefully acknowledges funding in the Emmy-Noether program of the German Research Foundation DFG (MA 5392/3-1). B. M. S. acknowledges support by the DFG (SM 199/9-1).Notes and references
- M. Graetzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef CAS
.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS
- L. F. Brown, Int. J. Hydrogen Energy, 2001, 26, 381–397 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191–195 CrossRef CAS PubMed
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS PubMed
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed
- R. Liu, Y. Lin, L.-Y. Chou, S. W. Sheehan, W. He, F. Zhang, H. J. M. Hou and D. Wang, Angew. Chem., 2011, 123, 519–522 CrossRef
- B. Cole, B. Marsen, E. Miller, Y. Yan, B. To, K. Jones and M. Al-Jassim, J. Phys. Chem. C, 2008, 112, 5213–5220 CAS
- E. L. Miller, B. Marsen, B. Cole and M. Lum, Electrochem. Solid-State Lett., 2006, 9, G248–G250 CrossRef CAS
- D. K. Bora, A. Braun and E. C. Constable, Energy Environ. Sci., 2013, 6, 407–425 CAS
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed
- D. A. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682–6702 CAS
- Y. Lin, G. Yuan, S. Sheehan, S. Zhou and D. Wang, Energy Environ. Sci., 2011, 4, 4862–4869 CAS
- M. Mishra and D.-M. Chun, Appl. Catal., A, 2015, 498, 126–141 CrossRef CAS
- X. Yang, A. Wolcott, G. Wang, A. Sobo, R. C. Fitzmorris, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2009, 9, 2331–2336 CrossRef CAS PubMed
- A. Wolcott, W. A. Smith, T. R. Kuykendall, Y. Zhao and J. Z. Zhang, Adv. Funct. Mater., 2009, 19, 1849–1856 CrossRef CAS
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed
- T. S. Sinclair, B. M. Hunter, J. R. Winkler, H. B. Gray and A. M. Müller, Mater. Horiz., 2015, 2, 330–337 RSC
- R. C. Vernon, J. Appl. Phys., 1962, 33, 2140–2141 CrossRef CAS
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Grey, M. D. Horne and J. A. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed
- J. R. Bolton, Sol. Energy, 1996, 57, 37–50 CrossRef CAS
- S. C. Warren, K. Voïtchovsky, H. Dotan, C. M. Leroy, M. Cornuz, F. Stellacci, C. Hébert, A. Rothschild and M. Grätzel, Nat. Mater., 2013, 12, 842–849 CrossRef CAS PubMed
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 CAS
- H. P. Maruska and A. K. Ghosh, Sol. Energy Mater., 1979, 1, 237–247 CrossRef CAS
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and P. R. Trevellick, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2027–2041 RSC
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., 2010, 122, 6549–6552 CrossRef
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed
- R. Ostermann, J. Cravillon, C. Weidmann, M. Wiebcke and B. M. Smarsly, Chem. Commun., 2011, 47, 442–444 RSC
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed
- M. Einert, C. Wessel, F. Badaczewski, T. Leichtweiß, C. Eufinger, J. Janek, J. Yuan, M. Antonietti and B. M. Smarsly, Macromol. Chem. Phys., 2015, 216, 1930–1944 CrossRef CAS
- P. Voepel, C. Suchomski, A. Hofmann, S. Gross, P. Dolcet and B. Smarsly, CrystEngComm, 2016, 18, 316–327 RSC
- S. Chaudhari and M. Srinivasan, J. Mater. Chem., 2012, 22, 23049–23056 RSC
- N. Saveh-Shemshaki, M. Latifi, R. Bagherzadeh, M. M. Byranvand, N. Naseri and A. Dabirian, Polym. Adv. Technol., 2016, 27, 358–365 CrossRef CAS
- Y. Liu, H. Yu, S. Zhan, Y. Li, Z. Lv, X. Yang and Y. Yu, J. Sol-Gel Sci. Technol., 2011, 58, 716–723 CrossRef CAS
- N. Li, S. Jayaraman, S. Y. Tee, P. S. Kumar, C. J. J. Lee, S. L. Liew, D. Chi, T. A. Hor, S. Ramakrishna and H.-K. Luo, J. Mater. Chem. A, 2014, 2, 19290–19297 CAS
- Y. Cheng, B. Zou, C. Wang, Y. Liu, X. Fan, L. Zhu, Y. Wang, H. Ma and X. Cao, CrystEngComm, 2011, 13, 2863–2870 RSC
- E. H. Sanders, R. Kloefkorn, G. L. Bowlin, D. G. Simpson and G. E. Wnek, Macromolecules, 2003, 36, 3803–3805 CrossRef CAS
- A. V. Bazilevsky, A. L. Yarin and C. M. Megaridis, Langmuir, 2007, 23, 2311–2314 CrossRef CAS PubMed
- E. Cynthia, B. Arnaud, S. Vincent, P. Jean-Claude, A. Roy, M. Yves, K. Randa, K. Antonio and M. Philippe, Beilstein J. Nanotechnol., 2010, 21, 125701 Search PubMed
- M. T. Mayer, Y. Lin, G. Yuan and D. Wang, Acc. Chem. Res., 2013, 46, 1558–1566 CrossRef CAS PubMed
- N. J. Cherepy, D. B. Liston, J. A. Lovejoy, H. Deng and J. Z. Zhang, J. Phys. Chem. B, 1998, 102, 770–776 CrossRef CAS
- A. G. Joly, J. R. Williams, S. A. Chambers, G. Xiong, W. P. Hess and D. M. Laman, J. Appl. Phys., 2006, 99, 053521 CrossRef
- J. Li, Y. Qiu, Z. Wei, Q. Lin, Q. Zhang, K. Yan, H. Chen, S. Xiao, Z. Fan and S. Yang, Energy Environ. Sci., 2014, 7, 3651–3658 CAS
- Y.-F. Xu, H.-S. Rao, B.-X. Chen, Y. Lin, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, Adv. Sci., 2015, 2, 1500049 CrossRef
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS
- S. H. Nam, H.-S. Shim, Y.-S. Kim, M. A. Dar, J. G. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 2046–2052 CAS
- Y. Yu, L. Gu, C. Zhu, P. A. Van Aken and J. Maier, J. Am. Chem. Soc., 2009, 131, 15984–15985 CrossRef CAS PubMed
- C. Grote, T. Cheema and G. Garnweitner, Langmuir, 2012, 28, 14395–14404 CrossRef CAS PubMed
- C. Grote, K. J. Chiad, D. Vollmer and G. Garnweitner, Chem. Commun., 2012, 48, 1464–1466 RSC
- J. Ba, D. F. Rohlfing, A. Feldhoff, T. Brezesinski, I. Djerdj, M. Wark and M. Niederberger, Chem. Mater., 2006, 18, 2848–2854 CrossRef CAS
- T. Wahl, S. Zellmer, J. Hanisch, G. Garnweitner and E. Ahlswede, Thin Solid Films, 2016, 616, 419–424 CrossRef CAS
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555–560 CrossRef CAS
- S. Lahiri and S. Sarkar, Appl. Radiat. Isot., 2007, 65, 387–391 CrossRef CAS PubMed
- R. Hill and C. Howard, J. Appl. Crystallogr., 1987, 20, 467–474 CrossRef CAS
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CAS
- K. Brezesinski, J. Haetge, J. Wang, S. Mascotto, C. Reitz, A. Rein, S. H. Tolbert, J. Perlich, B. Dunn and T. Brezesinski, Small, 2011, 7, 407–414 CrossRef CAS PubMed
- D. Bersani, P. Lottici and A. Montenero, J. Raman Spectrosc., 1999, 30, 355–360 CrossRef CAS
- A. A. Tahir, K. G. U. Wijayantha, S. Saremi-Yarahmadi, M. Mazhar and V. McKee, Chem. Mater., 2009, 21, 3763–3772 CrossRef CAS
- W. B. White and V. G. Keramidas, Spectrochim. Acta, Part A, 1972, 28, 501–509 CrossRef CAS
- C. Kranert, R. Schmidt-Grund and M. Grundmann, J. Phys. D: Appl. Phys., 2014, 8, 554–559 CAS
- O. M. Berengue, A. D. Rodrigues, C. J. Dalmaschio, A. J. Lanfredi, E. R. Leite and A. J. Chiquito, J. Phys. D: Appl. Phys., 2010, 43, 045401 CrossRef
- D. Bersani, P. P. Lottici and X.-Z. Ding, Appl. Phys. Lett., 1998, 72, 73–75 CrossRef CAS
- D. Bersani and P. P. Lottici, Phys. Status Solidi B, 1992, 174, 575–582 CrossRef CAS
- J. Xu, W. Ji, Z. Shen, W. Li, S. Tang, X. Ye, D. Jia and X. Xin, J. Raman Spectrosc., 1999, 30, 413–415 CrossRef CAS
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids, Academic Press, San Diego 1999 Search PubMed.
- S. Kaskel, K. Schlichte, G. Chaplais and M. Khanna, J. Mater. Chem., 2003, 13, 1496–1499 RSC
- A. Bhattacharya, A. Hartridge, K. Mallick, C. Werrett and J. Woodhead, J. Mater. Sci., 1996, 31, 4479–4482 CrossRef CAS
- K. P. Pramoda, T. Liu, Z. Liu, C. He and H.-J. Sue, Polym. Degrad. Stab., 2003, 81, 47–56 CrossRef CAS
- L. A. Marusak, R. Messier and W. B. White, J. Phys. Chem. Solids, 1980, 41, 981–984 CrossRef CAS
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS
- H. Fan, G. You, Y. Li, Z. Zheng, H. Tan, Z. Shen, S. Tang and Y. Feng, J. Phys. Chem. C, 2009, 113, 9928–9935 CAS
- N. Beermann, L. Vayssieres, S. E. Lindquist and A. Hagfeldt, J. Electrochem. Soc., 2000, 147, 2456–2461 CrossRef CAS
- A. Kleiman-Shwarsctein, M. N. Huda, A. Walsh, Y. Yan, G. D. Stucky, Y.-S. Hu, M. M. Al-Jassim and E. W. McFarland, Chem. Mater., 2009, 22, 510–517 CrossRef
- V. Senthilkumar, P. Vickraman, M. Jayachandran and C. Sanjeeviraja, Vacuum, 2010, 84, 864–869 CrossRef CAS
- M. Alam and D. Cameron, Thin Solid Films, 2000, 377, 455–459 CrossRef
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC
- U. Bjoerksten, J. Moser and M. Graetzel, Chem. Mater., 1994, 6, 858–863 CrossRef CAS
- M. J. Alam and D. C. Cameron, Thin Solid Films, 2000, 377–378, 455–459 CrossRef CAS
- M. J. Alam and D. C. Cameron, Surf. Coat. Technol., 2001, 142–144, 776–780 CrossRef CAS
- Y. Liang, C. S. Enache and R. van de Krol, Int. J. Photoenergy, 2008, 2008, 7 CrossRef
- H. O. Finklea, J. Electrochem. Soc., 1982, 129, 2003–2008 CrossRef CAS
- J. Schoonman, K. Vos and G. Blasse, J. Electrochem. Soc., 1981, 128, 1154–1157 CrossRef CAS
- S. Wilhelm, K. Yun, L. Ballenger and N. Hackerman, J. Electrochem. Soc., 1979, 126, 419–424 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of α-Fe2O3 fibres prepared by calcination of Fe(NO3)3·9H2O/PVP fibres, SEM images of the as-spun α-Fe2O3 nanofibres and as-prepared ITO nanoparticles, table of Raman phonon modes for the α-Fe2O3 and ITO system, TGA of PVP nanofibres, BJH-pore size distribution and cumulative pore volume of the ITO nanoparticles, Kubelka–Munk spectra of dense, hollow and composite α-Fe2O3 fibres as well as of as-prepared and 550 °C annealed ITO nanoparticles, Mott–Schottky plot at distinct frequencies, IPCE spectra at a potential of 430 mV and 600 mV for dense α-Fe2O3 fibres, IPCE spectrum of hollow α-Fe2O3 fibres for the frontside and backside illumination. See DOI: 10.1039/c6ta06979g |
| This journal is © The Royal Society of Chemistry 2016 |