
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Parallel bulk heterojunction photovoltaics based on all-conjugated block copolymer additives†
Jorge W.
Mok
a,
Dylan
Kipp
b,
Luis R.
Hasbun
a,
Andrei
Dolocan
c,
Joseph
Strzalka
d,
Venkat
Ganesan
*b and
Rafael
Verduzco
 *ae
*ae
aDepartment of Chemical and Biomolecular Engineering, Rice University, Houston, Texas 77005, USA. E-mail: rafaelv@rice.edu
bDepartment of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: venkat@che.utexas.edu
cDepartment of Materials Science and Texas Materials Institute, University of Texas at Austin, Austin, Texas 78712, USA
dX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA
eDepartment of Materials Science and NanoEngineering, Rice University, Houston, Texas 77005, USA
First published on 23rd August 2016
Abstract
In recent studies, we demonstrated that the addition of block copolymers to binary donor–acceptor blends represents an effective approach to target equilibrium, co-continuous morphologies of interpenetrating donors and acceptors. Here, we report a study of the impact of all-conjugated poly(thieno[3,4-b]-thiophene-co-benzodithiophene)-b-polynaphthalene diimide (PTB7-b-PNDI) block copolymer additives on the electronic properties and photovoltaic performance of bulk heterojunction organic photovoltaic active layers comprised of a PTB7 donor and a phenyl-C61-butyric acid methyl ester (PCBM61) acceptor. We find that small amounts of BCP additives lead to improved performance due to a large increase in the device open-circuit voltage (VOC), and the VOC is pinned to this higher value for higher BCP additive loadings. Such results contrast prior studies of ternary blend OPVs where either a continuous change in VOC or a value of VOC pinned to the lowest value is observed. We hypothesize and provide evidence in the form of device and morphology analyses that the impact of VOC is likely due to the formation of a parallel bulk heterojunction made up of isolated PCBM and PNDI acceptor domains separated by intermediate PTB7 donor domains. Altogether, this work demonstrates that all-conjugated block copolymers can be utilized as additives to both dictate morphology and modulate the electronic properties of the active layer.
Introduction
Bulk heterojunction (BHJ) organic photovoltaic (OPV) devices are comprised of an interpenetrating network of donor and acceptor semiconductors. Systematic materials design and processing optimization has produced tremendous progress in device performance, and recent studies have reported single-junction device efficiencies exceeding 10%.1,2 However, a key challenge in further development of these devices is finding reliable strategies for controlling and directing the blend morphologies, which exert a significant impact on the electronic properties, stability, mechanical robustness, and overall functionality. A number of strategies have been implemented to optimize and control the nanoscale active layer morphology, including the use of processing additives, solvent annealing, surface treatment, and others, as detailed in recent reviews and studies.3–13The use of block copolymer additives represents an effective approach to control the donor–acceptor morphology. Indeed, properly designed block copolymer additives can be utilized to improve thermal stability and potentially produce equilibrium, bicontinuous phases of interpenetrating donor and acceptor semiconductors. As detailed in recent reviews, a number of studies have reported the design and implementation of conjugated block copolymers in bulk heterojunction active layers and demonstrated clear benefits to using BCP additives.14–17 For example, Chen et al. studied poly(3-hexylthiophene)-b-poly(ethylene oxide) (P3HT-b-PEO) additives and found that these additives could be used to tune domain sizes in thermally annealed OPVs.18 Sun et al. reported P3HT-b-polystyrene (PS) additives that had a beneficial impact on stability, crystallinity, and photovoltaic performance.19 Kim et al. reported P3HT additives with poly(4-vinylpyridine) (P4VP) side-chains that improved thermal stability.20 However, a limitation of these prior studies is that they have focused primarily on block copolymers with optically and electronically inactive polymer blocks or backbones, which can be detrimental to electronic properties, especially at higher loadings. Furthermore, these studies have focused predominantly on BHJ OPVs with P3HT as the donor, while there is still less clarity on effects expected in BHJ OPVs based on the more recently explored non-crystalline, alternating copolymers as the donor.
Fully conjugated block copolymers with donor and acceptor blocks can be utilized as additives to both modulate the morphology and potentially improve the electronic properties of the active layer. However, only a few studies have investigated all-conjugated BCPs as additives to BHJ OPVs. Mulherin et al. demonstrated that all-conjugated BCP additives could suppress phase separation at 40 wt% loadings.21 Lee et al. found that all-conjugated BCP additives improved the performance of P3HT/phenyl-C61-butyric acid methyl ester (PCBM61) BHJ OPVs.22 Recently, we investigated the morphological impact of fully conjugated block copolymers as additives for dictating the morphology of poly(thieno[3,4-b]-thiophene-co-benzodithiophene) (PTB7) bulk heterojunction OPVs. We applied a combined simulation and experiment-based approach to investigate if block copolymer (BCP) additives can be used to intelligently control the self-assembled morphologies of PTB7/PCBM61 mixtures. Single chain in mean field (SCMF) simulations23,24 were used to identify regions within the PTB7/PCBM phase diagram where all-conjugated poly(thieno[3,4-b]-thiophene-co-benzodithiophene)-b-poly(naphthalene diimide) (PTB7-b-PNDI) block copolymer (BCP) additives would lead to co-continuous equilibrium morphologies desired for photovoltaic applications. These simulations predicted the formation of co-continuous phases at much higher block copolymer additive concentrations than that is typically studied (>40 wt%), and TEM and X-ray diffraction analysis of blend films produced qualitative agreement with computational predictions.25 In a subsequent study, we followed up with more detailed SCMF simulations that provided design rules for producing bicontinuous phases with BCP compatibilizers.26 These studies demonstrate that all-conjugated block copolymer additives are useful for dictating the morphology of BHJ OPVs, but the impact of these additives on electronic properties and performance is unclear.
Here, we report a study of the impact of all-conjugated PTB7-b-PNDI block copolymer additives on the electronic properties and photovoltaic performance of bulk heterojunction OPV active layers comprised of a PTB7 donor and a PCBM61 acceptor. We find that small amounts of BCP additives lead to a large increase in the device open-circuit voltage (VOC), and the VOC is pinned to this higher value for higher BCP additive loadings. By comparison, PNDI homopolymer additives produce a continuous increase in VOC with increasing loading. We hypothesize and provide evidence in the form of device and morphology analyses that the impact of VOC is due to the formation of a parallel bulk heterojunction27,28 made up of isolated PCBM-rich and PNDI-rich acceptor domains separated by intermediate PTB7 donor domains. Altogether, this work demonstrates that all-conjugated block copolymers can be utilized as additives to both dictate morphology and modulate the electronic properties of the active layer and may represent a novel approach for producing BHJ OPVs with enhanced VOC.
Experimental details
Materials
4,9-Dibromo-2,7-bis(2-octyldodecyl)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone, 2-ethylhexyl-4,6-dibromo-3-fluorothieno[3,4-b]thiophene-2-carboxylate, and 2,6-bis(trimethyltin)-4,8-bis(2-ethylhexyloxy)benzo[1,2-b:4,5-b′] were purchased from Sunatech Inc. All other reagents were purchased from commercial sources and used as received.Materials synthesis
1H NMR analysis of all synthesized materials is provided in ESI Fig. S1–S3.†
Results and discussion
The majority of all-conjugated block copolymers are comprised of one or more polythiophene blocks,29–32 in which the polythiophene block can be synthesized by controlled catalyst transfer polycondensation.33–35 Here, we target a conjugated block copolymer comprised of two alternating copolymers, PTB7 and PNDI (Scheme 4). Our overall approach to the synthesis of PTB7-b-PNDI block copolymers involves sequential Stille polycondensation reactions along with purification of reactive intermediates and the final product through selective solvent extraction. First, PTB7 was synthesized in a Stille polycondensation reaction followed by fractionation by solvent washing in a Soxhlet extractor. Low and high PTB7 molecular weight fractions were collected by washing with hexanes and chloroform, respectively. Next, a PTB7-b-PNDI block copolymer was synthesized in a Stille polycondensation reaction in the presence of PTB7 macroreagent from the low molecular weight fraction extracted from the first step. After polymerization, the final material was washed in a Soxhlet reactor to remove oligomers and unreacted PTB7 macroreagent before collecting in chloroform.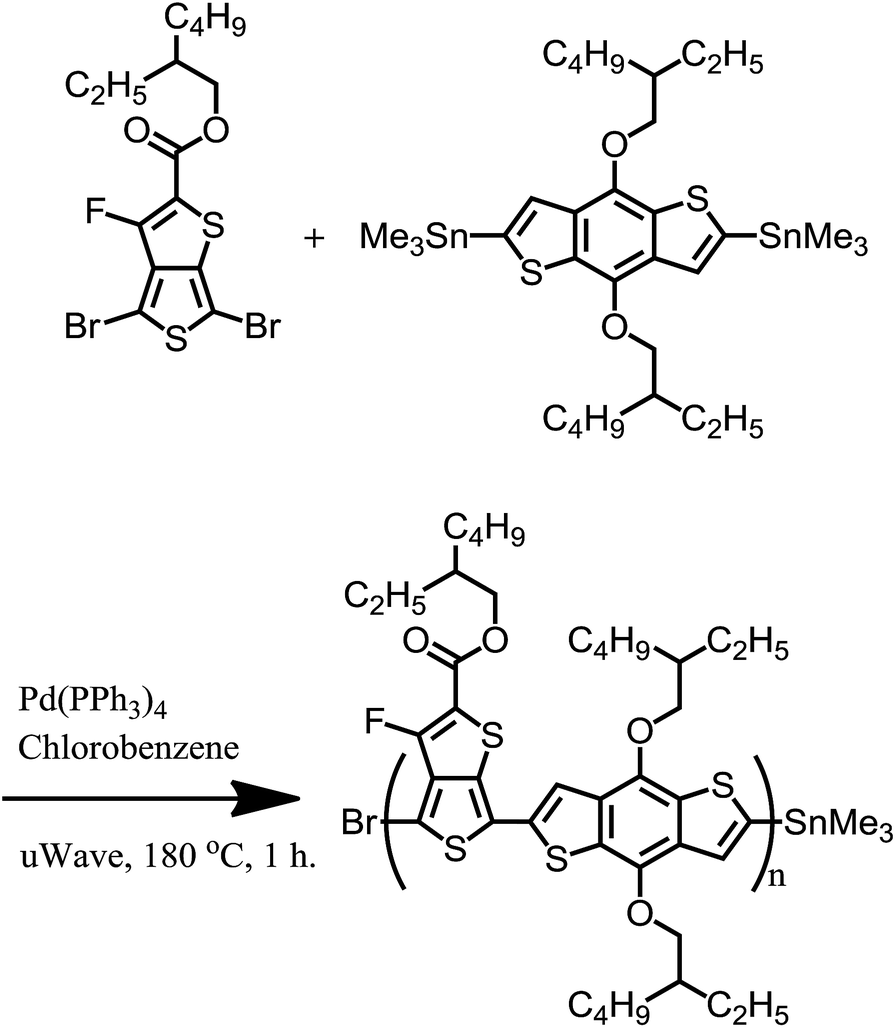 | ||
| Scheme 1 Synthetic scheme for the preparation of PTB7. | ||
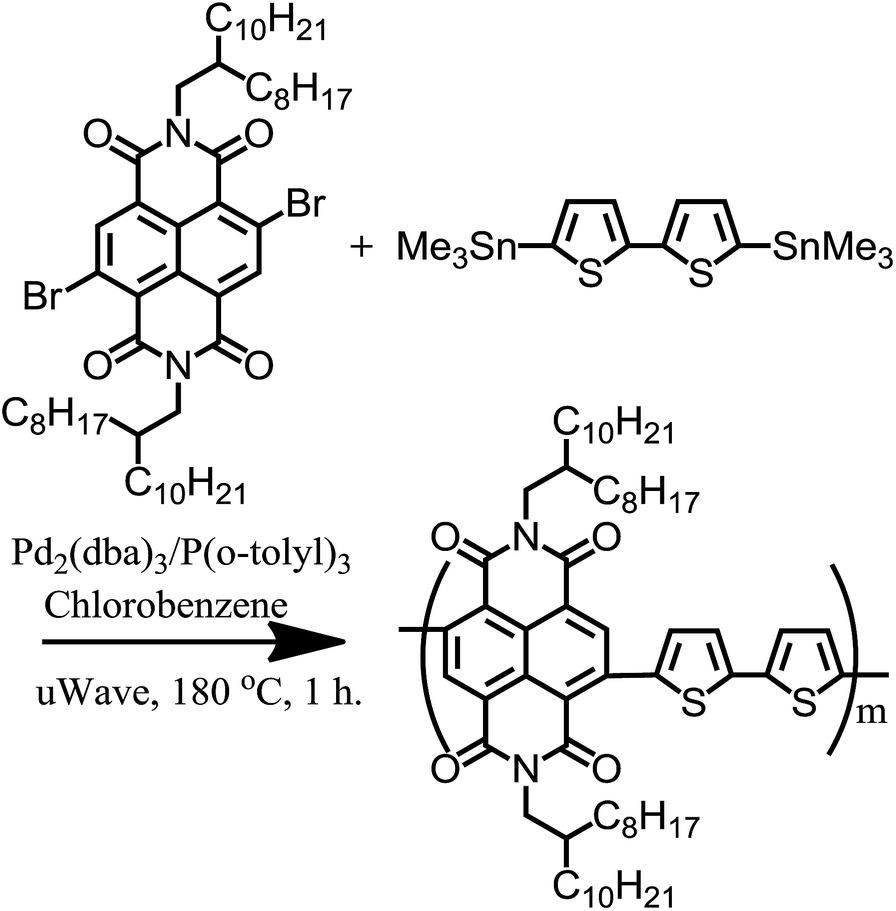 | ||
| Scheme 2 Synthetic scheme for the preparation of PNDI. | ||
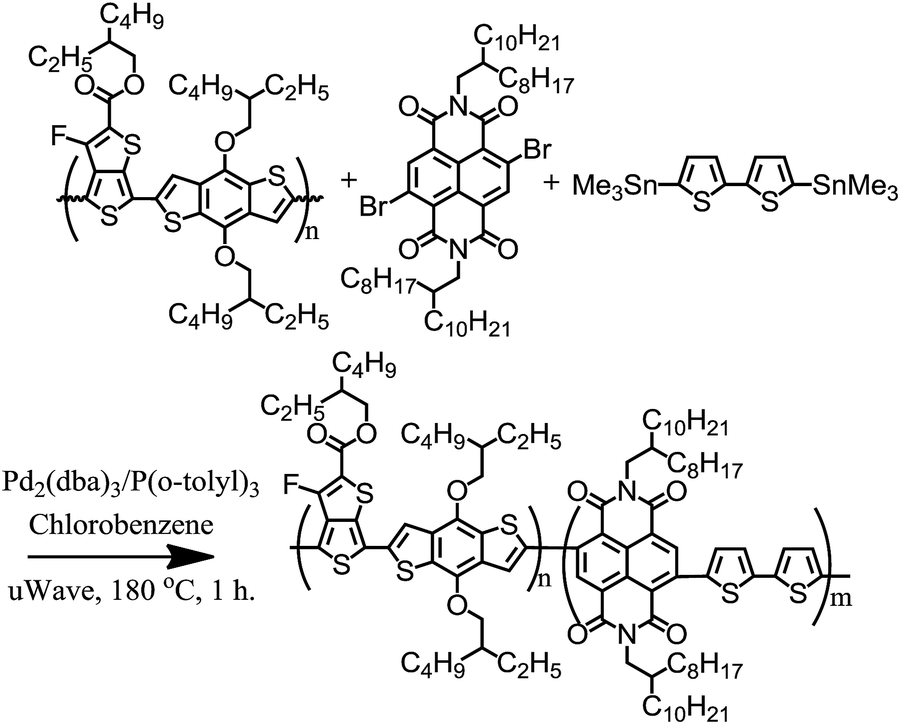 | ||
| Scheme 3 Synthetic scheme of PTB7-b-PNDI. | ||
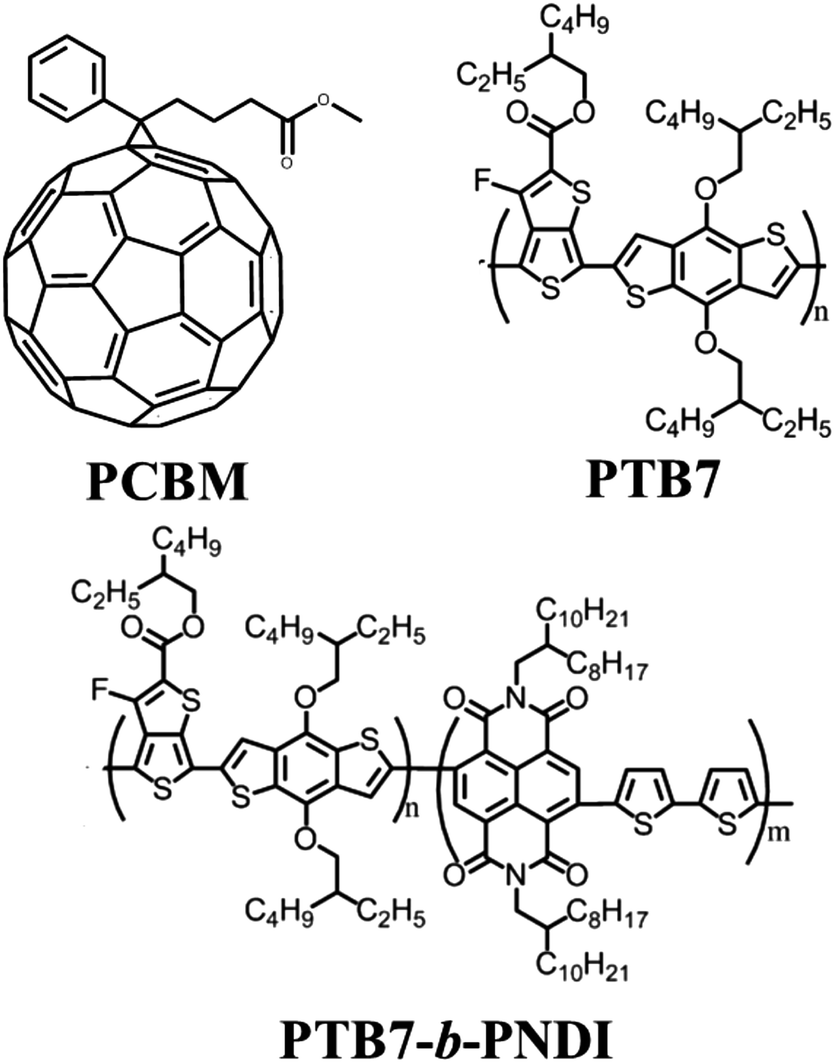 | ||
| Scheme 4 Structures of the PTB7 donor, PCBM acceptor, and all-conjugated PTB7-b-PNDI block copolymer additive. | ||
The molecular weight, composition, and polydispersity of the PTB7-b-PNDI block copolymer along with PTB7 and PNDI homopolymers were determined through a combination of size exclusion chromatography (SEC) (Fig. 1), 1H NMR (ESI Fig. S1–S3†), and steady state UV-Vis absorbance spectroscopy (ESI Fig. S4†), and are tabulated in Table 1. SEC analysis shows a clear shift to shorter retention times for the final block copolymer product, and 1H NMR analysis confirms the presence of both PTB7 and PNDI in the final product. Time of flight secondary ion mass spectroscopy (TOF-SIMS) also shows evidence of both PTB7 and PNDI in the final product, as shown in ESI Fig. S5.† Although the presence of some PNDI homopolymers cannot be excluded due to the overlap in the distributions, little or no PTB7 macro-reagent is present in the final product. SEC-UV-VIS analysis shows coincident peaks at both 380 and 630 nm, corresponding to peak absorbances for PNDI and PTB7 blocks, respectively, and greater absorbance intensity at 630 nm is observed for the block copolymer relative to the PNDI homopolymer. Based on 1H NMR analysis, the PTB7-b-PNDI block copolymer is comprised of 9.7 wt% PTB7 (see ESI† for details on analysis). Two additional estimates of the block copolymer composition are provided in the ESI† based on (i) SEC UV-Vis analysis (estimated composition 9.9 wt% of PTB7) and (ii) steady state solution UV-Vis absorption (estimated composition 9.5 wt% of PTB7). All three methods estimate comparable composition of the block copolymer. The discrepancy between the Mw of the initial PTB7 macro-reagent (11.6 kg mol−1) and that of the PTB7 block in the final block copolymer (approx. 5 kg mol−1) suggests fractionation during the second Stille polycondensation reaction. Lower molecular weight PTB7 macroreagent reacts faster, as would be expected, and unreacted PTB7 is washed out in the subsequent sample purification.
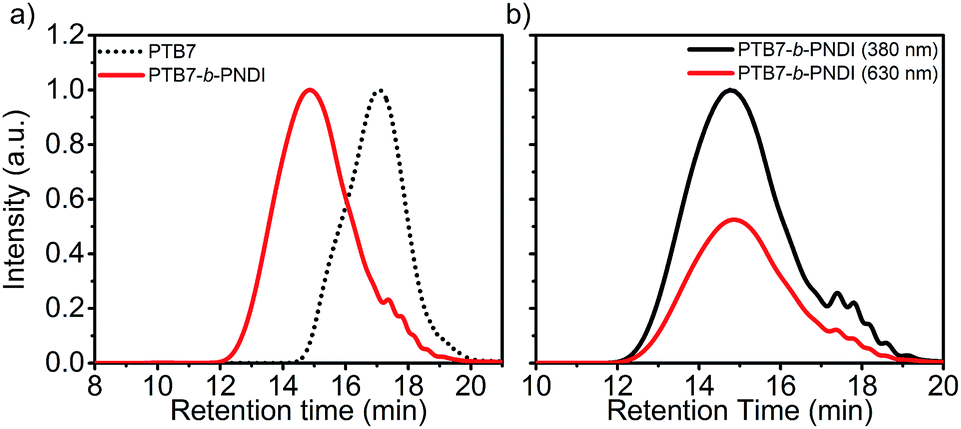 | ||
| Fig. 1 (a) Size exclusion chromatography (SEC) traces of PTB7 and PTB7-b-PNDI with (a) differential refractive index analysis and (b) UV-VIS analysis at 380 nm and 680 nm, corresponding to peak absorbances for PNDI and PTB7, respectively. | ||
Next, we examined the impact of PTB7-b-PNDI additives on the performance and electronic properties of PTB7/PCBM61 bulk heterojunction photovoltaic devices. The overall device architecture consisted of an inverted structure (ITO/ZnO/active layer/PEDOT:PSS/Ag), and the overall ratio between PTB7 and PCBM61 was held constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.75 consistent with prior optimization studies of PTB7/PCBM61 OPVs.11,36Fig. 2 shows current–voltage (J–V) curves for devices ranging from 0 to 15 wt% block copolymer added, and Table 2 provides quantitative data for the best performance for each blend. Device characteristics averaged over 10 devices exhibit similar trends and are provided in ESI Table S1.† The PCE measured for the base case of PTB7/PCBM61 with no additive is comparable to prior literature reports which use PCBM61 as the acceptor.11,36
:1.75 consistent with prior optimization studies of PTB7/PCBM61 OPVs.11,36Fig. 2 shows current–voltage (J–V) curves for devices ranging from 0 to 15 wt% block copolymer added, and Table 2 provides quantitative data for the best performance for each blend. Device characteristics averaged over 10 devices exhibit similar trends and are provided in ESI Table S1.† The PCE measured for the base case of PTB7/PCBM61 with no additive is comparable to prior literature reports which use PCBM61 as the acceptor.11,36
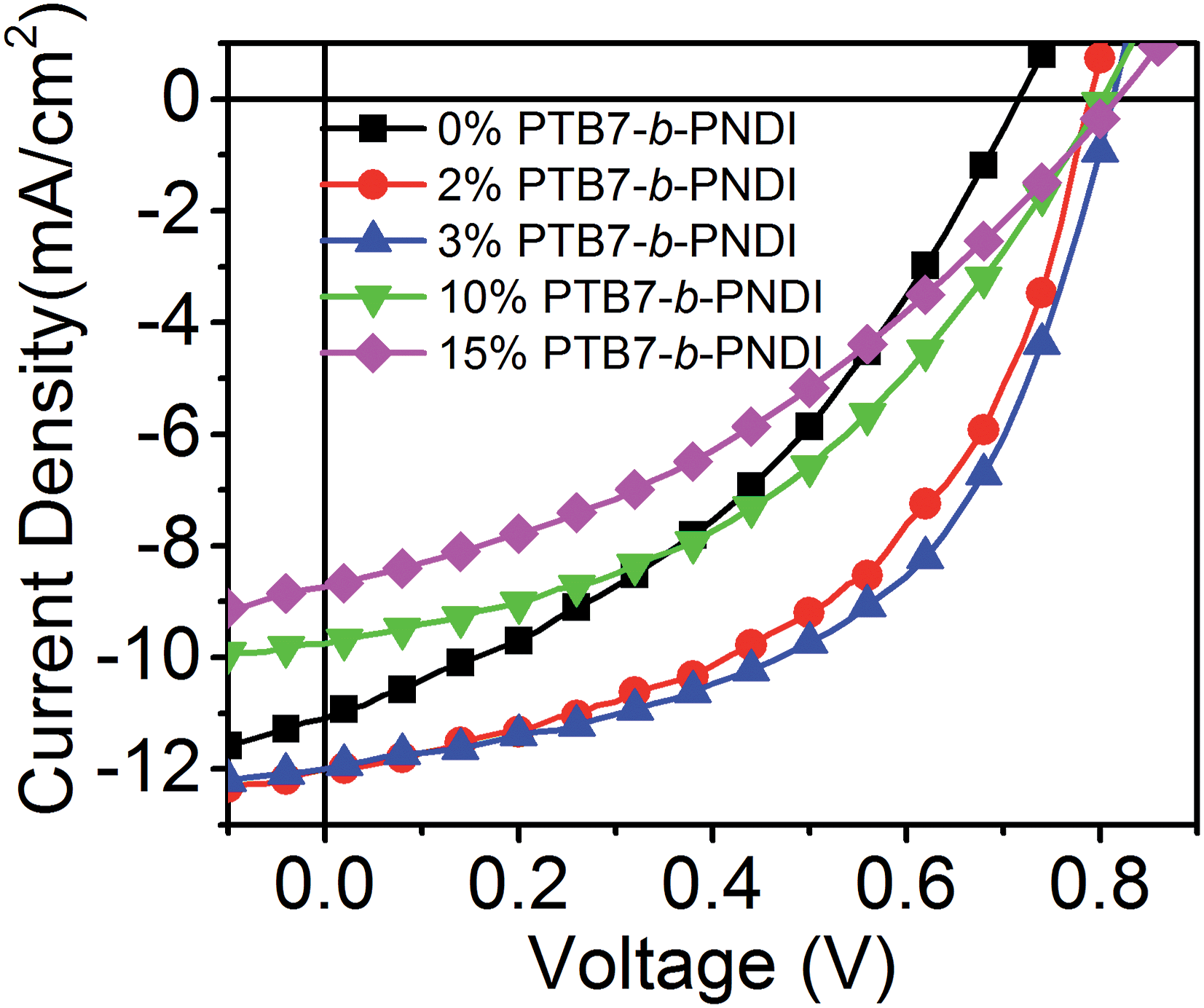 | ||
| Fig. 2 J–V curves of photovoltaic devices with PTB7-b-PNDI additives illuminated at AM 1.5 and 100 mW cm−2. | ||
| Additive | Additive conc. (wt%) | PCE (%) | V OC (V) | J SC (mA cm−2) | FF (%) |
|---|---|---|---|---|---|
| a PCE: power conversion efficiency; JSC: short circuit current; VOC: the open-circuit voltage; FF: fill factor. | |||||
| No additive | 0 | 3.1 | 0.72 | 10.9 | 38.9 |
| PTB7-b-PNDI | 1 | 4.2 | 0.76 | 12.3 | 44.6 |
| 2 | 5.0 | 0.82 | 12.1 | 50.6 | |
| 3 | 5.1 | 0.82 | 11.9 | 52.5 | |
| 10 | 3.3 | 0.82 | 9.7 | 41.4 | |
| 15 | 2.2 | 0.82 | 9.3 | 29.9 | |
| PNDI | 2 | 5.2 | 0.76 | 16.0 | 43.1 |
| 3 | 4.5 | 0.78 | 15.1 | 40.3 | |
| 5 | 3.1 | 0.78 | 9.7 | 40.6 | |
| 10 | 2.7 | 0.80 | 8.8 | 38.4 | |
| 15 | 2.7 | 0.84 | 9.3 | 34.5 | |
A significant improvement of PCE was observed with small amounts of BCP additives, from a PCE of 3.05% with no additive up to 5.1% with 3 wt% BCP additive. Most striking is the increase in the VOC with the addition of BCPs, from a value of 0.72 V to 0.82 V with only 2 wt% BCP added. The VOC remains constant at 0.82 V for higher contents of BCP beyond 2 wt%. This value of VOC is also higher than that reported for state-of-the art PTB7–PCBM61 and PTB7–PCBM71 OPV devices,5,37,38 including studies that incorporate a third component to enhance performance.39–43 A recent study by Lu et al. found a similar magnitude increase in VOC with the addition of 50 wt% of a second donor.43 Importantly, the observed trend of VOC is different from that typically observed for ternary blend systems, where a number of studies have reported either a linear change in VOC for blends with good compatibility or pinning of VOC to a low value for blends with poor compatibility.43–45 Here, we observe pinning of VOC to a higher value. The short-circuit current (JSC) and fill factor (FF) increase with the added BCP and then quickly drop for BCP contents above 10 wt%.
To further understand the impact the BCP additive has on electronic properties, we analyzed ternary PTB7/PCBM61/PNDI ternary blend OPVs. With a pure PNDI additive, the VOC exhibited a roughly linear increase with increasing additive concentration. The PNDI additive has a beneficial impact on device PCE predominantly through an increase in the short-circuit current and fill factor. The evolution of device characteristics with the PNDI additive was consistent with prior studies of ternary blend OPVs with two acceptors.46
Trends in device characteristics with different additive contents are shown in Fig. 3. The results shown for PTB7-b-PNDI and PNDI homopolymer additives reveal a qualitative and quantitative difference in the impact these additives have on VOC and JSC. The PTB7-b-PNDI additive has a dramatic impact on VOC, which reaches a plateau at 2 wt% and remains high for subsequent devices. In contrast, the VOC increases more gradually with the PNDI additive, eventually reaching a value of 0.82 V at 15 wt% additive, and PNDI additive has a more dramatic impact on the JSC.
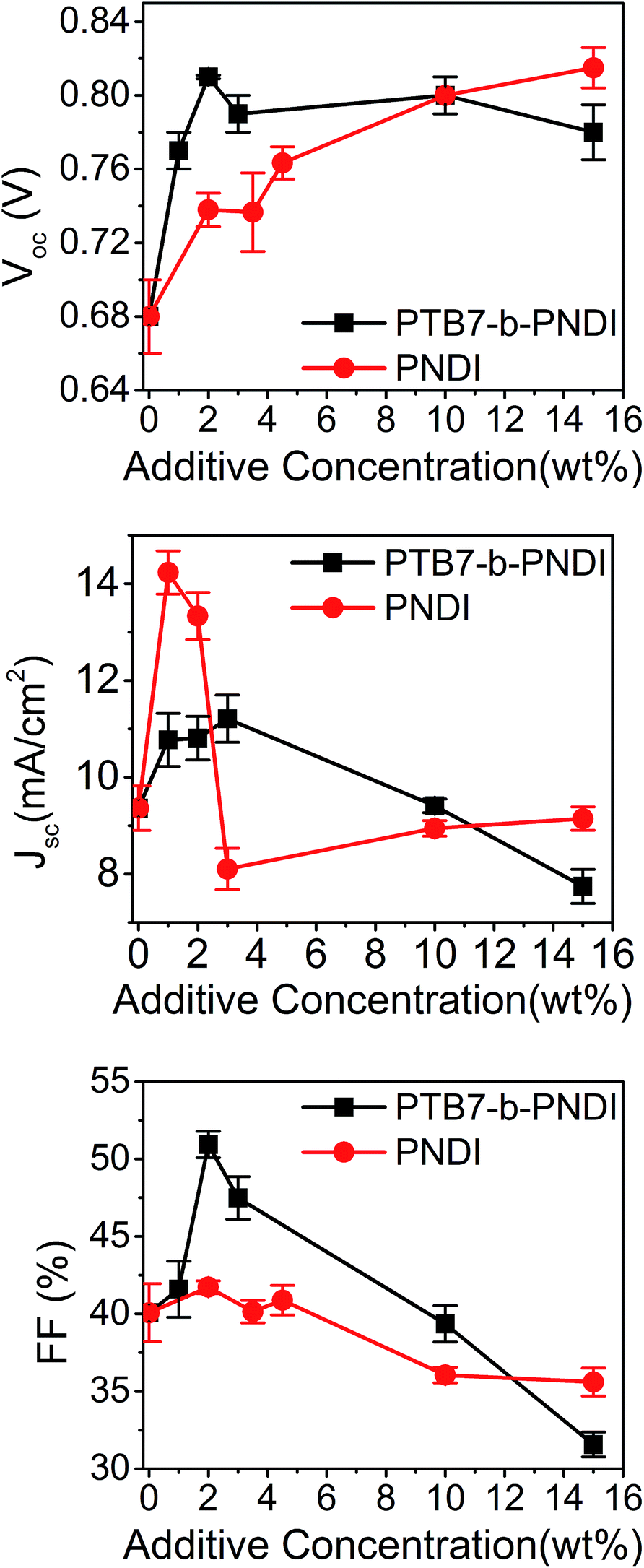 | ||
| Fig. 3 Device characteristics of PTB7/PCBM61/additive ternary blend cells as a function of additive composition. Averaged over 10 devices. | ||
There are several examples reported in the literature in which the VOC is pinned to the lowest value VOC due to poor compatibility between components, and, for example, varying the additive concentration from 0 to 90% does not change the VOC.44,45 In one example, Khlyabich et al. examined ternary blend OPVs comprised of two immiscible polymeric donors and found a low VOC pinned by the smallest HOMO of the two polymer donors and the LUMO of the acceptor.45 Lu et al. studied ternary blends of PTB7 a second donor polymer poly-3-oxothieno[3,4-d]isothiazole-1,1-dioxide/benzodithiophene (PID2) and PCBM71. EQE measurements showed improved light harvesting by PID2, enhancing PCE from 7.2% to 8.2% at optimal additive composition. However, VOC was pinned at 0.72 V for blend compositions up to 90% PID2, compared to 0.86 V for 100% PID2 as a donor.39 Here, in the case of PTB7-b-PNDI additives, we observe an increased VOC compared to that in the case of both PNDI-b-PTB7 and PNDI additives.
Given the relative ordering of the energy levels (see Fig. 4), and the electron affinity of PNDI,47–49 PNDI is expected to act as an electron acceptor (see ESI Fig. S6† for cyclic voltammetry measurements). This leads to the possibility that PNDI and PCBM61 form an alloyed acceptor blend.46 However, in the case of PTB7-b-PNDI the VOC is observed to be independent of composition above 2 wt% PTB7-b-PNDI. Furthermore, while devices with the pure PNDI additive show a continuous change in VOC which may reflect an alloyed acceptor, the change in VOC is larger than would be expected for just 15 wt% PNDI added, where the PCBM61 should still be dominant. Instead, the observed device characteristics are consistent with a parallel-type model proposed for ternary blend OPVs.27,28 In this picture, the third component forms a phase-segregated percolating channel rather than blending or alloying with the donor or acceptor phase.
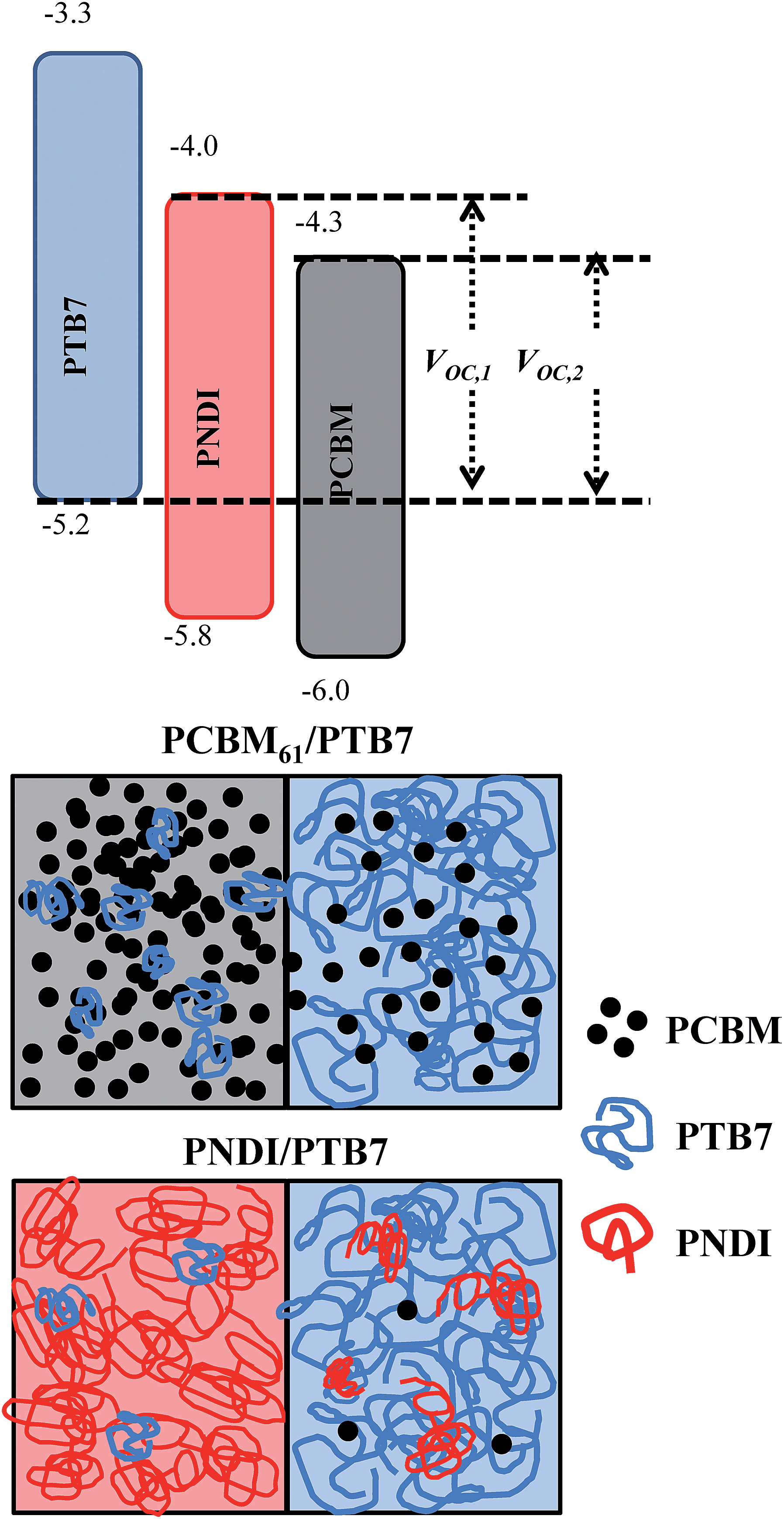 | ||
| Fig. 4 (Top) Energy level diagram for ternary blend OPVs and (bottom) schematic of domain interfaces for BHJ OPVs. Without any additive, the only domains are a PCBM61-rich and PTB7-rich domain. With the PNDI additive (PTB7-b-PNDI or PNDI homopolymer) phase segregated PNDI domains form a separate channel for electron collection and transport. For clarity, no mixing is shown between PNDI and PCBM61. | ||
To understand the predictions of this model for the particular system under study, we first note that the energy levels of PNDI are such that the HOMO and LUMO are positioned between those of PTB7 and PCBM, as shown in Fig. 4. In a perfectly mixed system, electrons would relax to the lowest energy state available, which would be either on PCBM or on an alloy of PCBM and PNDI. In the parallel-like model, with the addition of PTB7-b-PNDI, PNDI forms an independent percolating channel. As a result, the electrons can distribute to the LUMO of PNDI, resulting in the observed increase in the VOC.
Surface energy values provide an estimate of enthalpic interactions between different components and can provide insight into the expected morphological changes. The surface energies were previously measured through contact angle measurements and can used to estimate the enthalpic interactions between different components.25 The surface energies of the polymers and binary Flory–Huggins interaction parameters χ indicate that PNDI will be strongly segregated from both PTB7 and PCBM (χPTB7–PNDI = 1.20 and χPCBM–PNDI = 3.36), while mixing is expected between PTB7 and PCBM61 (χPTB7–PCBM = 0.77), as has been observed in prior morphological studies.5,11 As a result, PNDI should be segregated from both PTB7 and PCBM61, consistent with the expected morphology of a parallel-like BHJ OPV.27,28
For comparison with parallel bulk heterojunction photovoltaics, we fabricated true parallel bilayer OPVs. Bilayer OPVs provide a useful model system for studying electronic properties of the donor–acceptor interface.50 Bilayer devices with PCBM61/PTB7 or PNDI/PTB7 active layers were fabricated separately and then connected in parallel, as shown in ESI Fig. S7.† The device characteristics and J–V curves are shown in ESI Table S2 and Fig. S7.† The PTB7/PCBM61 bilayer device exhibits a VOC of 0.72 V while the PTB7/PNDI device has a much higher VOC of 0.80 V. When connected in parallel, the resulting VOC is 0.74 V, intermediate between the two bilayer devices and in good agreement with the expected VOC based on weighting each cell by its current production.28 Similarly, the parallel bulk heterojunction OPVs exhibit an increase in VOC with the addition of block copolymers. The higher value of VOC in the block copolymer devices indicates a larger current through the PNDI domains relative to the parallel bilayer OPVs.
To investigate phase separation and mixing in the ternary blend system, we analyzed model bilayer blends of PTB7, PNDI, and deuterated PCBM61 (d-PCBM61) by time of flight secondary ion mass spectroscopy (TOF-SIMS). TOF-SIMS has been applied to organic and inorganic films to perform compositional and isotopic analyses along with depth profiling with subnanometer resolutions.51–53 We analyzed bilayer films in order to investigate mixing and phase separation of d-PCBM from either PNDI or PTB7, as shown schematically in Fig. 5. d-PCBM61 was used in place of PCBM61 to enable compositional analysis through detection of 2H ions.
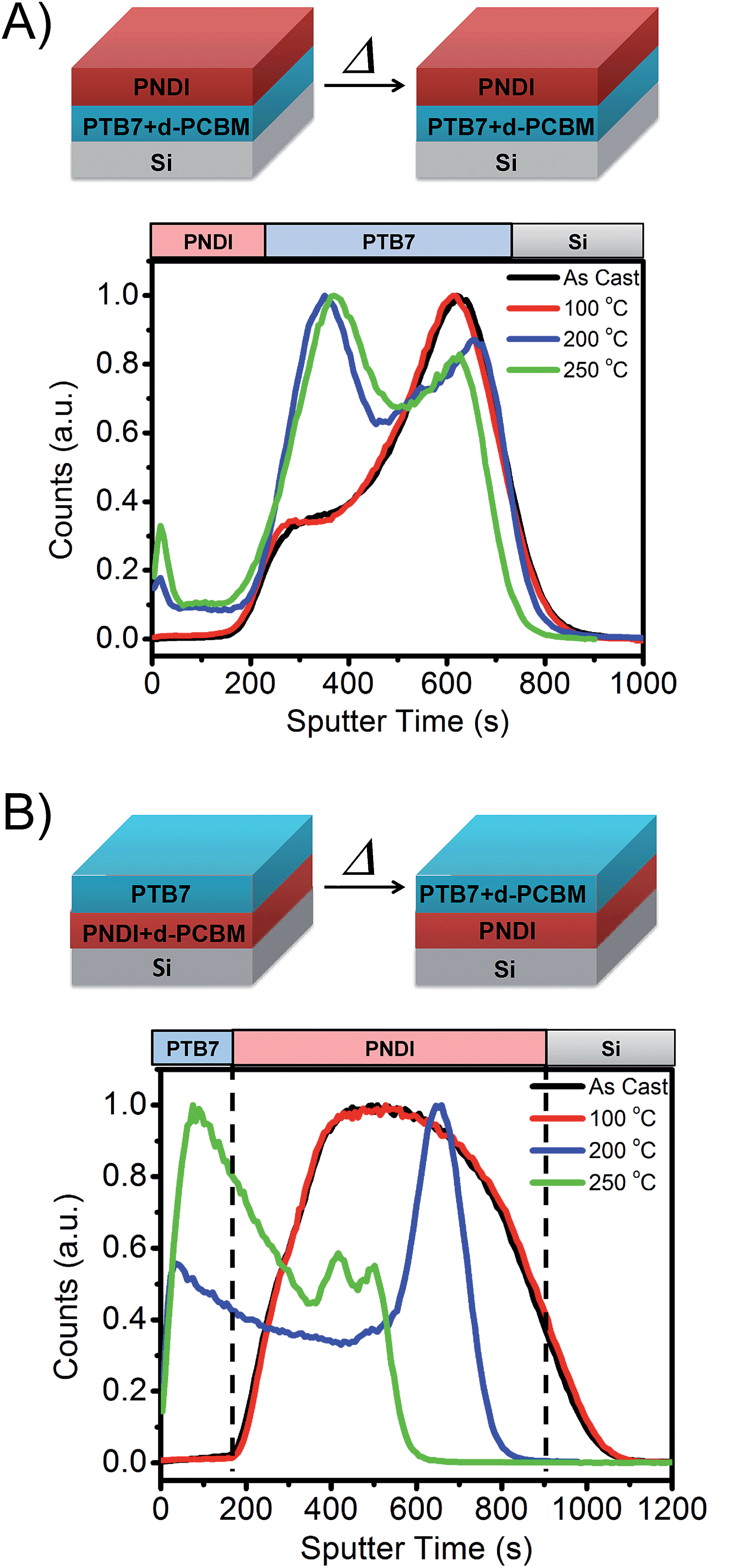 | ||
| Fig. 5 Schematics and TOF-SIMS analysis of 2H− distribution in bilayer films of (A) pure PNDI on top of a PTB7/d-PCBM blend and (B) pure PTB7 on top of a PNDI/d-PTB7 blend. The 2H− distribution corresponds to d-PCBM61 in the films. Samples were annealed at 100 °C, 200 °C and 250 °C for 10 minutes. | ||
In one experiment, a film of pure PNDI (70 nm) was deposited on top of a blend film (50:50 wt%) of PTB7:d-PCBM61 while in a second experiment a film of pure PTB7 (70 nm) was deposited on top of a blend film (50:50 wt%) of PNDI and d-PCBM61. The bilayers were prepared through film transfer at room temperature, ensuring that no mixing occurred prior to TOF-SIMS analysis and/or thermal annealing. Based on the interaction energies of each component, we hypothesized that d-PCBM61 would preferentially segregate to the PTB7 layer. In the first experiment, we thus expected d-PCBM61 to remain in the bottom PTB7 layer, while in the second experiment we expected diffusion of the d-PCBM61 to the top PTB7 layer during annealing. Due to the slow diffusion kinetics and incompatibility between PTB7 and PNDI, no mixing was expected during annealing. Indeed, this is supported through TOF-SIMS analysis of secondary ions corresponding to each component (see ESI Fig. S8†).
As shown in Fig. 5B, d-PCBM61 remains in the PTB7 layer even after annealing up to a temperature of 250 °C. Some diffusion of PCBM61 is observed towards the PNDI interface, but d-PCBM61 remains distributed only within the PTB7 film. In contrast, for the case where d-PCBM61 and PNDI are initially blended, segregation of d-PCBM61 to the top PTB7 film is indeed observed. The d-PCBM61 is initially broadly distributed within the bottom layer, and after annealing at 200 °C segregation towards the PTB7 and the bottom Si interface is observed. On annealing at a higher temperature of 250 °C, most of the d-PCBM61 is located within the PTB7 domain. TOF-SIMS analysis thus demonstrates that the PNDI will segregate strongly from both PTB7 and PCBM61 domains.25
To quantify morphological changes within bulk-heterojunction OPVs, ternary blend films were analyzed by transmission electron microscopy (TEM) and grazing-incidence wide angle X-ray scattering (GIWAXS). TEM and GIWAXS samples were prepared under similar conditions to those of devices, by spin-coating blends from a mixture of chlorobenzene:DIO (97:3 v/v) and annealing at 100 °C for 10 minutes. TEM results suggest that, indeed, a layer of PTB7 may insulate the PNDI from the PCBM (Fig. 6 and ESI Fig. S9†). The TEM analysis of PTB7:PCBM61 blends is shown in ESI Fig. S10† for reference. In the TEM micrographs for blends with PTB7-b-PNDI and PNDI additives, we observe evidence of three-phase coexistence: black domains, gray domains, and white domains. The PCBM is the highest density component, and dark areas indicate the preferential distribution of the PCBM material. Accordingly, we propose that the black domains are rich in PCBM. The relative ordering of the interaction parameters indicates that the strength of segregation between the PNDI and PCBM is stronger than the strength of segregation between the PTB7 and PCBM. Accordingly, it is likely that the PTB7 should distribute preferentially to the space between the PNDI and PCBM (effectively screening the PNDI and PCBM from interacting with one another). Given this explanation, we propose that the gray domains are PTB7 and PCBM intermixed while the white domains are rich in PNDI.
 | ||
| Fig. 6 TEM image of PTB7/PCBM blends with PTB7-b-PNDI (top) and PNDI (bottom) as the additive. Scale bar = 200 nm. | ||
GIWAXS analysis shown in Fig. 7 and in ESI Fig. S11–S14† also supports this interpretation of morphology. Pristine PTB7/PCBM blends exhibit an intense peak along the qz = 0.30 Å−1 characteristic from PTB7 and a ring at q = 1.40 Å−1 from amorphous PCBM. PNDI crystallization is suppressed below 2 wt% PTB7-b-PNDI additive, but at 3 wt% PNDI and higher, a diffraction peak at q = 0.48 Å−1 reflects PNDI crystallization. This suggests enrichment of PNDI into a distinct phase. Similar trends are found for the PNDI homopolymer, except that evidence for crystallization is not observed up to concentrations higher than 3 wt% PNDI additive, suggesting some mixing with PCBM at these low concentrations.
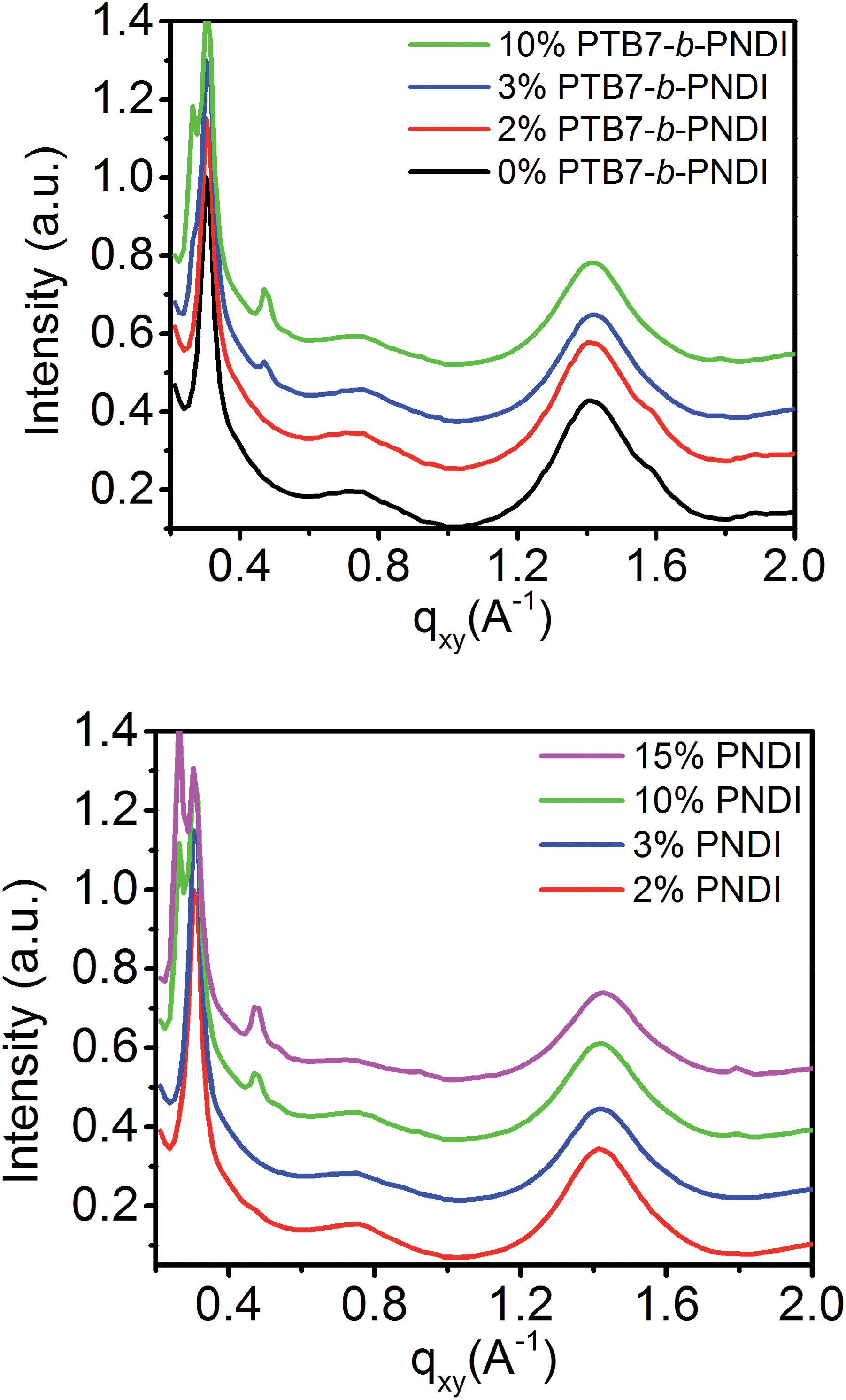 | ||
| Fig. 7 Linecuts of the grazing incidence wide angle X-ray scattering profile of PTB7/PCBM + PTB7-b-PNDI blends and PTB7/PCBM + PNDI blends. | ||
The morphological studies therefore support the picture of phase separated PNDI domains, which supports the hypothesis a parallel-like BHJ OPV with the addition of additives. This also implies that PCBM61 mixes preferentially with PTB7 domains. Selective mixing of PCBM61 with one polymer of a ternary blend system has been recently observed in a ternary blend system. Hartmeier et al. showed that preferential solubility was detrimental to the performance of a ternary blend OPV consisting of two donor polymers, and the authors proposed a guideline for the design of ternary blend OPVs that the miscibility of PCBM61 with both polymers should be matched.54 The present system is comprised of a ternary blend with one donor and one acceptor polymer, and as a result matching PCBM61 solubility for both polymers is likely not to be an important design principle. In comparing the VOC trends for BCP and PNDI homopolymer loadings, we propose that the VOC pinning may be sharper for the case of the BCP loading because the PNDI polymer is attached to a PTB7 block. This PTB7 block is likely to shield the PNDI from the PCBM or drive the BCP into the PTB7-rich phase rather than the PCBM-rich domains, which enhances the formation of a parallel-like morphology.
Conclusion
In conclusion, we studied the effect of an all-conjugated block copolymer PTB7-b-PNDI in PTB7/PCBM ternary blends solar cells. PTB7-b-PNDI was synthesized by sequential Stille polycondensation, in which a low molecular weight PTB7 was used as a macroend-capper. Combination of size-exclusion chromatography, UV-VIS absorption measurements and 1H NMR confirms the success of the sequential polymerization. Small amounts (2 wt%) of BCP lead to significant impact on photovoltaic performance by enhancing open-circuit voltages by 100 mV. We also compared the results with PTB7-b-PNDI additives with those with homopolymer PNDI additives, which exhibit a smaller but still significant impact on the VOC. Based on surface energy calculations and morphological analysis through TOF-SIMS and TEM, we conclude that the ternary blend systems studied are consistent with parallel-like OPVs, in which the PNDI forms a distinct acceptor phase. These results point to novel effects with both PNDI and PNDI-b-PTB7 additives and suggest that the development of all-conjugated block copolymers with donor and acceptor blocks may be a viable way to tune electronic properties and enhance the performance of BHJ OPVs.Acknowledgements
J. W. M., L. R. H., and R. V. acknowledge the support of the National Science Foundation (CBET-1264703 and DMR-1352099) and the Welch Foundation for Chemical Research (C-1888) for support of this research. V. G. and D. K. acknowledge support in part by grants from the Robert A. Welch Foundation (Grant F1599), the National Science Foundation (CBET-1264583), and the US Army Research Office (W911NF-13-1-0396). We acknowledge Krüss for providing support and access to DSA 100 for contact angle measurements. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge the NSF MRI grant DMR-0923096 for access to TOF-SIMS instrument at the Texas Materials Institute at the University of Texas at Austin.References
- N. Li and C. J. Brabec, Energy Environ. Sci., 2015, 8, 2902–2909 CAS.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- N. E. Jackson, B. M. Savoie, T. J. Marks, L. X. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2015, 6, 77–84 CrossRef CAS PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2014, 44, 78–90 RSC.
- F. Liu, W. Zhao, J. R. Tumbleston, C. Wang, Y. Gu, D. Wang, A. L. Briseno, H. Ade and T. P. Russell, Adv. Energy Mater., 2014, 4, 1301377 CrossRef.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- H. Chen, Y.-C. Hsiao, B. Hu and M. Dadmun, Adv. Funct. Mater., 2014, 24, 5129–5136 CrossRef CAS.
- H. Chen, J. Peet, S. Hu, J. Azoulay, G. Bazan and M. Dadmun, Adv. Funct. Mater., 2013, 24, 140–150 CrossRef.
- W. Yin and M. Dadmun, ACS Nano, 2011, 5, 4756–4768 CrossRef CAS PubMed.
- J. T. Rogers, K. Schmidt, M. F. Toney, E. J. Kramer and G. C. Bazan, Adv. Mater., 2011, 23, 2284–2288 CrossRef CAS PubMed.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
- D. Chen, A. Nakahara, D. Wei, D. Nordlund and T. P. Russell, Nano Lett., 2010, 11, 561–567 CrossRef PubMed.
- G. Li, Y. Yao, H. Yang, V. Shrotriya, G. Yang and Y. Yang, Adv. Funct. Mater., 2007, 17, 1636–1644 CrossRef CAS.
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS.
- S. B. Darling, Energy Environ. Sci., 2009, 2, 1266–1273 CAS.
- I. Botiz and S. B. Darling, Mater. Today, 2010, 13, 42–51 CrossRef CAS.
- K. Yuan, L. Chen and Y. Chen, Polym. Int., 2014, 63, 593–606 CrossRef CAS.
- J. Chen, X. Yu, K. Hong, J. M. Messman, D. L. Pickel, K. Xiao, M. D. Dadmun, J. W. Mays, A. J. Rondinone, B. G. Sumpter and S. M. Kilbey Ii, J. Mater. Chem., 2012, 22, 13013–13022 RSC.
- Z. Sun, K. Xiao, J. K. Keum, X. Yu, K. Hong, J. Browning, I. N. Ivanov, J. Chen, J. Alonzo, D. Li, B. G. Sumpter, E. A. Payzant, C. M. Rouleau and D. B. Geohegan, Adv. Mater., 2011, 23, 5529–5535 CrossRef CAS PubMed.
- H. J. Kim, K. Paek, H. Yang, C.-H. Cho, J.-S. Kim, W. Lee and B. J. Kim, Macromolecules, 2013, 46, 8472–8478 CrossRef CAS.
- R. C. Mulherin, S. Jung, S. Huettner, K. Johnson, P. Kohn, M. Sommer, S. Allard, U. Scherf and N. C. Greenham, Nano Lett., 2011, 11, 4846–4851 CrossRef CAS PubMed.
- Y.-H. Lee, W.-C. Chen, C.-J. Chiang, K.-C. Kau, W.-S. Liou, Y.-P. Lee, L. Wang and C.-A. Dai, Nano Energy, 2015, 13, 103–116 CrossRef CAS.
- G. Pandav and V. Ganesan, Macromolecules, 2013, 46, 8334–8344 CrossRef CAS.
- G. Pandav and V. Ganesan, J. Chem. Phys., 2013, 139, 214905 CrossRef PubMed.
- D. Kipp, J. Mok, J. Strzalka, S. B. Darling, V. Ganesan and R. Verduzco, ACS Macro Lett., 2015, 4, 867–871 CrossRef CAS.
- D. Kipp, O. Wodo, B. Ganapathysubramanian and V. Ganesan, ACS Macro Lett., 2015, 4, 266–270 CrossRef CAS.
- L. Yang, H. Zhou, S. C. Price and W. You, J. Am. Chem. Soc., 2012, 134, 5432–5435 CrossRef CAS PubMed.
- B. M. Savoie, S. Dunaisky, T. J. Marks and M. A. Ratner, Adv. Energy Mater., 2015, 5, 1400891 CrossRef.
- A. Yassar, L. Miozzo, R. Gironda and G. Horowitz, Prog. Polym. Sci., 2013, 38, 791–844 CrossRef CAS.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, Adv. Mater., 2013, 5686–5700 CrossRef CAS PubMed.
- M. He, F. Qiu and Z. Lin, J. Mater. Chem., 2011, 21, 17039–17048 RSC.
- Y. Lee and E. D. Gomez, Macromolecules, 2015, 48, 7385–7395 CrossRef CAS.
- A. Kiriy, V. Senkovskyy and M. Sommer, Macromol. Rapid Commun., 2011, 32, 1503–1517 CrossRef CAS PubMed.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC.
- S. Foster, F. Deledalle, A. Mitani, T. Kimura, K.-B. Kim, T. Okachi, T. Kirchartz, J. Oguma, K. Miyake, J. R. Durrant, S. Doi and J. Nelson, Adv. Energy Mater., 2014, 4, 1400311 CrossRef.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- L. Lu, T. Xu, W. Chen, E. S. Landry and L. Yu, Nat. Photonics, 2014, 8, 716–722 CrossRef CAS.
- H. Wang, J. Huang, S. Xing and J. Yu, Org. Electron., 2016, 28, 11–19 CrossRef CAS.
- L. Lu, M. A. Kelly, W. You and L. Yu, Nat. Photonics, 2015, 9, 491–500 CrossRef CAS.
- Y. Ohori, T. Hoashi, Y. Yanagi, T. Okukawa, S. Fujii, H. Kataura and Y. Nishioka, J. Photopolym. Sci. Technol., 2014, 27, 569–575 CrossRef.
- L. Lu, W. Chen, T. Xu and L. Yu, Nat. Commun., 2015, 6, 7327 CrossRef CAS PubMed.
- S. A. Mollinger, K. Vandewal and A. Salleo, Adv. Energy Mater., 2015, 5, 1501335 CrossRef.
- P. P. Khlyabich, A. E. Rudenko, B. C. Thompson and Y.-L. Loo, Adv. Funct. Mater., 2015, 25, 5557–5563 CrossRef CAS.
- R. A. Street, D. Davies, P. P. Khlyabich, B. Burkhart and B. C. Thompson, J. Am. Chem. Soc., 2013, 135, 986–989 CrossRef CAS PubMed.
- Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- W. Lee, C. Lee, H. Yu, D.-J. Kim, C. Wang, H. Y. Woo, J. H. Oh and B. J. Kim, Adv. Funct. Mater., 2016, 26, 1543–1553 CrossRef CAS.
- M. M. Durban, P. D. Kazarinoff and C. K. Luscombe, Macromolecules, 2010, 43, 6348–6352 CrossRef CAS.
- K. Nakano, K. Suzuki, Y. Chen and K. Tajima, Sci. Rep., 2016, 6, 29529 CrossRef PubMed.
- R. N. S. Sodhi, Analyst, 2004, 129, 483–487 RSC.
- A. Benninghoven, Angew. Chem., Int. Ed. Engl., 1994, 33, 1023–1043 CrossRef.
- H. Chou, A. Ismach, R. Ghosh, R. S. Ruoff and A. Dolocan, Nat. Commun., 2015, 6, 7482 CrossRef CAS PubMed.
- B. F. Hartmeier, M. A. Brady, N. D. Treat, M. J. Robb, T. E. Mates, A. Hexemer, C. Wang, C. J. Hawker, E. J. Kramer and M. L. Chabinyc, J. Polym. Sci., Part B: Polym. Phys., 2015, 54, 237–246 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, average values for device characteristics, 1H NMR spectra of polymers, TEM images of ternary blends, and GIWAXS data for ternary blends. See DOI: 10.1039/c6ta06502c |
| This journal is © The Royal Society of Chemistry 2016 |