
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnionic porous polymers with tunable structures and catalytic properties†
Wuxue
Zhao
a,
Fan
Zhang
*a,
Lingyun
Yang
b,
Shuai
Bi
a,
Dongqing
Wu
a,
Yefeng
Yao
b,
Manfred
Wagner
c,
Robert
Graf
c,
Michael Ryan
Hansen
 cd,
Xiaodong
Zhuang
*ae and
Xinliang
Feng
ae
cd,
Xiaodong
Zhuang
*ae and
Xinliang
Feng
ae
aSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Key Laboratory of Electrical Insulation and Thermal Ageing, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China. E-mail: fan-zhang@sjtu.edu.cn; zhuang@sjtu.edu.cn
bPhysics Department & Shanghai Key Laboratory of Magnetic Resonance, East China Normal University, North Zhongshan Road 3663, 200062 Shanghai, China
cMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany
dInstitute of Physical Chemistry, Westfälische Wilhelms-Universität Münster, Corrensstr. 28/30, D-48149 Münster, Germany
eCenter for Advancing Electronics Dresden (cfaed), Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 2nd September 2016
Abstract
A series of boron-containing conjugated microporous polymers with hierarchical porous structures have been readily prepared via typical transition metal-catalyzed coupling reactions. The distribution of micro- and mesopores in the networks as well as the specific surface areas are tunable via tailoring the structures of the building blocks. The distinct capability of the resulting Lewis acid-based neutral porous polymers to selectively capture fluoride ions provides a high-efficiency conversion into stable anionic porous polymers. For the first time, fluoride anion binding to boron atoms in a solid sample was essentially characterized by solid-state 11B MAS NMR spectroscopy, clearly revealing such an efficient conversion from a neutral network to a negatively charged one only through Lewis acid–base adduct formation. Upon a simple ion-exchange process, various heavy metal cations were facile to be loaded into the networks of the anionic porous polymers. Furthermore, the cobalt(II)-loaded porous polymers were shown to promote the stoichiometric homocoupling reactions of the different aryl Grignard regents, and exert distinct size selectivities for the homocoupling products, highly dependent on their porous structures. Such a successful loading strategy might be used for design and synthesis of new types of zeolite-like porous polymers with desirable catalytic properties for a certain organic transformation, as well as other functional materials.
Introduction
Porous materials1 have attracted tremendous academic and commercial attention in numerous fields, such as membrane separation,2 industrial catalysis,3 and energy conversion and storage.4 Among them are metal organic frameworks (MOFs),5 consisting of organic linkers and coordinated metal centers, that have cationic networks with extra-framework counter anions. In contrast, zeolites and MOFs6 possess anionic networks balanced with extra-network counter-cations to maintain the electroneutrality of the materials. The unique structural and porous features of zeolites facilitate their use as heterogeneous catalysts in many organic transformations, such as Friedel–Crafts alkylation,7 alkylaromatic isomerization8 and disproportionation,9 realized on an industrial scale. However, due to the lack of controlled and versatile functionalization at the molecular level, the structures of zeolites cannot be finely tailored. A lot of work remains to be done to understand the relationship between the catalytic mechanism and a material's structure.10Porous polymer networks are generally built up from light elements, such as carbon, nitrogen, boron and oxygen, and are formed by cross-linking organic-directing and building units through covalent bonds.11 Their well-defined structure can be tailored at the molecular level to optimize their textural and chemical properties. Compared with MOFs, porous organic polymers exhibit much higher chemical and thermal stabilities.12 In general, porous polymers feature a neutral character, but in the past few years, some cationic porous polymers have been reported,13 while the anionic porous polymers remain rarely explored. Very recently, anionic microporous polymers were prepared by using tetraaryl boron anion units, suitable for being used as catalysts, solid electrolytes or absorbents, thus arousing much more attention to the charged porous polymers.14
In this paper, we present a concise synthetic strategy toward a series of neutral Lewis acid boron-containing porous polymers, which can be readily converted into anionic porous polymers via Lewis acid–base interactions with fluoride ions. Further, various alkali or heavy metals can be selectively loaded on such anionic porous polymers by a simple ion-exchange process, representing the first example of the successful loading approach to functional porous materials. The as-prepared neutral and anionic porous polymers exhibit high permanent surface areas and hierarchical porous structures. Remarkably, as an example, the cobalt(II)-loaded porous polymers can effectively promote stoichiometric homocoupling reactions of aryl Grignard regents with molecular size selectivity depending on the porous features of the networks.
Experimental section
All starting materials were purchased from Aladdin and Aldrich. All air-sensitive reactions were carried out under a nitrogen atmosphere and performed using Schlenk techniques. All solvents were dried before use. Tetrahydrofuran (THF) was refluxed with sodium. Triethylamine (TEA) and dichloromethane were refluxed with calcium hydroxide. Tris(bromoduryl)borane,15 4,4-diethynyl-1,1-biphenyl,16 1,4-diethynylbenzene,16 and 1,3,5-triethynylbenzene,17 were synthesized according to the reported procedures.General protocol for the synthesis of neutral B-Ph-ae-n
All polymerization reactions were carried out at 75 °C for 72 h. The molar ratio of ethynyl to bromo functionalities in the monomer feed was set at 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The typical experimental procedure is given as follows:
:1. The typical experimental procedure is given as follows:
General protocol for the synthesis of anionic porous polymers MF@B-Ph-ae-n
Results and discussion
The synthetic route toward Lewis acid boron-based porous polymers B-Ph-ae-n (n = 1, 2, and 3) is presented in Scheme 1. The key monomers of tris(bromoduryl)borane (1), 1,4-diethynylbenzene (2), 4,4-diethynyl-1,1-biphenyl (3), and 1,3,5-triethynylbenzene (4) were synthesized according to previous reports (see the ESI†). Under a typical Sonogashira cross-coupling condition, monomer 1 was reacted with monomers 2–4, to afford the porous polymers B-Ph-ae-1, B-Ph-ae-2 and B-Ph-ae-3, all as yellow powders in high yields (from 87% to 91%). The as-synthesized porous polymers were revealed to exhibit excellent stabilities after treatment with water (Fig. S1†). The duryl moiety containing four sterically bulky methyl groups at the 2, 3, 5, and 6-positions of the phenyl ring in monomer 1 is clearly beneficial for achieving high polymerization efficiency as well as to improve the inertness of the resulting porous polymers to moisture and oxygen.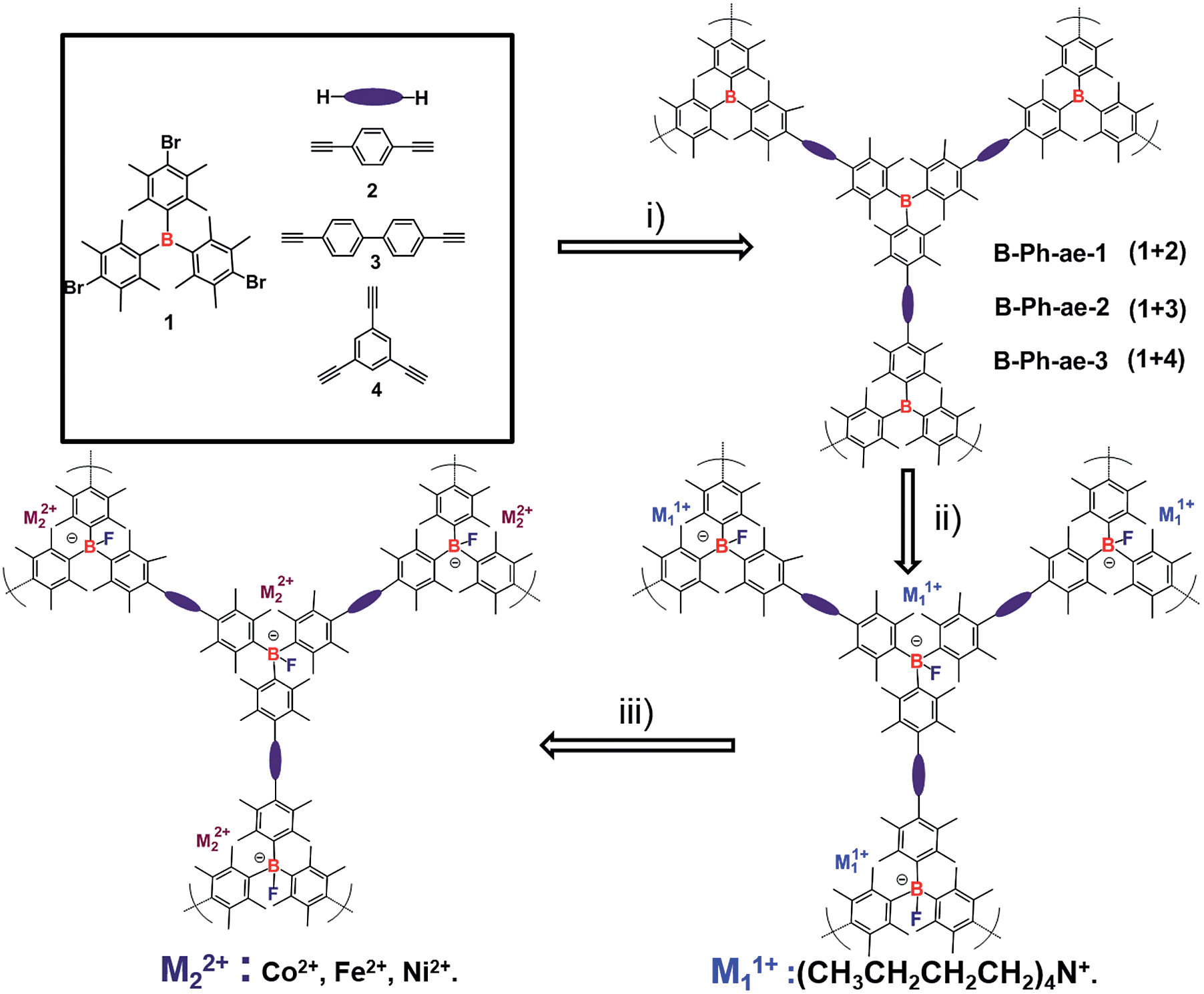 | ||
| Scheme 1 Synthesis of Lewis acid-based neutral porous polymers and corresponding ionic porous polymers. (i) Et3N, Pd(PPh3)4, CuI, 75 °C, 72 h, tetrahydrofuran. (ii) Tetrabutylammonium fluoride (Bu4NF), THF, room temperature (RT). (iii) Ethanol and THF, cobalt acetate, nickel acetate, and iron nitrate, at RT. | ||
The structures of the polymeric networks B-Ph-ae-1, B-Ph-ae-2, and B-Ph-ae-3 were characterized by Fourier transform infrared (FTIR) spectroscopy (Fig. S2a†) and 13C solid-state nuclear magnetic resonance (NMR) spectroscopy (Fig. 1a). In the FTIR spectra, a typical C–B stretching signal at around 1105 cm−1 was observed for all three porous polymers. A peak with a very low intensity at 2110 cm−1 corresponds to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– stretching. The broad peaks at 3300 cm−1 can be attributed to the –CC– linkages. The signals at 2870–3050 cm−1 originate from aromatic C–H stretching.18 In the solid-state 13C {1H} cross-polarization/magic angle spinning (CP/MAS) NMR spectra, the well-resolved resonances at around 20.0 ppm are ascribed to the methyl carbons (Car–CH3). The peaks at ∼136.3 and 131.0 ppm can be attributed to the substituted aromatic carbons (Car–CH3) and the hydrogen-bearing aromatic carbons (Car–H), respectively. The signals located at ∼123.3 and 91.6 ppm can be assigned to (Car–CC–Car′) (Car and Car′ are the carbon atoms of duryl and phenyl rings, respectively; they cannot be distinguished under the current measurement conditions) and the carbon atoms of the quaternary alkynes (Car–CC–Car), respectively. There are no detectable resonances at about 83.0 and 77.0 ppm from the terminal acetylenic groups (Car–CC–H), suggesting a high polymerization efficiency for the porous polymers.19
C– stretching. The broad peaks at 3300 cm−1 can be attributed to the –CC– linkages. The signals at 2870–3050 cm−1 originate from aromatic C–H stretching.18 In the solid-state 13C {1H} cross-polarization/magic angle spinning (CP/MAS) NMR spectra, the well-resolved resonances at around 20.0 ppm are ascribed to the methyl carbons (Car–CH3). The peaks at ∼136.3 and 131.0 ppm can be attributed to the substituted aromatic carbons (Car–CH3) and the hydrogen-bearing aromatic carbons (Car–H), respectively. The signals located at ∼123.3 and 91.6 ppm can be assigned to (Car–CC–Car′) (Car and Car′ are the carbon atoms of duryl and phenyl rings, respectively; they cannot be distinguished under the current measurement conditions) and the carbon atoms of the quaternary alkynes (Car–CC–Car), respectively. There are no detectable resonances at about 83.0 and 77.0 ppm from the terminal acetylenic groups (Car–CC–H), suggesting a high polymerization efficiency for the porous polymers.19
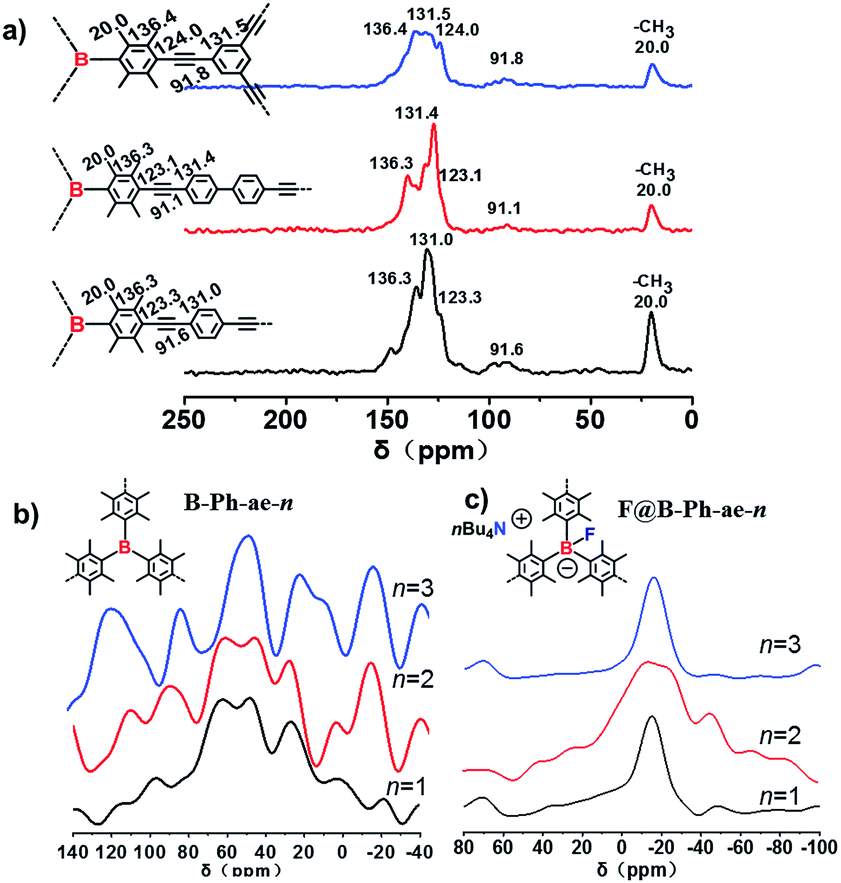 | ||
| Fig. 1 (a) Solid-state 13CCP/MAS NMR spectra of B-Ph-ae-1 (black), B-Ph-ae-2 (red), and B-Ph-ae-3 (blue); (b) solid-state 11B MAS NMR spectra of B-Ph-ae-n; (c) solid-state 11B MAS NMR spectra of F@B-Ph-ae-n. | ||
X-ray diffraction (XRD) (Fig. S2b†) demonstrates the amorphous features that are typical for porous polymers built up from metal-catalyzed coupling reactions. Thermogravimetric analysis (TGA) (Fig. S2c†) under a nitrogen atmosphere manifests a weight loss of only 5% at 315, 312 and 267 °C for B-Ph-ae-1, B-Ph-ae-2, and B-Ph-ae-3, respectively, suggesting good thermal stability for these polymers. The morphology and microstructure of the polymer networks were investigated by transmission electron microscopy (TEM) (Fig. S3†). The TEM images of polymers show the presence of a vermicular texture. The alternating areas of light and dark contrast in the TEM images disclose a disordered porous structure for the porous polymers.
The porous nature of the polymer networks was further confirmed by nitrogen physisorption measurements. It was found that these porous polymers exhibit type IV isotherms.20 The distinct hysteresis phenomena between the adsorption and desorption cycles at relatively low pressure for B-Ph-ae-1, and, in particular, for B-Ph-ae-2, stem from the deformation and swelling of the polymer networks.21 The Brunauer–Emmett–Teller (BET) surface areas of these polymer networks increased in the sequence of B-Ph-ae-2 (226 m2 g−1) < B-Ph-ae-1 (432 m2 g−1) < B-Ph-ae-3 (935 m2 g−1). The pore size distribution curves for these polymers from the adsorption branches of the isotherms were calculated using non-local density functional theory (NL-DFT) (Fig. 2). A set of peaks from 0.88 to 1.94 nm are found for B-Ph-ae-2, demonstrating its distinct microporous features. However, two main peaks at 1.62 and 3.85 nm for B-Ph-ae-1 and three narrowly distributed peaks with maxima at 1.70, 2.70, and 3.90 nm for B-Ph-ae-3 were observed, signifying the presence of both micro- and meso-pores in the networks. This result is in good agreement with the profiles of N2 absorption isotherms. Compared with B-Ph-ae-3, B-Ph-ae-2 is constructed from the conjugated monomer with a longer chain length, and B-Ph-ae-1 exhibits a lower surface area and smaller pore sizes, which can be ascribed to the possible formation of an interpenetrating network and a coiled structure as the backbone rigidity decreased.
 | ||
| Fig. 2 Nitrogen adsorption (open symbols) and desorption (solid symbols) isotherms of (a) B-Ph-ae-n, (b) F@B-Ph-ae-n, and (c) CoF@B-Ph-ae-n measured at 77 K. Pore size distribution curves of (d) B-Ph-ae-1 and CoF@B-Ph-ae-1, (e) B-Ph-ae-2 and CoF@B-Ph-ae-2, and (f) B-Ph-ae-3 and CoF@B-Ph-ae-3 as calculated by NL-DFT. | ||
Given that the Lewis-acid boron center in tridurylborane has a highly selective and strong binding capacity for F−,22 we consider that the as-prepared neutral porous polymers can be readily converted into corresponding anionic porous polymers, as shown in Scheme 1. Thus, the neutral polymers B-Ph-ae-n (n = 1, 2, and 3) were suspended in the THF solution of tetrabutylammonium fluoride (Bu4NF), and stirred at room temperature for 2 days to afford the anionic porous polymers F@B-Ph-ae-n (n = 1, 2, and 3), respectively.
In order to reveal the difference between the samples before and after nBu4NF treatment, B-Ph-ae-n and F@B-Ph-ae-n were studied by X-ray photoelectron spectroscopy (XPS). For the neutral B-Ph-ae-n, all XPS spectra of these samples show a B1s region of 188.4 eV, which can be attributed to typical B–C bonds (Fig. S4†). After ionization, two B1s peaks at 190.3 and 188.4 eV can be observed for F@B-Ph-ae-n,23 which can be ascribed to B–F and B–C bonds, respectively (Fig. S5†). B-Ph-ae-n and F@B-Ph-ae-n were further characterized by 11B solid-state nuclear magnetic resonance (SS-NMR) spectroscopy (see Fig. 1b and c). From the 11B MAS NMR spectra of B-Ph-ae-n, the broad signal centered at around 55 ppm can be assigned to B incorporated into the conjugated polymer networks at regular positions located in symmetric environments.11d As a contrast, in the 11B MAS NMR spectra of F@B-Ph-ae-n (Fig. 1c), a new relatively sharp signal at −15.8 ppm in the high field regions can be observed and the peak at around 55 ppm almost disappeared in the case of F@B-Ph-ae-3, essentially suggestive of approximately all boron atoms coordinated with F− in the network. This result coincides well with the 11B NMR chemical shift of the model compound (Fig. S6†). Notably, for F@B-Ph-ae-1 or F@B-Ph-ae-2, the main signal at −15.8 ppm, with the tailed peaks in the low field regions, might be attributed to the presence of no F−-binding boron atoms. Such a phenomenon is highly related to the porous structure character in the different porous polymer samples. Furthermore, the binding of F− to boron atoms in the porous polymers and model compound was also verified by 19F NMR spectroscopy (Fig. S7†). The signal at −148.8 ppm can be clearly attributed to F atoms in F–B bonds, highly in line with the 19F NMR chemical shift of the model compound (Fig. S8†) and the reported literature.24 All these characterizations strongly indicate that the ionic porous polymers F@B-Ph-ae-n have been successfully synthesized by the simple treatment of the neutral porous polymers B-Ph-ae-n with nBu4NF in some common organic solvents.
The loading degree of F− was next examined by fluorescence spectroscopy. A decreased intensity of the fluorescence emission was found (Fig. S9†), provided the residual empty orbitals of boron atoms in the polymer networks were gradually occupied by the fluoride anions.25 The very low dissociation constant of the F− bonded triduryl boron moiety26 essentially ensures that the framework consisting of such a kind of moiety is able to be kept in a relatively stable charged state. The content of F− remained unchanged after washing many times with THF for thoroughly removing off the free nBu4NF. Furthermore, considering that the exact content of fluoride in a sample is normally difficult to be determined directly by elemental analysis,27 alternatively, we can measure the content of nitrogen (from the counter cation tetrabutyl ammonium ions, equal molar amount relative to the fluoride binding to boron) in the as-prepared F@B-Ph-ae-n samples. As an example, F@B-Ph-ae-3 was examined to have a content of N (1.52 wt%); thus the content of F (2.05 wt%) can be evaluated. Meanwhile, by comparing with the content of B (1.48 wt%) from elemental analysis, we can confirm that F and B exist nearly in a 1:1 molar ratio in F@B-Ph-ae-3. The detailed B, N and F contents of F@B-Ph-ae-n are summarized in Table S1.† After ionization, the surface areas of F@B-Ph-ae-n decreased by 10.4% (n = 1), 22.6% (n = 2) and 54.7% (n = 3) in comparison with those of B-Ph-ae-n. This result can be easily attributed to the increased weight after ionization. The relatively poor binding performance of B-Ph-ae-1 and B-Ph-ae-2 can be attributed to their lower surface area and higher ratio microporous structural characters. These results are well in agreement with the 11B NMR spectra analyses above. The N2 adsorption isotherm manifested significantly the reduced surface areas and pore volumes for the anionic F@B-Ph-ae-n compared with the neutral B-Ph-ae-n (Table 1). This can be explained by the increased molecular mass and space filling resulting from the loaded potassium ions. Interestingly, the pore volume of 0.62 cm3 g−1 for F@B-Ph-ae-3 is much higher than that of 0.48 cm3 g−1 for recently reported anionic porous polymers with the counter cation Na+.14
| Sample | S BET (m2 g−1) | V micro (cm3 g−1) | V tot (cm3 g−1) |
|---|---|---|---|
| a Surface areas calculated from the N2 adsorption isotherm using the BET method. b The micropore volume derived using the non-local density functional theory. c Total pore volume at p/p0 = 0.99. | |||
| B-Ph-ae-1 | 432 | 0.14 | 0.26 |
| B-Ph-ae-2 | 226 | 0.12 | 0.42 |
| B-Ph-ae-3 | 935 | 0.20 | 0.71 |
| F@B-Ph-ae-1 | 387 | 0.10 | 0.15 |
| F@B-Ph-ae-2 | 175 | 0.08 | 0.22 |
| F@B-Ph-ae-3 | 424 | 0.13 | 0.47 |
| CoF@B-Ph-ae-1 | 424 | 0.11 | 0.17 |
| CoF@B-Ph-ae-2 | 186 | 0.10 | 0.26 |
| CoF@B-Ph-ae-3 | 490 | 0.15 | 0.53 |
Ion exchange is a favorable strategy to incorporate various ions into the charged materials, e.g. zeolites, for achieving the desired properties, such as catalysis28 and gas adsorption.13,29 As expected, the tetrabutyl ammonium cations in the networks of the as-prepared F@B-Ph-ae-n can be readily exchanged with the different heavy metal ions by the simple treatment with the corresponding heavy metal salt in a solvent mixture of ethanol and THF, resulting in heavy metal ion loaded porous polymers MF@B-Ph-ae-n (see the ESI†). Here, CoF@B-Ph-ae-3 was selected as a typical example for the detailed investigation. According to ICP-OES analysis, the loading content of cobalt in Co@B-Ph-ae-3 is 2.43 wt%, which is nearly half the molar amount of B (1.11 wt%) in the corresponding F@B-Ph-ae-3 sample, suggesting that tetrabutyl ammonium ions have been almost completely replaced by Co(II) ions to maintain the charge balance. In addition, the acetate anion signal was not observed in the FTIR spectra (Fig. S10†), confirming that there is no free Co(Ac)2 left in the networks.
The compositions of the resulting Co(II) loaded anionic polymers were further analyzed via X-ray photoelectron spectroscopy (XPS) (Fig. 3 and S11, S12†). There are two characteristic peaks at 782.2 and 798.5 eV, corresponding to the 2p3/2 and 2p1/2 spin–orbit peaks of Co(II). The peaks at around 787.2 and 803.3 eV are associated with the shake-up type peaks of the 2p3/2 and 2p1/2 edges, respectively.30 XPS also indicates a cobalt content of 2.40 wt%, which is in good agreement with the value obtained from ICP-OES analysis. Of note, elemental mapping images show the homogeneous distribution of carbon, boron, and cobalt elements in CoF@B-Ph-ae-3 (Fig. 3). The surface area and pore volume of CoF@B-Ph-ae-3 were determined to be 468 m2 g−1 and 0.51 cm3 g−1, respectively, which are slightly lower than those of F@B-Ph-ae-3 (Table 1) due to the lower molecular weight of Co2+ (58.93) in comparison with Bu4N+ (242.28). Therefore, one can recognize the remarkable differences in N2 isotherm profiles, surface areas, and pore volumes for B-Ph-ae-n, F@B-Ph-ae-n, and CoF@B-Ph-ae-n (Fig. 2 and Table 1), implying that the physisorption properties of porous polymers can be tuned by introducing the fluoride anion and different metal cations, without changing the backbone of the polymer networks.
 | ||
| Fig. 3 (a) Typical scanning electron microscopy (SEM) image of CoF@B-Ph-ae-3 and the corresponding elemental mapping images of (b) carbon, (c) boron, and (d) cobalt. The XPS spectra of CoF@B-Ph-ae-3: (e) Co 2p core-level spectrum and (f) the survey spectrum. | ||
Encouraged by the success in the loading of cobalt cations, we further investigated the inclusion of other metal cations in these anionic porous polymers. For instance, nickel(II) acetate or iron(II) nitrate was added to the dispersion of F@B-Ph-ae-3 in THF, followed by stirring for 2 days, respectively. The resulting Ni or Fe containing porous polymers (NiF@B-Ph-ae-3 and FeF@B-Ph-ae-3) exhibit a loading amount of 1.05 wt% and 3.11 wt% respectively. Moreover, the capability of neutral porous polymers B-Ph-ae-n to selectively capture metal ions from a mixture system was further examined (Fig. S13†). Reasonably, the ratios of the transition metals loaded in an anionic sample were governed by the corresponding ion exchange constants.31 Therefore, the as-synthesized Lewis acid-based neutral porous polymers might have potential application to selectively collect F− and heavy metal ions in the polluted environment through a one-pot process.
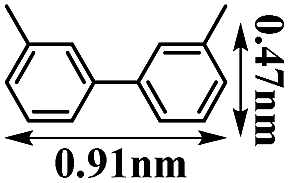
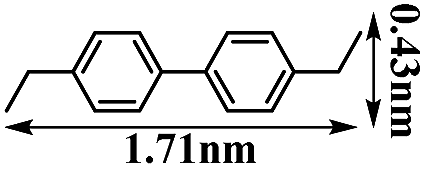
It is well known that transition metal ion-loaded porous materials are promising heterogeneous catalysts.33 The size and shape selectivity3a,34 for a porous catalyst plays a key role in the controlled synthesis of molecules with a certain size or configuration. Thus, the as-prepared MF@B-Ph-ae-n porous polymers, loaded with different transition metal ions, presumably possess heterogeneous catalytic properties highly related to their unique porous structures. With these in our mind, homocoupling reactions of aromatic Grignard reagents catalyzed by employing CoF@B-Ph-ae-n loaded with a stoichiometric amount of cobalt cations (calculated on the basis of the Grignard reagent) were carried out. To elucidate the catalytic activity and size selectivity of CoF@B-Ph-ae-n, a series of substrates of different sizes were examined: phenyl Grignard reagent (0.43 × 0.84 nm2), 3-methylphenyl Grignard reagent (0.47 × 0.84 nm2), 4-methylphenyl Grignard reagent (0.43 × 0.93 nm2), and 4-ethylphenyl Grignard reagent (0.43 × 1.10 nm2). The homocoupling products were isolated, and the catalytic data are summarized in Table 2. CoF@B-Ph-ae-3, with the largest mesoporous structure, provided the homocoupling products of biphenyl, 4,4′-dimethylbiphenyl, and 3,3′-dimethylbiphenyl in high yields of 91%, 84% and 81%, respectively, but a poor yield of 14% for 4,4′-diethylbiphenyl with a larger molecular size. While the microporous CoF@B-Ph-ae-2 only resulted in a high conversion of 80% for biphenyl, but no larger size molecule 4,4′-dimethylbiphenyl or 4,4′-diethylbiphenyl was found and isolated as the product. Interestingly, using 3-methylphenyl Grignard reagent as the substrate, which has a smaller size than 4-methylphenyl Grignard reagent, the microporous CoF@B-Ph-ae-2 enables delivering a remarkable catalytic activity up to a yield of 68%. As a comparison, CoCl2 (its amount was calculated relative to the content of cobalt in CoF@B-Ph-ae-n) was used as a catalyst for the homocoupling of the aromatic Grignard reagents under the same reaction conditions, which offered the coupling products biphenyl, 4,4′-dimethylbiphenyl, 3,3′-dimethylbiphenyl or 4,4′-diethylbiphenyl in good yields without efficient size selectivity. Therefore, the porous structures had the major impact on the catalytic behavior through spatially controlling the formation of the species in the process of homocoupling reactions (e.g. the intermediates formed in the transmetallation step).
| Reactant |
|
|
|
|
|---|---|---|---|---|
| Productb |
|
|
|
|
| a Reaction conditions: 1.6 mmol Grignard reagents, CoF@B-Ph-ae-n containing 0.8 mmol cobalt, 10 mL of THF at 55 °C, 1 h. b Isolated yield after chromatographic purification.32 | ||||
| CoF@B-Ph-ae-1 | 85% | 43% | 71% | n. d. |
| CoF@B-Ph-ae-2 | 80% | n. d. | 68% | n. d. |
| CoF@B-Ph-ae-3 | 91% | 84% | 81% | 14% |
| CoCl2 | 89% | 87% | 83% | 86% |





To further probe the catalytic activities and selectivities with respect to the porous structures, a mixture containing phenyl, 4-methylphenyl, and 4-ethylphenyl Grignard reagents in 1:1:1 eq. was treated with CoF@B-Ph-ae-n containing a stoichiometric amount of cobalt cations in THF under mild conditions (55 °C). Gas chromatography mass-spectrometry (GC-MS) analyses revealed that the distribution of coupling products is highly dependent on the pore size of the as-made anionic porous polymers, and the microporous CoF@B-Ph-ae-2 showed the best size selectivity for the formation of biphenyl homocoupling products (Table S2, Fig. S14–S18†). Undoubtedly, such results will be helpful for developing new catalytic systems with high size and shape selectivity.
Conclusions
In conclusion, we have developed a series of Lewis acid boron-containing conjugated polymers with hierarchical porous structures via typical transition metal-catalyzed cross-coupling reactions. The distinct capability of the resulting neutral porous polymers to selectively capture fluoride ions provides a high-efficiency conversion into stable anionic porous polymers. Importantly, the fluoride anion binding boron in a solid sample, for the first time, was clearly characterized through solid-state 11B MAS NMR spectroscopy, which would provide a very valuable reference for the corresponding investigation. Upon a simple ion-exchange process, various heavy metal cations were able to be loaded into the networks of the anionic porous polymers. Generally, such a synthetic strategy might represent an efficient approach to versatile functional porous materials via a successful loading process. As an example, the cobalt(II)-loaded porous polymers exhibit a distinct size selectivity for homocoupling reactions of aryl Grignard reagents, which is highly related to the porous structures. Using such a simple ion exchange strategy, various catalytically active metal ions can be rationally incorporated into the as-prepared anionic porous polymers, resulting in new heterogeneous catalytic systems. One can expect that the catalytic behavior is tunable not only by the pore structures of the porous polymers, but also through introduction of other ligands by a “ship in bottle” technique. Moreover, simultaneous integration of multi-metals into the porous polymers may provide synergetic effects for certain catalytic organic transformations with the size and shape control. On the other hand, the pronounced capability to selectively recycle and reuse environmentally harmful fluoride anions and heavy metals makes the as-prepared boron-based porous polymers promising candidates for potential applications in the comprehensive management of the environment.Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 Program: 2013CBA01602 and 2012CB933404), the Natural Science Foundation of China (21574080, 51403126 and 21102091), the Shanghai Committee of Science and Technology (15JC1490500) and an ERC grant on 2DMATER. We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for providing some measurements.Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 8075 CrossRef PubMed.
- (a) X. Zhu, C. Tian, S. M. Mahurin, S.-H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478 CrossRef CAS PubMed; (b) J. Zhang, S. H. Chai, Z. A. Qiao, S. M. Mahurin, J. Chen, Y. Fang, S. Wan, K. Nelson, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 932 CrossRef CAS PubMed.
- (a) Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878 CrossRef CAS PubMed; (b) R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schuth, Angew. Chem., Int. Ed., 2009, 48, 6909 CrossRef CAS PubMed; (c) H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohen, J. Am. Chem. Soc., 2014, 136, 4965 CrossRef CAS PubMed.
- (a) F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819 RSC; (b) S. Jin, H. J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 955 CrossRef CAS PubMed; (c) Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753 CrossRef CAS PubMed.
- (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) Z. J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867 RSC; (c) Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS PubMed; (d) A. Corma, H. García and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed.
- (a) W. J. Roth, P. Nachtigall, R. E. Morris and J. Cejka, Chem. Rev., 2014, 114, 4807 CrossRef CAS PubMed; (b) M. E. Davis, Chem. Mater., 2013, 26, 239 CrossRef; (c) A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313 CrossRef CAS.
- (a) G. Sartori and R. Maggi, Chem. Rev., 2011, 111, 181 CrossRef PubMed; (b) P. H. Espeel, B. Janssens and P. A. Jacobs, J. Org. Chem., 1993, 58, 7688 CrossRef CAS.
- S. Saravanamurugan, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 135, 5246 CrossRef CAS PubMed.
- S. Tontisirin and S. Ernst, Angew. Chem., Int. Ed., 2007, 46, 7304 CrossRef CAS PubMed.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880 CrossRef CAS PubMed.
- (a) D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS PubMed; (b) R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530 CrossRef CAS; (c) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC; (d) X. M. Liu, Y. W. Zhang, H. Li, S. A, H. Xia and Y. Mu, RSC Adv., 2013, 3, 21267 RSC; (e) Q. Sun, Z. Dai, X. Meng and F. S. Xiao, Chem. Soc. Rev., 2015, 44, 6018 RSC; (f) C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11 RSC; (g) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- Y. Yuan, F. Sun, L. Li, P. Cui and G. Zhu, Nat. Commun., 2014, 5, 4260 CAS.
- (a) S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174 CrossRef CAS PubMed; (b) Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733 CrossRef CAS PubMed; (c) J. F. V. Humbeck, M. L. Aubrey, A. Alsbaiee, R. Ameloot, G. W. Coates, W. R. Dichtel and J. R. Long, Chem. Sci., 2015, 6, 5499 RSC.
- Y. Liu, X. Xu, F. Zheng and Y. Cui, Angew. Chem., Int. Ed., 2008, 120, 4614 CrossRef.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. Jones and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS PubMed.
- S. H. Lim, Y. Su and S. M. Cohen, Angew. Chem., Int. Ed., 2012, 51, 5106 CrossRef CAS PubMed.
- (a) M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511 CrossRef CAS; (b) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS PubMed.
- (a) W. Lu, Z. Wei, D. Yuan, J. Tian, S. Fordham and H.-C. Zhou, Chem. Mater., 2014, 26, 4589 CrossRef CAS; (b) J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed; (c) X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668 CrossRef CAS PubMed; (d) X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081 CrossRef CAS PubMed; (e) X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789 CrossRef CAS PubMed; (f) Y. Su, Z. Yao, F. Zhang, H. Wang, Z. Mics, E. Cánovas, M. Bonn, X. Zhuang and X. Feng, Adv. Funct. Mater., 2016, 26, 5893 CrossRef CAS; (g) K. Yuan, X. Zhuang, H. Fu, G. Brunklaus, M. Forster, Y. Chen, X. Feng and U. Scherf, Angew. Chem., Int. Ed., 2016, 55, 6858 CrossRef CAS PubMed.
- (a) S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723 CrossRef CAS; (b) P. B. Balbuena and K. E. Gubbins, Langmuir, 1993, 9, 1801 CrossRef CAS.
- J. Weber, J. Schmidt, A. Thomas and W. Bohlmann, Langmuir, 2010, 26, 15650 CrossRef CAS PubMed.
- (a) F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286 CrossRef CAS PubMed; (b) H. Pan, G.-L. Fu, Y.-H. Zhao and C.-H. Zhao, Org. Lett., 2011, 13, 4830 CrossRef CAS PubMed; (c) X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475 CrossRef CAS PubMed; (d) W. M. Wan, F. Cheng and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 8934 CrossRef CAS PubMed.
- (a) L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li and P. M. Ajayan, Nat. Mater., 2010, 9, 430 CrossRef CAS PubMed; (b) H. Li, J. Xing, Z. Xia and J. Chen, Electrochim. Acta, 2014, 139, 331 CrossRef CAS.
- (a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372 CrossRef CAS PubMed; (b) Y. Kubo, M. Yamamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 115, 2082 CrossRef; (c) X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 118, 5601 CrossRef.
- (a) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142 CrossRef CAS PubMed; (b) Z. M. Hudson, X.-Y. Liu and S. Wang, Org. Lett., 2010, 13, 300 CrossRef PubMed.
- S. Sole and F. P. Gabbai, Chem. Commun., 2004, 1284 RSC.
- X. D. Hou and B. T. Jones, Encyclopedia of Analytical Chemistry, Wiley Online Library, 2000, p. 9468 Search PubMed.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- E. Quartapelle Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS PubMed.
- (a) G. Yang, D. Gao, Z. Shi, Z. Zhang, J. Zhang, J. Zhang and D. Xue, J. Phys. Chem. C, 2010, 114, 21989 CrossRef CAS; (b) C. Moreno-Castilla, F. J. Maldonado-Hódar and A. F. Pérez-Cadenas, Langmuir, 2003, 19, 5650 CrossRef CAS.
- (a) W. Meng, H. Li, Z. Xu, S. Du, Y. Li, Y. Zhu, Y. Han, H. Hou, Y. Fan and M. Tang, Chem.–Eur. J., 2014, 20, 2945 CAS; (b) S. Kocaoba, Energy Sci. Eng., 2008, 25, 697 CAS.
- The molecular length was calculated by density functional theory (RB3LYP/6-311G** (d,p) level).
- (a) D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515 RSC; (b) A. Nag, D. S. Chung, D. S. Dolzhnikov, N. M. Dimitrijevic, S. Chattopadhyay, T. Shibata and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 13604 CrossRef CAS PubMed; (c) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819 CrossRef CAS; (d) J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702 CrossRef CAS.
- (a) M. Shakeri, R. J. Klein Gebbink, P. E. de Jongh and K. P. de Jong, Angew. Chem., Int. Ed., 2013, 52, 10854 CrossRef CAS PubMed; (b) D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2007, 46, 1311 CrossRef CAS PubMed; (c) L. Li, R. Matsuda, I. Tanaka, H. Sato, P. Kanoo, H. J. Jeon, M. L. Foo, A. Wakamiya, Y. Murata and S. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7543 CrossRef CAS PubMed; (d) L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta04917f |
| This journal is © The Royal Society of Chemistry 2016 |