
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient electro-reduction of CO2 to formic acid by nano-copper†
Kalyani
Gupta
,
Marco
Bersani
and
Jawwad A.
Darr
*
Department of Chemistry, Christopher Ingold Laboratories, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: j.a.darr@ucl.ac.uk
First published on 11th August 2016
Abstract
Ultra-fine copper(II) oxide nanoparticles were used for the electrocatalytic reduction of CO2 to formic acid at high Faradaic efficiencies. The nanoparticles were directly synthesised via continuous hydrothermal flow synthesis (CHFS) process, which used water as a solvent and reagent. The as-prepared nanoparticles were subsequently formulated into Nafion based inks. For the electroreduction of CO2, the influence of Nafion fraction on the Faradaic efficiencies and overpotential (for formic acid production), was explored over a wide potential range. The highest Faradaic efficiency for formic acid production (61%) was observed with a 25 wt% Nafion fraction, at a potential of −1.4 V vs. Ag/AgCl. Some insights into the significant increase in Faradaic efficiency for the production of formic acid with the optimum Nafion content, was elucidated with electrochemical impedance spectroscopy.
Introduction
Global warming and climate change have been strongly linked to the increasing concentration of CO2 in the atmosphere.1 Over the past 100 years, the amount of CO2 has risen from 150 ppm to >400 ppm by 2016.2 In tandem with CO2 sequestration and storage, there is a strong drive towards utilising CO2 to form useful chemicals, as CO2 is an inexpensive, relatively non-toxic and an abundant carbon feedstock.3 The electrochemical reduction of CO2 into formic acid, methanol, syngas or hydrocarbons, is an attractive way to utilise captured CO2 whilst making valuable chemicals.4Copper is a well-known catalyst for converting CO2 to various products such as methane, formic acid, methanol and CO.4–6 However, both polycrystalline copper and single crystal copper electrodes, can also suffer from low activity and require high overpotentials.7 In particular, nanostructured copper and its oxides, have been investigated for the electrochemical conversion of CO2 to chemicals.7 The use of oxides, particularly Cu(I), is also of interest as it has been shown to require a relatively low overpotential to convert CO2 to chemicals.8 Typically, nano-Cu (and its oxides) can be synthesised by a range of methods including electrochemical deposition,9 batch hydrothermal syntheses,10,11 precipitation reactions,12 microwave assisted13 and solid state syntheses.14 Whilst such methods can produce small quantities of catalyst, successful and reproducible scale-up syntheses of nanoparticles are generally more difficult to achieve in batch processes. In contrast to batch processes, continuous synthesis methods can offer several advantages over batch methods in terms of scalability, ease of processing and allowing independent control over reaction parameters such as reaction time, temperature and pressure. In particular, Continuous Hydrothermal Flow Synthesis (CHFS) using supercritical water is a promising and scalable method for the rapid and controlled synthesis of high surface area metal and metal oxide nanoparticles.15 A typical CHFS process involves the mixing of superheated water (generally above its critical point Tc = 374 °C and pc = 22.1 MPa) in flow with a stream of an aqueous metal salt solution (at ambient temperatures) in a controlled manner. Upon mixing of the two feeds, nanoparticles are instantly formed via simultaneous hydrolysis and dehydration under supersaturated conditions. Recently, the use of a confined jet mixer (CJM) was reported by the authors for mixing in CHFS, under highly turbulent conditions.16–20 Due to the instant mixing of the reactants and supercritical water within the CJM, nanoparticles can be made consistently and with a narrow size distribution at kg per day rates.21–23
In the electroreduction of CO2 using nanoparticles, typical electrodes are prepared via deposition of the catalyst nanopowder (in the form of a suspended ink) onto a microporous substrate or glassy carbon electrode. When considering the performance of such electrodes, the final microstructure of the resulting catalyst layer can often be overlooked. Recently, proton exchange membrane fuel cell (PEMFC)-like designs were adopted for the electrocatalytic reduction of CO2, where Nafion ionomer was incorporated in the catalyst layer to help improve CO2 reduction.24 Nafion can act as a binder, creating a continuously connected ionic conductive matrix, which allows the reactants and products to be transported between the active catalytic sites and the reactant.25 Several studies have shown that the performance of PEMFCs is often sensitive to the Nafion fraction, where an optimal fraction is typically in the range 25 to 40 wt% (depending on Pt fraction and Pt particle size in the case of PEMFCs). Recent studies have also investigated the effect of Nafion and PTFE as binders on the electrochemical reduction of CO2 on Sn-based catalysts.26–28 To the author's knowledge, no systematic study considering the Nafion fraction with nano-copper based catalysts for CO2 reduction has been previously investigated. As the microstructure of the final catalyst layer plays a significant role in the catalytic performance, it is imperative to understand the correlation between Nafion content and electrocatalytic activity for CO2 reduction. This also becomes critical when scaling up from conventional three electrode cells typically used in electroreduction of CO2, to fuel-cell like designs, which have the potential to convert CO2 to chemicals at pilot scale level.29
Herein, we report the scalable synthesis of nano-sized, high surface area nano copper(II) oxide using copper(II) nitrate trihydrate and KOH in a continuous hydrothermal process. The resulting nanoparticles were characterised by a range of analytical methods including powder XRD, TEM, SEM and XPS. The ultrafine CuO nanopowder was subsequently formulated into Nafion based inks with varying Nafion content in the range 1 to 66 wt% (vs. total solid fraction). The effect of the Nafion fraction on the Faradaic efficiency and overpotential for the conversion of CO2 to formic acid, was studied. The effect of the applied potential (in the range −0.6 to −1.4 V) on the performance of the CuO catalysts, was also explored. Electrochemical impedance spectroscopy (EIS) was used to provide an insight into the influence of the Nafion fraction on the performance of CO2 reduction to formic acid.
Experimental section
Materials
Copper(II) nitrate trihydrate [Cu(NO3)2·3H2O, technical grade, >98%] and potassium hydroxide pellets (KOH) were used in the synthesis of ultrafine CuO. All reactions were conducted in deionised water (>10 MΩ). Nafion (5 wt% in a mixture of lower aliphatic alcohols and 45% water) and high purity LC-MS chromasolv isopropanol (IPA) and methanol were used in the ink formulation. All chemicals, unless stated otherwise, were supplied by Sigma Aldrich, (Dorset, UK). High purity KHCO3 was supplied by Alfa Aesar, (Lancashire, UK). All chemicals were used as purchased.Synthesis of CuO
Pure copper(II) oxide nanoparticles were synthesised using a lab scale CHFS system, which uses four high-pressure diaphragm pumps (Fig. 1), the details of which are described elsewhere.17 Briefly, in the process, a room temperature aqueous solution of 0.1 M copper nitrate trihydrate was pumped (via pump 2 at 40 mL min−1) to mix with an aqueous flow of 0.2 M potassium hydroxide solution (via pump 3 at 40 mL min−1) under pressure in a dead volume T-piece. The resulting mixture was then mixed with a superheated water feed at 400 °C (via pump 1 at 80 mL min−1) inside a confined jet mixer, (CJM) (Fig. 1). This CJM was designed to mix the reagents without blockages under a turbulent mixing regime (Reynolds number ≈ 6079). The calculated mixing temperature was ca. 305 °C, with a residence time of ca. 0.6 s before the nanoparticle slurry was quenched rapidly as it mixed with a feed of room temperature deionised water (via pump 4 at 160 mL min−1) in a second CJM (Reynolds number ≈ 3000). The design and sizes of the two CJMs are given in more detail in the ESI† and in previous publications.30 The newly formed and partially cooled nanoparticle laden slurry was then cooled further via a pipe-in-pipe heat exchanger and was then continuously collected from the exit of the back-pressure regulator (BPR), which maintained the pressure in the system at 24.1 MPa. The particle slurry was cleaned via dialysis and repeated centrifugation until the conductivity of the slurry was <50 μS, as measured by a conductivity probe (Hanna Instruments HI98311, Bedfordshire UK). The concentrated slurry was then freeze-dried with slow heating from −40 °C to 25 °C using a VirTis Genesis 35 XL Lyophilizer at ca. 10−7 MPa for 24 hours.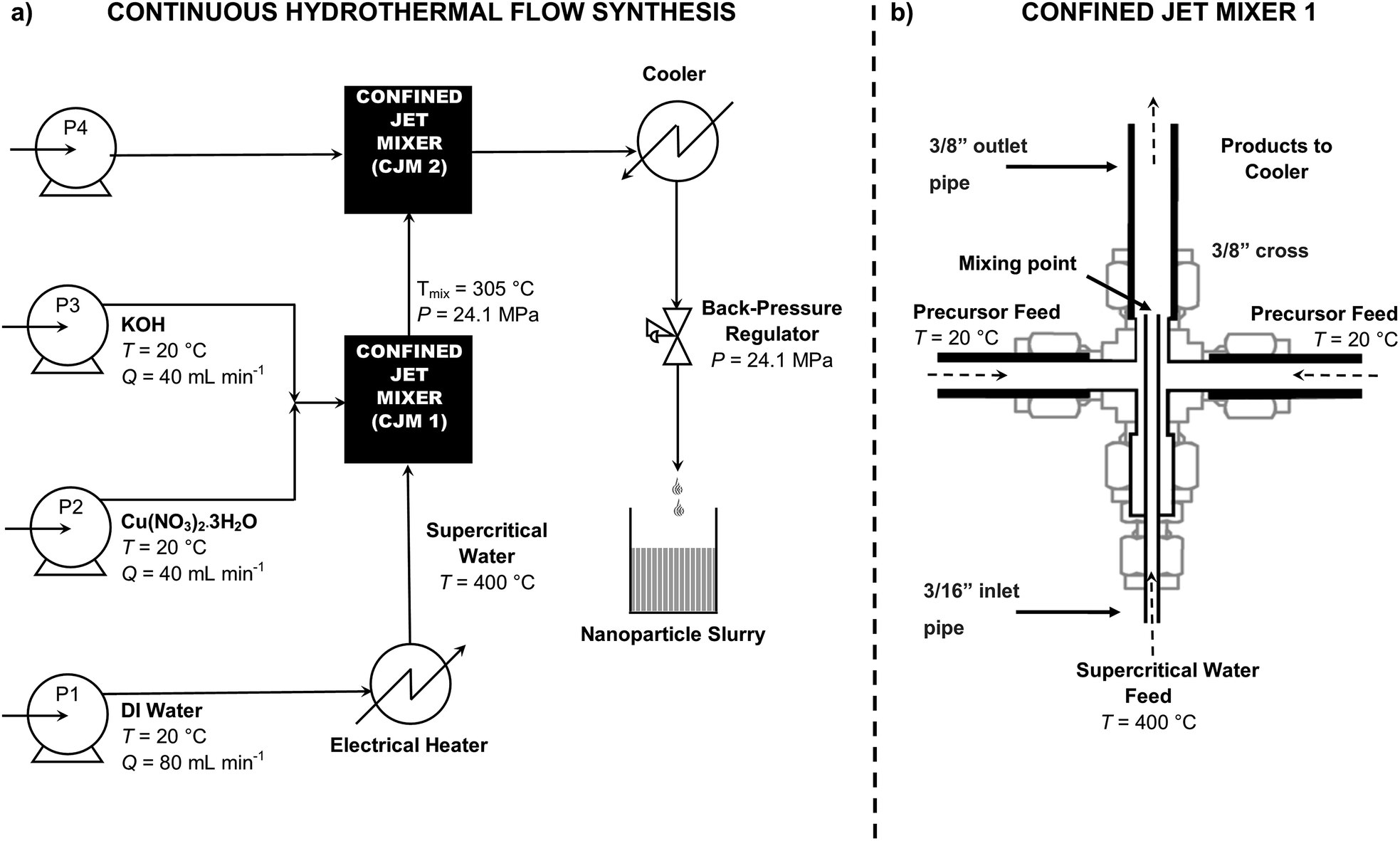 | ||
| Fig. 1 (a) Schematic representation of the Continuous Hydrothermal Flow Synthesis (CHFS) process using supercritical water for the synthesis of ultrafine copper oxide nanoparticles in flow. Pumps are denoted by P. Water at room temperature is drawn from P1 and is heated in flow before it enters the first confined jet mixer in the supercritical state. The metal salt feed is drawn from P2 and the base feed from P3. P4 is used as a quench where chemical additive or D.I. water is added. The metal and base feed first meet in a tee piece, then travel to meet the supercritical water feed in a confined jet mixer (denoted as CJM1). The nanoparticles form instantaneously and travel to meet the quench feed inside CJM2. (b) Detailed view of confined jet mixer 1 (CJM1). See ESI† for CJM sizes and design. | ||
Materials characterisation
The freeze-dried CuO nanopowder was examined by powder X-ray diffraction (Bruker D4 diffractometer, Cu Kα1, λ = 1.54 Å). Brunauer–Emmett–Teller (BET) surface area measurements were carried out using N2 in a Micrometrics TriStar II plus apparatus. The sample was degassed up to 150 °C for 12 hours in a nitrogen flow before measurements were undertaken. Surface analysis was carried out using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer. Survey scans were conducted at a pass energy of 150 eV and high-resolution region scans were conducted at 50 eV. The XPS spectra were processed using Casa™ software. The binding energy scale was calibrated by the C 1s peak at 285.0 eV. Detailed morphology and size analysis was carried out using high-resolution transmission electron microscopy (HRTEM). Images were obtained on a JEOL JEM 2100 TEM with 200 keV accelerating voltage and a LaBF6 filament. HRTEM samples were prepared by dispersing the particles in methanol 99.9% (Sigma Aldrich, Dorset, UK) by ultra-sonication before dropping the resulting dispersion onto 300 mesh Holey copper film grids (Agar Scientific, Stansted UK). Particle size distribution was determined by measuring the diameter of 300 particles from TEM images.Ink formulation
50 mg of CuO nanoparticles were suspended in a mixture of isopropanol (IPA), methanol and DI water in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:6, respectively. The Nafion content was varied at values of 1, 10, 25 and 66 wt% (vs. total solid fraction). The suspension was sonicated using a high power dispersion sonic tip (Branson Sonifiers 250D, Geneva, CH) for 30 minutes at 20% amplitude. The ink suspension was immersed in an ice bath to avoid solvent evaporation and conversion of CuO to copper carbonates.
:6:6, respectively. The Nafion content was varied at values of 1, 10, 25 and 66 wt% (vs. total solid fraction). The suspension was sonicated using a high power dispersion sonic tip (Branson Sonifiers 250D, Geneva, CH) for 30 minutes at 20% amplitude. The ink suspension was immersed in an ice bath to avoid solvent evaporation and conversion of CuO to copper carbonates.
Electrode preparation
The glassy carbon sheet (1 × 1 cm) was cleaned by polishing using a 0.05 μm alumina slurry (Buehler, Stuttgart, DE) prior to drop casting. Electrodes were then prepared by casting CuO inks on a glassy carbon sheet (Alfa Aesar, Lancashire UK) to have a loading of approximately 0.3 mg cm−2 with a thickness range of 10–15 μm. Subsequently, the glassy carbon was dried overnight in a vacuum oven at 40 °C to give a homogeneous CuO film, denoted as a catalyst layer (CL). Samples were thus named CL1, CL10, CL25 and CL66 for catalyst layers with 1, 10, 25 and 66 wt% Nafion fraction, respectively. The catalyst film morphology was observed using SEM (JEOL JSM-6700F at 10 kV).Electrochemical characterisation
The catalysts were tested in a conventional three-electrode set up; a (0.5 mm OD) platinum coil (Alfa Aesar, Lancashire UK) was used as a counter electrode, a Ag/AgCl electrode (BASi, West Lafayette, US) was used as a reference electrode and the working electrode was a CuO coated glassy carbon sheet. A Gamry 1000 potentiostat (Gamry, Warminster, US) was used for all electrochemical experiments. All experiments were conducted at ambient temperature. The inks were characterised by cyclic voltammetry in 0.5 M KHCO3 saturated with CO2 for 30 minutes. The electrode was cycled in the range 0.7 to −0.8 V vs. Ag/AgCl at 100, 50, 20 and 5 mV s−1. Constant potential electrolysis measurements were conducted at potentials in the range −0.6 to −1.4 V vs. Ag/AgCl. Headspace analysis of the cell was conducted by extracting aliquots with a gas tight syringe and analysed via a Shimadzu GC 2014 equipped with TCD and FID detectors and a packed CTR-1 column. Liquid phase products were analysed post electrolysis using a 600 MHz Bruker 1H NMR using DSS in H2O/D2O as a reference control. Surface area measurements were conducted by cycling between −0.25 and −0.45 V in 0.1 M H2SO4 after holding the CuO coated glassy carbon electrodes at a constant potential of −1.0 V for 1 hour in a CO2 saturated 0.5 M KHCO3 electrolyte. Electrochemical impedance spectroscopy (EIS) was performed over a frequency range of 10 kHz to 0.1 Hz with an AC perturbation of 5 mV at an applied potential of −1.4 V. The EIS data was analysed by Gamry EChem Analyst software. All potentials are referenced and reported against an Ag/AgCl electrode.Results and discussion
Synthesis of CuO
Ultrafine CuO nanoparticles were synthesised via continuous hydrothermal flow synthesis (CHFS) using 0.1 M copper nitrate and 0.2 M KOH, resulting in an aqueous nanoparticle slurry with a pH of 5. The particles in the slurry were cleaned by centrifugation and dialysis to remove any possible unreacted precursors and by-products. The wet solids were freeze-dried, resulting in a black powder (yield = 76%). The powder XRD (Fig. 2a), confirmed that pure phase CuO was formed, with a monoclinic structure (similar to standard reference pattern tenorite, JCPDS 01-089-2529), with the most intense reflections arising from the (−111), (111) and (−202) peaks. The broad nature of the peaks suggested a small crystallite size for the CuO, which was estimated from application of the Scherrer equation to be ca. 15 nm.31 Surface analysis of the particles by XPS confirmed the presence of CuII species, as indicated by the XPS peaks for Cu 2p3/2 at 933.9 eV and Cu 2p1/2 at 953.7 eV (Fig. 2b). Additional confirmation of the CuII state was seen with the broad satellite peaks at higher binding energy than the main peaks. Shake up satellites are not normally seen with CuI or Cu0 oxidation states as reported previously in the literature, which suggested the presence of only CuII species on the surface.32,33 As synthesised nanopowders were also analysed by Transmission Electron Microscopy (TEM) (Fig. 3a). The CuO particles appeared to be well defined with some faceting, monocrystallinity and a rhombic morphology, with an average size of 17 ± 5 nm (300 particles measured, Fig. 3b). The average particle size matched well with the crystallite size of ca. 15 nm as estimated from the Scherrer equation. Higher magnification analysis showed a lattice planar spacing of 0.23 nm, which was in agreement with literature value for the (111) plane (Fig. 3c) of 0.23 nm.13 The BET surface area was measured as 52 m2 g−1 and compared well to estimated BET surface area based on a hard sphere model (57 m2 g−1). The crystallinity and phase purity of the nanoparticles were consistent with previous observations reported for the formation of metal oxides synthesised via CHFS, which suggested the formation of CuO occurred via rapid hydrolysis of a Cu(II) salt to form Cu(OH)2 and subsequent dehydration to form CuO.19,34 Also, the small crystallite size, high yield and monodispersity of particles, suggested a nucleation dominated mechanism of nanoparticle formation where a supersaturation of small nuclei was maximised due to the decrease in dielectric constant and the low solubility of inorganic ions under the reaction conditions in the first CJM.15,30,35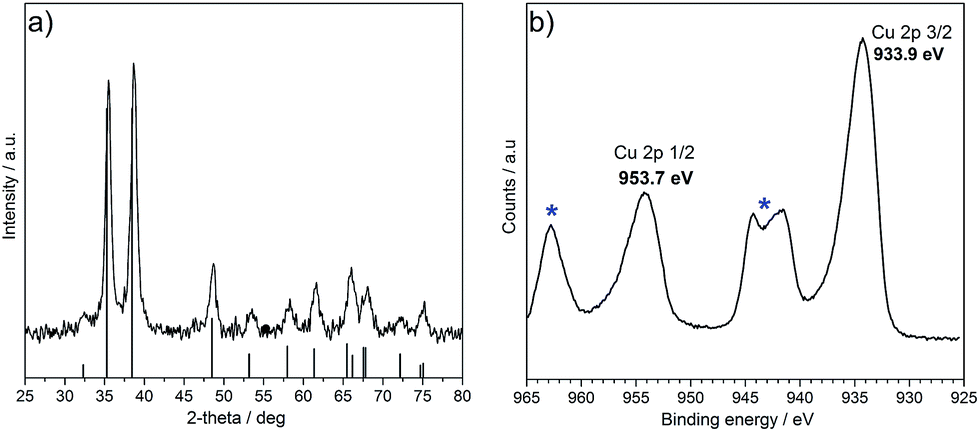 | ||
| Fig. 2 (a) Powder XRD pattern of CuO synthesised via CHFS (JCPDS 01-089-2529) and (b) high resolution Cu 2p scan of CuO as prepared via CHFS, Cu 2p3/2 at 933.9 eV and Cu 2p1/2 at 953.7 eV. Satellite peaks are highlighted by asterisks. | ||
 | ||
| Fig. 3 (a) TEM image of CuO nanoparticles as prepared by CHFS, (b) inset shows histogram of particle size distribution measured from 300 particles and (c) high resolution image of CuO nanoparticles showing (111) plane with a d spacing of 0.23 nm. | ||
Electrochemistry
The as-prepared nanoparticles were suspended into inks containing IPA, methanol and D.I. water in a volume ratio of 1:6:6 and 1, 10, 25 and 66 wt% Nafion (vs. total solid fraction). The inks were cast on glassy carbon electrodes and the redox behaviour of CuO to Cu (via Cu2O) was characterised by cyclic voltammetry in CO2 saturated 0.5 M KHCO3.36 Typically, CuO reduction followed a small peak for CuO to Cu2O reduction and the appearance of a nucleation loop and a crossover point characteristic of nuclei formation and growth process, could be observed in the potential range from −0.2 to −0.6 V vs. Ag/AgCl. This was typically higher than the equilibrium potential as the metal ions were deposited on a substrate that was crystallographically different, resulting in a substrate–metal misfit. Distinct cyclic voltammograms were observed in this case for glassy carbon coated with CuO inks with different Nafion fractions (Fig. 4).
 | ||
| Fig. 4 Cyclic voltammograms of (a) CL1, (b) CL10, (c) CL25 and (d) CL66 in 0.5 M KHCO3 saturated with CO2 at a scan rate 5 mV s−1, during the second cycle. | ||
A nucleation loop was observed for all samples, however, the position and size of the nucleation loop varied greatly between each sample. The initial reduction from CuO to Cu2O was observed at −0.1 V and is represented as I in Fig. 4. The onset of the crossover potential was similar for sample CL1 (Fig. 4a) and sample CL10 (Fig. 4b), and appeared at −0.3 V. However, the crossover potential for sample CL25 (Fig. 4c) was lower than the previous example, at −0.2 V, indicative of a lower overpotential requirement for Cu2O reduction to Cu on CuO. A considerable difference was observed for CL66 (Fig. 4d), where the crossover potential was shifted to a more negative potential at −0.6 V, indicative of a significant overpotential requirement for Cu2O reduction to Cu on CuO. The appearance of the sharp anodic stripping peak was consistent with the dissolution of the Cu layer that previously formed on CuO, represented by II. Small cathodic and anodic peaks were also observed in samples CL1, CL10 and CL66 that were attributed to the reduction or oxidation of copper species taking place on inhomogeneous sites, potentially arising from inconsistencies in the film deposition. Similarly, splitting of the anodic peak for sample CL66 was observed, where the secondary peak showed some asymmetric behaviour and was likely a contribution from the oxidation of Cu2O to CuO, as represented by peak III in Fig. 4d. CuO inks with different Nafion fractions were examined by constant potential electrolysis to determine the correlation between Nafion fraction, Faradaic efficiency and overpotential of CO2 reduction to formic acid. Glassy carbon electrodes prepared with CL1, CL10, CL25 and CL66 inks were held at various potentials (range of −0.6 to −1.4 V) and the effect on the Faradaic efficiency was studied (Fig. 5). The catalyst layer with 1 wt% Nafion fraction showed high Faradaic efficiency for applied potentials of −1.0 and −1.2 V. At −1.4 V, some delamination of the CuO/Cu film from the glassy carbon electrode was also observed and was attributed to excessive formation of hydrogen bubbles from water splitting. As the film was damaged during electrolysis, no product was observed via NMR or GC. The current density increased steadily with increasing applied potential, with a significant increase at −1.4 V (−16 mA cm−2) as a result of substantial water splitting.
 | ||
| Fig. 5 Faradaic efficiency of formic acid where the Nafion content is varied from 1 to 66 wt% in the catalyst layer, CL. | ||
Trace levels of formic acid were observed at −0.6 and −0.8 V, as the overpotential was not sufficient to drive the reduction at a rate, where measurable products were obtained. As the Nafion fraction was increased to 10 wt%, with increasing negative potential applied across the electrode, the Faradaic efficiency for formic acid production reached a maximum of 11% at −1.2 V. Increasing the potential to −1.4 V resulted in a sudden drop to a Faradaic efficiency of 1.8% as CO2 reduction competed further with water splitting. With increasing Nafion fraction in the catalyst layer to 25 wt%, at −0.6 V and −0.8 V, formic acid production appeared suppressed, with Faradaic efficiencies tending towards 0.6 and 1.8%, respectively. A significant increase in the Faradaic efficiency was observed at −1.4 V (61%), which to the author's knowledge is one of the highest Faradaic efficiency achieved on copper based catalysts for the formation of formic acid. The rate of formation of formic acid was also found to be very high, at 4.2 mmol g−1 h−1. Recently reported comparable results, using electrodeposited copper nanofoams have shown formic acid production at a Faradaic efficiency of 37% at −1.5 V vs. Ag/AgCl.7 In another report, copper oxide/copper electrocatalysts synthesised via batch hydrothermal methods showed high Faradaic efficiency for formic acid production of 59.3% at −0.9 V vs. Ag/AgCl and reached 62.2% at −1.3 V vs. Ag/AgCl after 20 hours of electrolysis time.37 High Faradaic efficiencies in this case were attributed to the distinct morphology of the electrocatalysts, however, the batch synthesis of nanostructured catalysts is inherently difficult to scale up reproducibly. Furthermore, there was no report of the rate of formation, so a direct comparison cannot be made regarding the kinetics of HCOOH formation.
Herein, a significant drop of Faradaic efficiency was observed as the Nafion fraction was increased to 66 wt%. No products were observed until −1 V was applied, suggesting a larger overpotential compared to lower Nafion fractions, was required to form any products. A shift to more negative overpotentials with increasing Nafion content, was also observed. Similarly, the current density decreased with increasing Nafion fraction in the catalyst layer. Thus, the best performing fraction of 25 wt% Nafion gave the highest Faradaic efficiency and good stability to the catalyst layer. The long term stability of sample CL25 at −1.4 V was examined over a 24 hour timescale. The chronoamperometric curve (ESI†) indicated excellent stability with minimal current loss over the time period tested, with an average current of −3.0 ± 0.5 mA, and a Faradaic efficiency of 53% for the production of formate.
Selected catalyst layers with different Nafion fraction held at −1.0 V for 3 hours, were examined by SEM post electrolysis (Fig. 6) and showed some significant changes to the morphology and size of the particles before electrolysis (see ESI†) also confirmed further by surface analysis via XPS (see ESI†). The initial CuO phase had undergone electrochemical reduction to Cu during the electrolysis process (Fig. 6a–d). From the structural characterisation and cyclic voltammetry, the active catalyst is inferred to be the copper phase, as the initial CuO phase is reduced in situ to Cu. The Cu particles remained nano-sized with a narrow size distribution. Some growth in size was observed from CuO electroreduction to Cu (from sub 20 nm to ca. 50 nm). The catalyst layer with a Nafion fraction of 66 wt%, showed dendritic growth of Cu during the electrolysis process, which was not observed with the other samples. The growth of this type of morphology may be facilitated by negatively charged sulfonic acid groups present in Nafion, which would inhibit the mass transport of Cu nuclei, allowing the growth of dendritic branched structures.38 With the presence of excess Nafion in the catalyst layer, dendritic structures may be more favourably formed in this instance, rather than at lower Nafion fraction. Despite a dendritic structure, the Faradaic efficiency was significantly lower compared to sample CL25 due to the presence of excess Nafion in the catalyst layer. Cross sectional SEM of the as prepared CuO ink with a fraction of 66 wt% Nafion was conducted to investigate the distribution of fluorine originating from the perfluorocarbon backbone in Nafion and copper (Fig. 7). A homogeneous distribution of fluorine, copper and oxygen was observed within the film layer. The high C count derived from the film was due to the film being supported on glassy carbon electrodes. This is in agreement with a film deposition that resulted in consistent distribution of Nafion within the catalyst layer without the formation of Nafion layers blocking access to the copper surface or contact of the catalyst with the electrode surface, even at significantly high fractions of Nafion.
 | ||
| Fig. 6 Post electrolysis SEM images of catalyst layer on glassy carbon electrode with (a) 1 wt%, (b) 10 wt%, (c) 25 wt% and (d) 66 wt% Nafion fraction. | ||
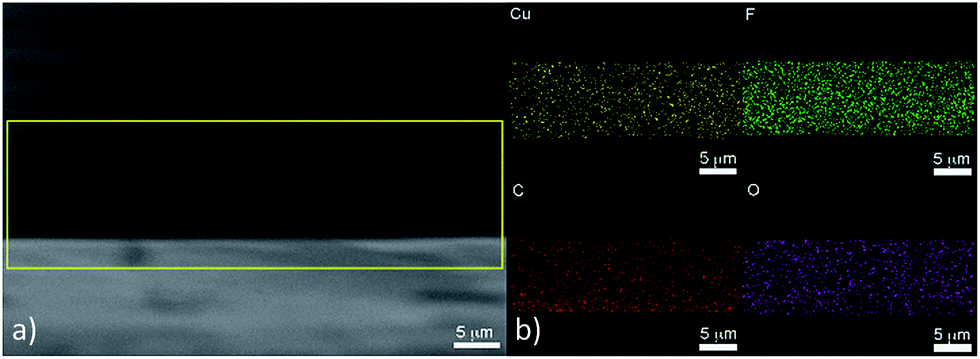 | ||
| Fig. 7 (a) Cross section of CuO film with 66 wt% Nafion fraction showing (b) elemental mapping of Cu, F, C and O. | ||
The surface roughness factor was calculated for samples with 1, 10, 25 and 66 wt% Nafion fraction, using capacitance where only the non-Faradaic region was explored. The CuO inks were reduced at −1.0 V in CO2 saturated 0.5 M KHCO3 for 1 hour before conducting cyclic voltammetry in the non-Faradaic region in 0.1 M H2SO4. The capacitance was calculated from a linear plot of current vs. scan rate (see ESI† for plot). The capacitance and surface roughness factor (Table 1) indicated the surface roughness factor increased with increasing Nafion content up to 10 wt% Nafion fraction, however, a sharp decrease was observed at 25 wt% Nafion content. The bare glassy carbon had a low capacitance value, indicative of low surface area as expected for a glassy carbon surface, and the incorporation of the catalyst film increased the surface roughness factor.
| Nafion fraction (wt%) | Capacitance (μF cm−2) | Surface roughness factor |
|---|---|---|
| Glassy carbon | 0.03 | 1 |
| 1 | 1.14 | 37 |
| 10 | 1.37 | 44 |
| 25 | 0.70 | 23 |
| 66 | 0.39 | 13 |
A decrease in the surface area was observed for catalysts with >10 wt% Nafion fraction, however, the Faradaic efficiency for formic acid production was still particularly high for sample CL25 at 61% Faradaic efficiency at −1.4 V, which suggested the high activity at CL25 was due to other factors and not entirely due to the high surface area of nano-sized Cu. Some insightsinto the mechanism of formic acid production on copper have been highlighted in theoretical calculations by Nørskov et al., which suggest that a formate pathway (F-intermediated pathway, eqn (1)) on copper surfaces, leads to exclusive formation of formic acid whereas a carboxyl pathway (C-intermediate), leads to the production of formic acid and some other higher order hydrocarbons.39,40
| CO2 + H+(aq) + e− + * → HCOO* (F intermediate) | (1) |
| HCOO* + H+(aq) + e− → HCOOH* | (2) |
| HCOOH* → HCOOH + * | (3) |
Headspace analysis via gas chromatography (GC), did not reveal the presence of hydrocarbons such as methane, ethane, or CO. Formic acid was the major product observed in 1H NMR experiments with trace levels of methanol and acetic acid. The dominance of (111) and (100) surfaces on Cu are likely to be linked to the production of formic acid via the formate pathway, where the presence of (200) surfaces leads to the production of formic acid via the carboxyl intermediate. Post electrolysis analysis of the copper particles via TEM (see ESI†) showed the presence of (111) copper surfaces (d111 spacing = 0.21 nm), which is in agreement with 0.21 nm for d111 spacing in literature, which may explain the high selectivity for formic acid over other products.41
The particularly high Faradaic efficiency observed herein was also attributed to the 3-dimensional network that was formed when using optimum Nafion, under certain conditions between the Nafion, catalyst and reactants. Electrochemical impedance spectroscopy (EIS) was conducted to further elucidate the effect of Nafion content within the catalyst layer. The CuO catalyst layers with different Nafion fractions were subjected to an AC perturbation of 5 mV over a frequency range from 10 kHz to 0.1 Hz at a DC voltage of −1.4 V. Nyquist plots of CuO catalyst layers with different Nafion fractions were compared (Fig. 8) and the equivalent circuit was modelled (see ESI†) to obtain simulated values of charge transfer resistance, Rct and mass transport resistance, Rmt. The CuO catalyst layer with 1 wt% was not considered due to mechanical degradation of the film at −1.4 V, which would have resulted in poor impedance data. A significant Rct difference was observed between the CuO catalyst layer with 25 wt% (14 Ω) and 66 wt% (63 Ω) Nafion, whilst the CL10 sample had a Rct of 17 Ω. The Rmt was also significantly higher for the 66 wt% Nafion fraction (10 Ω) compared to the catalyst layers with lower Nafion content. The Nafion content is a crucial factor when considering the performance of the catalyst and has previously been explained through the percolation threshold.
 | ||
Fig. 8 Nyquist plot comparing different Nafion content ( 10 wt%, 10 wt%,  25 wt% and 25 wt% and  66 wt%) at a DC voltage of −1.4 V. 66 wt%) at a DC voltage of −1.4 V. | ||
Suzuki et al. modelled a three-dimensional (3D) meso-scale structure of the catalyst layer in the context of a proton exchange membrane fuel cell (PEMFC), where three different percolating pathways were modelled.42 A network of catalyst grain contacts is necessary for electronic conduction, the Nafion network that forms around the particles allows ionic conduction and the resultant channels formed in the catalyst layer allow for gas diffusion. This is significant as the channels allow the transport of CO2 and products (formate). The Nafion can promote H+ conduction and the copper oxide derived catalyst can provide good electronic conduction. At a low Nafion fraction, although the particles form a percolating pathway for electronic conduction there is likely to be poor catalyst utilisation, which is also observed in the increased charge transfer resistance for CL10 (Fig. 8) as there was not enough Nafion to allow good ionic, including proton conduction. Subsequently, when the Nafion fraction was too high, the Nafion can form aggregates or micelle structures, which are more thermodynamically stable at certain ionomer content.43 This resulted in particle separation at large distances where good electronic conduction was likely to have been inhibited. The presence of excess Nafion also impeded gas transport and increased mass transport resistance within the catalyst layer leading to a reduced performance. At the optimal Nafion fraction of 25 wt%, the three percolating pathways resulted in good catalyst utilisation for electronic conduction, sufficient Nafion for proton conduction and gas channels for gas transport without adverse mass transport issues. As CO2 reduction is sensitive to the competing water splitting reaction, the presence of Nafion was essential in proton transportation. Protons are also crucial for the synthesis of formic acid (eqn (1)–(3)). At −1.4 V, in the case of CL25, significant water splitting could be advantageous as the significant fraction of Nafion assisted proton conduction through the ionomer, providing protons without impeding the flow of electrons to form formic acid at high Faradaic efficiencies.
Conclusions
In summary, ultrafine CuO nanoparticles were successfully synthesised in a large scale (kg per day) continuous hydrothermal flow synthesis reactor. The resulting 17 nm nanoparticles were subsequently formulated in to homogenous Nafion based inks that were employed to prepare stable catalyst layers on glassy carbon electrodes. The ink composition was optimised with varying Nafion content to maximise the performance of the electrocatalytic reduction of CO2 to formic acid on copper surfaces derived from the CuO nanoparticles. The highest measured Faradaic efficiency was >60% which was significantly higher than the majority of comparable reports for copper catalysts in the literature and makes the process potentially attractive to larger PEMFC-like designs for CO2 reduction, especially considering the scale up capabilities, green nature and ease of formulation of the proposed method. The high Faradaic efficiency for formic acid was explained by the presence of Cu (111) planes, observed by TEM, which favoured the formation of formic acid on copper via the formate pathway. Further evidence of the importance of controlling the Nafion fraction on the electrocatalytic reduction of CO2 to formic acid was indicated by electrochemical impedance spectroscopy where the charge transfer and mass transport resistance were found to be minimal for the optimised Nafion fraction, highlighting the correlation between the catalyst ink formulation and the consequent layer structure on the electrode electrocatalytic performance.Acknowledgements
EPSRC financially supported this study as part of ‘Bio-inspired sulfide nanocatalysts: from proof of concept to ‘real’ catalysis’ grant EP/K035355/1.Notes and references
- R. Cicerone and P. Nurse, Climate Change Evidence and Causes, National Academy of Sciences, Washington D.C., 2014 Search PubMed.
- Earth System Research Laboratory – NOAA, U.S. Dep. Commer. |Natl. Ocean. Atmos. Adm.| NOAA Res., 2016.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- J. P. Jones, G. K. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef CAS.
- G.-Q. Yuan, H.-F. Jiang, C. Lin and S.-J. Liao, J. Cryst. Growth, 2007, 303, 400–406 CrossRef CAS.
- M. P. Neupane, Y. K. Kim, S. Park, K. A. Kim, M. H. Lee and T. S. Bae, Surf. Interface Anal., 2009, 41, 259–263 CrossRef CAS.
- M. Outokesh, M. Hosseinpour, S. J. Ahmadi, T. Mousavand, S. Sadjadi and W. Soltanian, Ind. Eng. Chem. Res., 2011, 50, 3540–3554 CrossRef CAS.
- J. Zhu, D. Li, H. Chen, X. Yang, L. Lu and X. Wang, Mater. Lett., 2004, 58, 3324–3327 CrossRef CAS.
- A. V. Nikam, A. Arulkashmir, K. Krishnamoorthy, A. A. Kulkarni and B. L. V. Prasad, Cryst. Growth Des., 2014, 14, 4329–4334 CAS.
- C. C. Vidyasagar, T. G. Venkatesha and R. Viswanatha, Nano-Micro Lett., 2012, 73–77 CrossRef CAS.
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117–166 CrossRef CAS.
- C. J. Tighe, R. I. Gruar, C. Y. Ma, T. Mahmud, X. Z. Wang and J. A. Darr, J. Supercrit. Fluids, 2012, 62, 165–172 CrossRef CAS.
- R. I. Gruar, C. J. Tighe and J. A. Darr, Ind. Eng. Chem. Res., 2013, 52, 5270–5281 CrossRef CAS.
- C. J. Tighe, R. Q. Cabrera, R. I. Gruar and J. A. Darr, Ind. Eng. Chem. Res., 2013, 52, 5522–5528 CrossRef CAS.
- J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495–541 CrossRef CAS PubMed.
- J. A. Darr, C. J. Tighe and R. Gruar, US Pat., 9192901, 2015.
- S. Elouali, L. G. Bloor, R. Binions, I. P. Parkin, C. J. Carmalt and J. A. Darr, Langmuir, 2012, 28, 1879–1885 CrossRef CAS PubMed.
- X. Weng, J. K. Cockcroft, G. Hyett, M. Vickers, P. Boldrin, C. C. Tang, S. P. Thompson, J. E. Parker, J. C. Knowles, I. Rehman, I. Parkin, J. R. G. Evans and J. A. Darr, J. Comb. Chem., 2009, 11, 829–834 CrossRef CAS PubMed.
- P. Boldrin, A. K. Hebb, A. A. Chaudhry, L. Otley, B. Thiebaut, P. Bishop and J. A. Darr, Ind. Eng. Chem. Res., 2007, 46, 4830–4838 CrossRef CAS.
- J. Wu, F. G. Risalvato, P. P. Sharma, P. J. Pellechia, F.-S. Ke and X.-D. Zhou, J. Electrochem. Soc., 2013, 160, 953–957 CrossRef.
- T. T. Ngo, T. L. Yu and H. L. Lin, J. Power Sources, 2013, 225, 293–303 CrossRef CAS.
- Q. Wang, H. Dong and H. Yu, RSC Adv., 2014, 4, 59970–59976 RSC.
- Q. Wang, H. Dong, H. Yu and H. Yu, J. Power Sources, 2015, 279, 1–5 CrossRef CAS.
- J. Wu, P. P. Sharma, B. H. Harris and X. Zhou, J. Power Sources, 2014, 258, 189–194 CrossRef CAS.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- C. J. Denis, C. J. Tighe, R. I. Gruar, N. M. Makwana and J. A. Darr, Cryst. Growth Des., 2015, 15, 4256–4265 CAS.
- B. Cullity, Elements of X-Ray Diffraction, Pearson, 3rd edn, 2001 Search PubMed.
- A. S. Ethiraj and D. J. Kang, Nanoscale Res. Lett., 2012, 7, 70 CrossRef PubMed.
- T. Y. Chang, R. M. Liang, P. W. Wu, J. Y. Chen and Y. C. Hsieh, Mater. Lett., 2009, 63, 1001–1003 CrossRef CAS.
- T. Adschiri, K. Kanazawa and K. Arai, J. Am. Ceram. Soc., 1992, 75, 1019–1022 CrossRef CAS.
- K. Byrappa, S. Ohara and T. Adschiri, Adv. Drug Delivery Rev., 2008, 60, 299–327 CrossRef CAS PubMed.
- Y. Lan, C. Gai, P. J. A. Kenis and J. Lu, ChemElectroChem, 2014, 1, 1577–1582 CrossRef CAS.
- J. Qiao, M. Fan, Y. Fu, Z. Bai, C. Ma, Y. Liu and X.-D. Zhou, Electrochim. Acta, 2015, 153, 559–565 CrossRef CAS.
- W. Yi, J. Liu, H. Chen, Y. Gao and H. Li, J. Solid State Electrochem., 2015, 1511–1521 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- X. Su, J. Zhao, H. Bala, Y. Zhu, Y. Gao, S. Ma and Z. Wang, J. Phys. Chem. C, 2007, 111, 14689–14693 CAS.
- A. Suzuki, U. Sen, T. Hattori, R. Miura, R. Nagumo, H. Tsuboi, N. Hatakeyama, A. Endou and H. Takaba, Int. J. Hydrogen Energy, 2010, 1–9 Search PubMed.
- S. M. Andersen and L. Grahl-Madsen, Int. J. Hydrogen Energy, 2016, 41, 1892–1901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for confined jet mixer, morphology of CuO catalyst layers, analytical characterisation of electrolysis products and impedance spectroscopy model included. See DOI: 10.1039/c6ta04874a |
| This journal is © The Royal Society of Chemistry 2016 |