
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A photoluminescent covalent triazine framework: CO2 adsorption, light-driven hydrogen evolution and sensing of nitroaromatics†
Asamanjoy
Bhunia
*ab,
Dolores
Esquivel
ac,
Subarna
Dey
b,
Ricardo
Fernández-Terán
 d,
Yasutomo
Goto
e,
Shinji
Inagaki
e,
Pascal
Van Der Voort
a and
Christoph
Janiak
*b
d,
Yasutomo
Goto
e,
Shinji
Inagaki
e,
Pascal
Van Der Voort
a and
Christoph
Janiak
*b
aDepartment of Inorganic and Physical Chemistry, Centre for Ordered Materials, Organometallics and Catalysis, Ghent University, Krijgslaan 281-S3, 9000 Ghent, Belgium. E-mail: asamanjoy.bhunia@gmail.com
bInstitut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
cDepartment of Organic Chemistry, Nanochemistry and Fine Chemistry Research Institute (IUIQFN), Faculty of Sciences, University of Córdoba, Campus of Rabanales, Marie Curie Building, Ctra. Nal. IV, km 396, 14071 Córdoba, Spain
dDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, SE-751 20, Uppsala, Sweden
eToyota Central R&D Laboratories, Inc., Nagakute, Aichi 480-1192, Japan
First published on 2nd August 2016
Abstract
A highly photoluminescent (PL) porous covalent triazine-based framework (PCTF-8) is synthesized from tetra(4-cyanophenyl)ethylene by using trifluoromethanesulfonic acid as the catalyst at room temperature. Due to triazine units in the framework, the PCTF-8 exhibits excellent thermal stability (>400 °C). The Brunauer–Emmett–Teller (BET) specific surface area of PCTF-8 is 625 m2 g−1 which is lower than the one obtained from the synthesis under Lewis acid conditions (ZnCl2). At 1 bar and 273 K, the PCTF-8 adsorbs a significant amount of CO2 (56 cm3 g−1) and CH4 (17 cm3 g−1) which is highly comparable to nanoporous 1,3,5-triazine frameworks (NOP-1-6, 29–56 cm3 g−1). This nitrogen rich framework exhibits good ideal selectivity (61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (85% N2:15% CO2) at 273 K, 1 bar). Thus, it can be used as a promising candidate for potential applications in post-combustion CO2 capture and sequestration technologies. In addition, photoluminescence properties as well as the sensing behaviour towards nitroaromatics have been demonstrated. The fluorescence emission intensity of PCTF-8 is quenched by ca. 71% in the presence of 2,4,6-trinitrophenol (TNP). From time-resolved studies, a static quenching behaviour was found. This high photoluminescence property is used for hydrogen evolving organic photocatalysis from water in the presence of a sacrificial electron donor and a cocatalyst.
:1 (85% N2:15% CO2) at 273 K, 1 bar). Thus, it can be used as a promising candidate for potential applications in post-combustion CO2 capture and sequestration technologies. In addition, photoluminescence properties as well as the sensing behaviour towards nitroaromatics have been demonstrated. The fluorescence emission intensity of PCTF-8 is quenched by ca. 71% in the presence of 2,4,6-trinitrophenol (TNP). From time-resolved studies, a static quenching behaviour was found. This high photoluminescence property is used for hydrogen evolving organic photocatalysis from water in the presence of a sacrificial electron donor and a cocatalyst.
Introduction
Porous organic materials have attracted considerable interest because of their unique physicochemical properties in various technological and energy-related applications such as gas storage, separation, catalysis, etc.1,2 In particular, π-conjugated porous organic polymers are an integral part of research for light emission, light harvesting and chemo-sensing.3 Due to their rigidity at the molecular level, they have a high affinity to aggregate in solution and the solid state, resulting typically in aggregation caused quenching (ACQ) effects.4 The aggregation of conjugated polymers can be prevented by molecular approaches based on site isolation with bulky polymeric matrices.5 This approach can improve the luminescence properties of the polymeric materials. At the same time, π-conjugated materials are also a promising platform for CO2 capture because of the interaction between the quadrupole moments of CO2 and the π-clouds in the porous organic materials.6 The incorporation of high amounts of nitrogen into the porous materials can facilitate CO2 capture. Such multifunctionality has been a long term challenge for the development of porous photofunctional materials which have attracted great interest as chemical sensors, e.g. to detect explosive nitroaromatic compounds.7 The sensitivity and selectivity in the detection of these compounds by using porous materials suffer from analyte–host interactions, chemical stability and so on. π-Conjugated triazine based porous materials may be attractive chemical sensors because of their high stability and high porosity. Due to the high electron mobilities and electron withdrawing character of the triazine ring, these materials have been widely used in synthetic chemistry and optoelectronics.8Currently, these types of materials have attracted great interest as organic photocatalysts for hydrogen production via photocatalytic water splitting under visible light.9 Their chemical structures, analogous to that of g-C3N4, in combination with their tunable porosity make them attractive candidates for light-induced hydrogen evolution.10 Lostch et al. reported the first example of a covalent organic framework (COF) which was active in visible light-induced hydrogen production from water using platinum as a cocatalyst.9a More recently, a yellow coloured phenyl-triazine oligomer with high crystallinity exhibited the highest activity obtained for metal-free organic photocatalysts in the absence of noble metal catalysts.9b
Herein we report a new strategy for the construction of photoluminescent materials based on π-conjugated building blocks. We employed tetra(4-cyanophenyl)ethylene as a single component for the synthesis of a porous covalent triazine-based framework (PCTF) using trifluoromethanesulfonic acid (TFMS) as the catalyst at room temperature, in which the tetraphenylethylene (TPE) units are directly connected to strong electron accepting triazine rings. Recently, some of us reported PCTFs with the TPE unit by using the ionothermal ZnCl2 route.11 The structures of CTFs from ionothermal reactions with ZnCl2 are in-between well-defined COFs and porous carbon materials.12 TFMS-catalyzed condensation frameworks13,14 offer significant advantages: (1) lower temperature and shorter reaction time, (2) avoiding undesired decomposition and condensation reactions such as C–H bond cleavage and carbonization which otherwise lead to defects in the framework and (3) absence of ZnCl2 contamination in the porous materials. These advantages are attractive to obtain reasonably well-defined CTFs which can exhibit a broad variety of physical properties including chemical sensing, gas storage/separation and organic photocatalysis. So far, CTFs have been used mainly in gas storage and separation.11,13 There are also few reports on the use of CTFs for heterogeneous catalysis including light-induced hydrogen evolution,9,15 as catalytic supports in liquid phase reactions,16 and as adsorbents in liquids,17 due to their chemical and thermal stabilities. In spite of their many applications, their photoluminescence was only briefly noted14 to the best of our knowledge. Thus, we show in this paper that PCTF-8 can not only be used for gas adsorption/separation but also exhibits high selectivity for the detection of 2,4,6-trinitrophenol (TNP) over other nitroaromatic analytes such as 4-nitrotoluene (NT), nitrobenzene (NB), 2,6-dinitrotoluene (2,6-DNT), 2,4-dinitrotoluene (2,4-DNT), 2,4-dinitrophenol (DNP) together with light-induced hydrogen evolution.
Experimental section
Materials
All chemicals were purchased from commercial suppliers (Sigma-Aldrich, Acros Organics, and Alfa Aesar chemical company) and used without further purification, unless stated otherwise. Tetra(4-cyanophenyl)ethylene was synthesized according to the reported procedures.11 Chloroform was distilled from P2O5 under a nitrogen atmosphere.Instrumentation
Infrared (IR) spectra were obtained on a Bruker FT-IR Tensor 37 Spectrometer in the 4000–550 cm−1 region with 2 cm−1 resolution as KBr disks. UV-vis absorption and fluorescence emission spectra were obtained using Jasco V-670 and FP-6500 spectrometers, respectively. Fluorescence quantum yields were evaluated using an absolute photoluminescence quantum yield measurement system equipped with a calibrated integrating sphere and a multi-channel spectrometer (C9920-02, Hamamatsu Photonics). The excitation wavelength was 395 nm. 1H and 13C spectra were recorded on a Bruker Avance DRX-500 instrument. 1H and 13C NMR chemical shifts are given in ppm relative to SiMe4 (δ = 0.0 ppm) with calibration against the residual protonated solvent signal (CDCl3: 7.26 (1H) and 77.0 (13C)). 13C cross-polarization (CP) MAS NMR measurements were performed at 100.6 MHz at a sample spinning frequency of 5 or 6 kHz using a Bruker Avance 400 spectrometer with a 7 mm zirconia rotor. For the 13C CP-MAS NMR measurements, the repetition delay was 5 s, the contact time was 1.75 ms, and the pulse width was 4.5 μs (1H 90° pulse). Elemental (CNH) analyses were carried out with a PerkinElmer 2400 series 2 elemental analyser. Powder X-ray diffraction (PXRD) data were collected on a Bruker D2 Phaser diffractometer using a flat sample holder (also a flat silicon, low background sample holder) and Cu Kα1/α2 radiation with λ = 1.5418 Å at 30 kV covering 2theta angles 5–80° over a time period of 2 h, that is 0.01° s−1. Diffractograms were obtained on flat layer sample holders where at low angles, the beam spot is strongly broadened so that only a fraction of the reflected radiation reaches the detector which leads to low relative intensities measured at 2θ < 7°. For hygroscopic or air-sensitive samples, the sample holder can be sealed with a dome. Scanning electron microscopy (SEM) images were obtained using an ESEM Quanta 400 FEG SEM equipped with a secondary electron detector. Thermogravimetric analyses (TGA) were carried out at a ramp rate of 5 °C min−1 under air with a Netzsch Thermo-Microbalance Apparatus TG 209 F3 Tarsus. A coulometric Karl–Fischer titration (KFT) for the determination of the water content was carried out with a Karl–Fischer titration apparatus AQUA 40.00 with a headspace module. The solid CTF sample was heated to 170 °C in the head space module and the liberated water was transferred to the measurement cell.Sorption isotherms were measured using a Micromeritics ASAP 2020 automatic gas sorption analyser equipped with oil-free vacuum pumps (ultimate vacuum <10−8 mbar) and valves, which guaranteed contamination free measurements. The sample was connected to the preparation port of the sorption analyser and degassed under vacuum until the outgassing rate, i.e. the rate of pressure rise in the temporarily closed manifold with the connected sample tube, was less than 2 μTorr min−1 at the specified temperature of 200 °C. After weighing, the sample tube was then transferred to the analysis port of the sorption analyser. All used gases (H2, He, N2, CO2, and CH4) were of ultra-high purity (UHP, grade 5.0, 99.999%) and the STP volumes are given according to the NIST standards (293.15 K, 101.325 kPa). Helium gas was used for the determination of the cold and warm free spaces of the sample tubes. H2 and N2 sorption isotherms were measured at 77 K (liquid nitrogen bath), whereas CO2 and CH4 sorption isotherms were measured at 293 ± 1 K (passive thermostating) and 273.15 K (ice/deionized water bath). The heat of adsorption values and the DFT pore size distributions (‘N2 DFT slit pore’ model) were calculated using the ASAP 2020 v3.05 software.
Photocatalytic H2 production experiments were performed with a Solar Box 3000 equipped with a Xe lamp (300 W). For each experiment, the polymer powder (10 mg) was suspended in a phosphate buffer solution (pH-7, 9 mL) containing triethanolamine (TEoA) or MeOH as a sacrificial donor (1 mL). Dihydrogen hexachloroplatinate (5 μL, 8 wt% aqueous solution) was added as the precursor for the in situ formation of a platinum cocatalyst. The solution was ultrasonicated for 15 min before being degassed by nitrogen bubbling for 30 min. The flask was illuminated at a 90° angle at a distance of 22 cm with a 300 W Xe lamp for the time specified. Gas samples were taken with a gas-tight syringe and quantified by using a gas chromatograph (Shimadzu GC-2010 Plus) equipped with a barrier discharge ionization detector (BID).
Time-correlated single photon counting (TCSPC) measurements were performed using a pulsed diode laser source (Edinburgh Instruments EPL405) operating at 404.6 nm with a pulse FWHM of ca. 77.1 ps. A neutral density filter was used to attenuate the beam to obtain photon counts of approx. 1% or less of the incoming light intensity. The detector used was a Hamamatsu MCP-photomultiplier tube R3809U-51 (cooled to ca. −40 °C), the signal was passed to a discriminator (Ortec 9307) and then into a TAC (Ortec 566, 50 ns time range used). The electrical trigger signal from the laser was also passed through a discriminator (Tennelec TC454) and onto the TAC (Ortec 566). The TAC output was read by a DAQ-1 MCA computer card using 4096 channels and collected with Horiba Jobin Yvon DataStation 2.3 software. All measurements were done in the reverse mode at 10 MHz and under magic angle polarisation. A cut-off filter (GG3) was used to block scattered excitation light. The instrument response function (IRF) was collected by using a dilute solution of Ludox as a standard scattering sample. Fluorescence lifetimes were obtained by iterative reconvolution of the IRF and the collected decay curves, by fitting to a multiexponential decay model using in-house scripts.
Synthesis
A 50 mL Schlenk flask was charged with trifluoromethanesulfonic acid (0.45 g, 3.0 mmol) in 8 mL dry CHCl3 under a N2 atmosphere. The reaction mixture was stirred and cooled to 0 °C. Tetra(4-cyanophenyl)ethylene (217 mg, 0.5 mmol) in 30 mL dry CHCl3 was added dropwise into the acid solution with stirring over 30 min and the temperature of 0 °C was maintained for another 2 h. Then, the resulting solution was stirred for 12 h at room temperature. An NH3/H2O solution (0.5 mol L−1) was added until the reaction mixture became neutral and the mixture was stirred for another 2 h. The yellow product was isolated by filtration and washed with water (3 × 50 mL), THF (3 × 30 mL), acetone (3 × 30 mL), chloroform (3 × 30 mL) and further purified by Soxhlet extraction for 24 h with acetone. The final product was dried in a vacuum. The yield was 180 mg, 83% for (C30H16N4)n.Results and discussion
Synthesis and spectroscopic characterization
PCTF-8 was synthesized by the TFMS catalyzed reaction from tetra(4-cyanophenyl)ethylene (1) at room temperature as a yellow powder in 83% yield (Scheme 1). In FT-IR, the carbonitrile band in the precursors around 2226 cm−1 has significantly decreased in intensity which indicates only a few free nitrile groups in PCTF-8 (Fig. 1, top). The intense bands around 1503, 1362 and 800 cm−1 correspond to the formed triazine rings.12 In the solid-state 13C CP/MAS NMR spectrum (Fig. 1, bottom), the major resonances at 174, 141 and 134 ppm can be assigned to the triazine ring (–CAr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), ethylene carbon (–CC–) and aromatic carbon (Ar) atoms, respectively.12 The peaks around 124 and 114 ppm were associated with residual cyano end groups.14 Powder X-ray diffraction of PCTF-8 confirmed its expected amorphous nature (Fig. S1, ESI†).
N–), ethylene carbon (–CC–) and aromatic carbon (Ar) atoms, respectively.12 The peaks around 124 and 114 ppm were associated with residual cyano end groups.14 Powder X-ray diffraction of PCTF-8 confirmed its expected amorphous nature (Fig. S1, ESI†).
 | ||
| Scheme 1 Synthesis of the nitrile linker and PCTF-8. | ||
 | ||
| Fig. 1 IR spectra of the monomer and PCTF-8 (top). Solid-state 13C CP/MAS NMR spectrum of PCTF-8 (bottom). Signals e and f are associated with residual cyano end groups and * belongs to sidebands. | ||
Thermogravimetric analysis (TGA) showed that PCTF-8 exhibited good thermal stability up to 420 °C under air (Fig. S2, ESI†). Elemental analysis (Table S1, ESI†) of PCTF-8 nearly matches the calculated values corrected with the results of KFT analysis which determined the water content of the sample (Fig. S3, ESI†). It became apparent that the PCTF-8 is hygroscopic and adsorbs about 10 wt% of moisture from ambient air upon storage under ambient conditions after vacuum drying at 120 °C. A similar hygroscopic behaviour of related CTFs was reported recently by Janiak et al.18 and may be a common phenomenon to CTFs which may also explain the frequently observed mismatch in theoretical and experimental elemental (CHN) analysis. Under SEM, the PCTF-8 particles are small and largely irregular as well as angular shaped with a wide range of sizes (greater than 1 μm) (Fig. 2).
 | ||
| Fig. 2 SEM images of PCTF-8. | ||
Gas sorption
The N2 sorption isotherm shows a steep slope at low P/P0 values and a type I character typical for microporous materials (Fig. 3 and Table S2, ESI†).19 The calculated BET surface area of 625 m2 g−1 is higher than for similar tetrakis(4-carboxyphenyl)ethylene linker-based fluorescent MOFs (244–317 m2 g−1).20 The surface area of PCTF-8 is in the range of other TFMS-catalysed CTFs with 2 to 1152 m2 g−1.14 The ratio of micropore volume to total pore volume (V0.1/Vtot) of 0.78 suggests that the material contains a very high fraction of micropores. A narrow distribution of micropores centered at 5, 5.9, 6.8, 11.8 and 14.8 Å was observed (insert in Fig. 3). PCTF-8 showed a H2 sorption of 112 cm3 g−1 at 77 K (with no hysteresis) that is very close to those of other CTFs.13 The CO2 adsorption capacities in activated PCTF-8 are 56 cm3 g−1 at 273 K and 35 cm3 g−1 at 293 K, respectively (Fig. 4). The CO2 uptake of PCTF-8 was comparable to that of CTF-type polymers synthesized by TFMS, which showed CO2 uptakes of 20–93 cm3 g−1 at 273 K and 1 bar.14 Such uptake capacity is also comparable to that of nanoporous 1,3,5-triazine frameworks (NOP-1-6, 29–56 cm3 g−1).21 The CO2 uptake of PCTF-8 is comparable to that of PCTF-1 which has about three times the BET surface area of PCTF-8.11 This is due to the higher nitrogen content and intact triazine units of PCTF-8 from the milder synthesis conditions as CO2 interacts with the triazine nitrogen due to its large polarizability and quadrupole moment. | ||
| Fig. 3 N2 sorption isotherms at 77 K and a NL-DFT pore size distribution profile (inset) of PCTF-8. | ||
 | ||
| Fig. 4 H2, CO2, and CH4 isotherms at 1 bar of PCTF-8. | ||
The isosteric heat of adsorption from the CO2 adsorption isotherms at 273 and 293 K (Fig. S4, ESI†) at zero loading is 37 kJ mol−1 and rapidly drops at adsorbate loadings of only a few cm3 g−1. The heat of adsorption stays around 29 kJ mol−1 for loadings between 3 and ∼30 cm3 g−1. When the loading is increased beyond 30 cm3 g−1 the heat of adsorption value appears to increase slightly which should not be over-interpreted however in view of the error margin of readily ±3 kJ mol−1.13,22 This heat of CO2 adsorption of ∼29 kJ mol−1 for PCTF-8 stays well above the heat of liquefaction of bulk CO2 with 17 kJ mol−123 or around the isosteric enthalpy of adsorption for CO2 on activated charcoals (e.g. BPL: 25.7 kJ mol−1, A10: 21.6 kJ mol−1, and Norit R1 Extra: 22.0 kJ mol−1).24 The high Qst value can be attributed to the high polar framework and the pore size effect. The high adsorption enthalpy at zero coverage is explained by the initial filling of the small ultramicropores with 4 Å diameter (Fig. S5, ESI†) with adsorbate–surface interactions to both sides or ends of the CO2 molecules.13
If the heat of adsorption would indeed exhibit an increase with CO2 uptake, this means that a simultaneous, exothermic process must take place, such as possibly the rearrangement of already adsorbed CO2 molecules towards a closer, energetically, more favorable configuration.
CH4 sorption values of PCTF-8 were 17 cm3 g−1 at 273 K and 10 cm3 g−1 at 293 K (Fig. 4) which are comparable with the values for CTFs from our previous work.11,13 From the available single-gas adsorption isotherms, the CO2 selectivity over N2 of PCTF-8 was calculated at 273 K by using the Henry equation and the ideal adsorbed solution theory (IAST) in order to understand greater affinity toward CO2. The calculated adsorption selectivity of CO2/N2 is 24 and 61 at 273 K using the Henry equation and the ideal adsorbed solution theory (IAST), respectively (Fig. S6 and Section 8 in the ESI†), which are comparable with the values for PCTF-1 to −7 from our previous work as well as other CTFs.11,13,14,25,26 This comparably large value of PCTF-8 for CO2 may be attributed to the presence of more basic nitrogen moieties.
Photophysical properties
Absorption and photoluminescence spectra of PCTF-8 and its monomer (1) were measured at room temperature (Fig. 5). The monomer 1 dispersed in CH2Cl2 shows several absorption bands below 400 nm attributed to the π–π* transitions of the TPE chromophore.27,28 Upon excitation at 350 nm, the monomer emits a weak blue emission with λmax = 389 nm (Fig. 5a). This type of subunit is known to show an aggregation induced emission in the solid state or concentrated solutions. The weak emission is due to the non-restricted rotation of phenyl rings in dilute solutions.29,30 On the other hand, a solid-state absorption spectrum of PCTF-8 exhibits a broad absorption band centred at around 400 nm ranging from 300 to 600 nm, which can be attributed to the TPE units (Fig. 5b). These different absorption profiles are responsible for the different colours; the monomer shows a light yellowish colour owing to absorbing UV light but reflecting all visible light, while PCTF-8 is a bright yellow powder due to absorbing blue light (Fig. 5).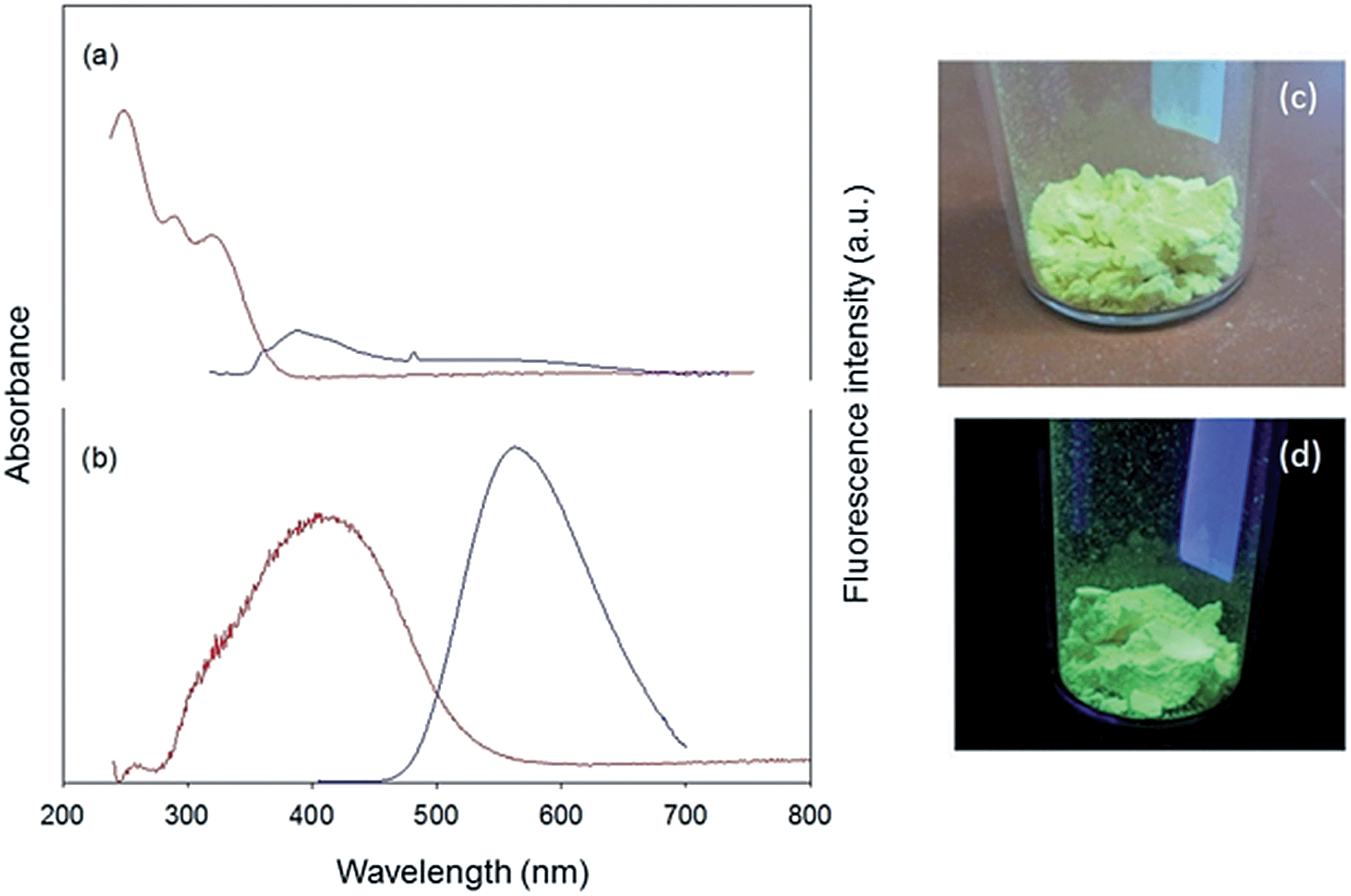 | ||
| Fig. 5 Absorption (red line) and fluorescence spectra (blue line) of (a) monomer 1 (in CH2Cl2, λexc 350 nm) and (b) the corresponding PCTF-8 solid (λexc 395 nm). Photos of PCTF-8 are shown under ambient light (c) and UV-light (d). | ||
To compare the emission behaviour of PCTF-8 synthesized under different conditions (Brønsted acid and ionothermal conditions), we have measured the emission spectrum of black coloured PCTF-1 that was synthesized under ionothermal conditions (Fig. S5, ESI†). PCTF-1 does not show appreciable emission because of partial carbonization in the framework. Therefore, highly emissive properties of CTFs seem only possible when avoiding carbonization by preparation at room temperature. In the solid-state, PCTF-8 shows a strong greenish-yellow fluorescence with an emission maximum at 562 nm when excited at 395 nm and a fluorescence quantum yield (Φf) of 31% at room temperature (Fig. 5b). This high value, compared to the extremely low quantum yield observed for the monomer in solution, is primarily attributed to the immobilization and restricted rotation of the aromatic rings in the tetraphenylethene units of the framework.30,31
From its ultraviolet-visible spectrum, the optical band gap of PCTF-8 was estimated to be 2.25 eV. In principle, this band gap is large enough to overcome the endothermic character of the water-splitting reaction. To investigate this property, photocatalytic hydrogen evolution experiments were performed with PTCF-8 and Pt-loaded PCTF-8 in the presence of a methanol or triethanolamine sacrificial electron donor (10 mol%) in buffered aqueous solutions. Without any platinum co-catalyst, no hydrogen evolution was observed. After loading with 2.3 wt% Pt, PCTF-8 showed H2 production from buffered (pH 7) aqueous solutions containing sacrificial electron donors under simulated sunlight (Fig. S8, ESI†). The total amount of H2 evolved of Pt-loaded PCTF-8 (10 mg) in a buffered aqueous methanol or TEoA solutions when irradiated for 20 h was 1780 and 2370 μmol g−1, respectively. These moderate values of hydrogen production could be due to the poor crystallinity of PCTF-8 which hinders the diffusion of light-induced charge carriers to the surface.9
To examine the sensing behaviour of PCTF-8, luminescence spectrometric quenching titration experiments were performed by gradual addition of nitroaromatic analytes (TNP, NT or NB, 2,4-DNP, 2,6-DNT, 2,4-DNT, cf.Fig. 6) to PCTF-8 dispersed in acetonitrile at room temperature (Fig. 6 and 7a, and Section 11 in the ESI†).
 | ||
| Fig. 6 Fluorescence quenching (%) of PCTF-8 with nitroaromatic analytes in acetonitrile dispersion. | ||
 | ||
| Fig. 7 (a) Fluorescence quenching of PCTF-8 by TNP (0 to 1 × 10−6 M) at room temperature (1 mg in 3 mL acetonitrile, λexc = 395 nm); (b) Stern–Volmer plots for the quenching of PCTF-8 by TNP in acetonitrile dispersion (λexc = 395 nm); (c) fluorescence decay profile of PCTF-8 (λexc = 405 nm) with different aliquots of TNP added, for a maximum quencher concentration of ca. 30 μM. | ||
Upon successive additions of nitroaromatic analytes, the fluorescence emission intensity was increasingly quenched, with TNP being the most effective quencher, for a maximum quenching of ca. 71% of the fluorescence emission intensity. The other analytes show similar quenching behaviour in the following order: TNP ≫ NT > NB > DNP > 2,6 DNT > 2,4 DNT (Fig. 6). Following the quenching experiment over time up to sixty minutes, it is evident that the quenching occurs very rapidly and does not increase over time (Fig. S16, ESI†).
To quantify the sensitivity towards the nitroaromatic analytes, the Stern–Volmer (SV) quenching constants (KSV) were calculated by using the standard linear curve-fitting of the fluorescence intensity (I0/I) in the presence of different concentrations of the analytes (1 × 10−5 to 5 × 10−5 mol L−1) (Fig. 7b, S17–S21 in ESI†). The obtained quenching constants (KSV) are in the order TNP ≫ DNP > NB > 2,6-DNT > 2,4-DNT > NT. The KSV of TNP was 1.32 × 105 L mol−1 (Fig. 7b), which is comparable with those of previously reported frameworks.32
To further understand the nature of the quenching process involved in “turn-off” fluorescence sensing by PCTF-8, we measured the emission decays by time-correlated single photon counting at different concentrations of TNP (added as aliquots of a stock solution in acetonitrile), as it has the highest quenching efficiency of the studied nitroaromatic compounds. From the decays shown in Fig. 7c, it can be observed that the lifetime is independent of the concentration of the quencher in the studied range, which strongly suggests that a static quenching mechanism takes place. The static quenching behaviour of PCTF-8 with TNP can be explained in terms of the formation of a ground-state complex due to a strong interaction with the analyte (see the ESI† for discussion).33 The lifetime of PCTF-8 was fitted to three exponential decay components (χ2 = 1.324, Fig. S22 in the ESI†): 0.5 ns (49%), 2.1 ns (42%) and 5 ns (1%). This decay behaviour can be rationalised in terms of the size distribution of the polymer, and due to the fact that emission comes from a heterogeneous suspension, where different aggregation behaviours can be observed.
Conclusions
In conclusion, we have used TPE units in the preparation of a porous and photoluminescent covalent triazine-based framework (PCTF) by using TFMS as the catalyst. This PCTF-8 showed photoluminescence behaviour under UV light, whereas the material PCTF-1 prepared by the high-temperature ZnCl2 route was completely non-emissive. PCTF-8 shows sensitive detection towards nitroaromatic analytes with trinitrophenol (TNP) quenching ca. 71% of the PCTF-8 fluorescence emission intensity. The quenching mechanism was found to be static in nature because the emission lifetimes did not change with increasing addition of the analytes. From optical transmission measurements, we estimated a band gap of about 2.25 eV which determines its efficiency as a photocatalyst for hydrogen evolution. The total amount of H2 evolved of Pt-loaded PCTF-8 in aqueous methanol or TEoA was 1780 and 2370 μmol g−1, when irradiated for 20 h. Hence, CTFs are not only interesting for gas adsorption/separation but also hold great potential for hydrogen production and for the development of new luminescent chemosensors.Acknowledgements
A. B. thanks Ghent Univ. (GOA nr. 01G00710). D. E. thanks the Scientific Research-Foundation Flanders (FWO), the Spanish Ministry of Economy and Competitiveness (Project MAT2013-44463-R), Junta de Andalucia (Project P10-FQM-6181 and Andalucia Talent Hub) and FEDER Funds. This work was partly supported by ACT-C, Japan Science and Technology Agency (JST), BMBF project OptiMat 03SF0492C and the HHU SFF fund. We thank Prof. Romero-Salguero of the University of Córdoba for his suggestions and support.Notes and references
- (a) A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS; (b) M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed; (c) J. R. Holst and A. I. Cooper, Adv. Mater., 2010, 22, 5212–5216 CrossRef CAS PubMed; (d) V. Guillerm, L. J. Weselinski, M. Alkordi, M. I. H. Mohideen, Y. Belmabkhout, A. J. Cairns and M. Eddaoudi, Chem. Commun., 2014, 50, 1937–1940 RSC; (e) M. H. Alkordi, Ł. J. Weseliński, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, R. G. AbdulHalim, J.-M. Basset and M. Eddaoudi, J. Mater. Chem. A, 2016, 4, 7453–7460 RSC.
- (a) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC; (b) Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- (a) J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed; (b) L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed; (c) A. Patra, J.-M. Koenen and U. Scherf, Chem. Commun., 2011, 47, 9612–9614 RSC; (d) X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed; (e) A. Patra and U. Scherf, Chem.–Eur. J., 2012, 18, 10074–10080 CrossRef CAS PubMed.
- I. A. Levitsky, K. Kishikawa, S. H. Eichhorn and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 2474 CrossRef CAS; S. Wang, W. J. J. Oldham, R. A. J. Hudack and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 5695 CrossRef.
- (a) Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed; (b) L. Martelo, A. Jimenez, A. J. M. Valente, H. D. Burrows, A. T. Marques, M. Forster, U. Scherf, M. Peltzer and S. M. Fonseca, Polym. Int., 2012, 61, 1023–1030 CrossRef CAS.
- V. M. Suresh, S. Bonakala, S. Roy, S. Balasubramanian and T. K. Maji, J. Phys. Chem. C, 2014, 118, 24369–24376 CAS.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed; (b) D. Banerjee, Z. Hu and J. Li, Dalton Trans., 2014, 43, 10668–10685 RSC; (c) S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed; (d) G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC; (e) L. Zhang, Z. Kang, X. Xin and D. Sun, CrystEngComm, 2016, 18, 193 RSC.
- (a) L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC; (b) S. Ren, R. Dawson, A. Laybourn, J.-X. Jiang, Y. Khimyak, D. J. Adamsa and A. I. Cooper, Polym. Chem., 2012, 3, 928–934 RSC; (c) T. Ishi-i, K. Yaguma, T. Thiemann, M. Yashima, K. Ueno and S. Mataka, Chem. Lett., 2004, 33, 1244–1245 CrossRef.
- (a) L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC; (b) K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC; (c) K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435–2439 CrossRef CAS PubMed; (d) K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed; (e) K. Kailasam, M. B. Mesch, L. Möhlmann, M. Baar, S. Blechert, M. Schwarze, M. Schröder, R. Schomäcker, J. Senker and A. Thomas, Energy Technol., 2016, 4, 744–750 CrossRef CAS; (f) R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 128, 1824–1828 CrossRef PubMed; (g) R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961–3963 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed; P. Kuhn, A. l. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef PubMed; M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef PubMed.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990–14999 CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- (a) C. E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537–541 CrossRef CAS PubMed; (b) J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- A. Bhunia, S. Dey, M. Bous, C. Zhang, W. von Rybinski and C. Janiak, Chem. Commun., 2015, 51, 484–486 RSC.
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6626 CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed.
- S. Xiong, X. Fu, L. Xiang, G. Yu, J. Guan, Z. Wang, Y. Du, X. Xiong and C. Pan, Polym. Chem., 2014, 5, 3424–3431 RSC.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC; F. Jeremias, V. Lozan, S. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC.
- S. Keskin, T. M. van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef CAS PubMed.
- S. Himeno, T. Komatsu and S. Fujita, J. Chem. Eng. Data, 2005, 50, 369–376 CrossRef CAS; K. B. Lee, M. G. Beaver, H. S. Caram and S. Sircar, Ind. Eng. Chem. Res., 2008, 47, 8048–8062 CrossRef.
- S. Xiong, X. Fu, L. Xiang, G. Yu, J. Guan, Z. Wang, Y. Du, X. Xiong and C. Pan, Polym. Chem., 2014, 5, 3424–3431 RSC.
- X. Liu, H. Li, Y. Zhang, B. Xu, S. A, H. Xia and Y. Mu, Polym. Chem., 2013, 4, 2445–2448 RSC.
- Y. Xu, A. Nagai and D. Jiang, Chem. Commun., 2013, 49, 1591 RSC.
- W. Lluo, Y. Zhu, J. Zhang, J. He, Z. Chi, P. W. Miller, L. Chen and C. Su, Chem. Commun., 2014, 50, 11942–11945 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- V. M. Suresh, S. Bonakala, S. Roy, S. Balasubramanian and T. K. Maji, J. Phys. Chem. C, 2014, 118, 24369–24376 CAS.
- Z. Wei, Z. Gu, R. K. Arvapally, Y. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, XRD, elemental analysis, KFT, isosteric heat of adsorption and pore size distribution of CO2, adsorption, IAST selectivity, emission spectra of PCTF-1, and detection of nitroaromatic explosives. See DOI: 10.1039/c6ta04623a |
| This journal is © The Royal Society of Chemistry 2016 |