
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcene-linked covalent organic frameworks as candidate materials for singlet fission†
Viktor
Laszlo
and
Tim
Kowalczyk
*
Department of Chemistry, Advanced Materials Science and Engineering Center, Institute for Energy Studies, Western Washington University, Bellingham, WA 98225, USA. E-mail: Tim.Kowalczyk@wwu.edu
First published on 14th June 2016
Abstract
Two-dimensional covalent organic frameworks (COFs) are a versatile class of porous materials that can be tailored for solar energy conversion applications through the incorporation of organic chromophores as linker units. In this work we examine whether such COFs can be engineered to support singlet fission (SF), the generation of two triplet excitons from a single photoexcitation. We simulate the structural and electronic properties of a series of COFs possessing embedded polyacene linkers from benzene through pentacene. Detailed electronic structure calculations suggest that necessary conditions on the energy and electronic coupling for SF can be met with COF architectures closely related to those already synthetically achieved. Molecular dynamics simulations reveal that the embedded acenes preferentially adopt orientations at an angle relative to the COF plane at room temperature, modifying the energy levels and electronic couplings relative to their minimum energy conformations at 0 K. Charge mobility through the COFs is quantified as a function of the length of the embedded acene through application of a recently introduced charge transport index. Implications for the future design of porous materials supporting singlet fission are discussed.
1 Introduction
Since the first covalent organic frameworks (COFs) were reported over a decade ago,1 the development of COF materials for gas storage2–4 and heterogeneous catalysis has blossomed due to their high surface area and porosity.5–8 Although the preparation of macroscopic single crystals of COF materials remains challenging, the pace of COF development for hydrogen uptake applications has been brisk, exceeding the 2015 DOE volumetric uptake target for H2 of 40 g L−1 in 2012 and rapidly approaching the 2050 target.9The porosity and ordered stacking of 2D COFs, combined with their synthetic tunability, offer important advantages for organic electronics applications. Several studies have proposed to employ electroactive COF materials, with organic chromophores embedded in the framework as linkers, as the active layer of a bulk heterojunction solar cell.10 Under illumination, singlet excitons can be generated on the embedded chromophores. These sites can then act as electron donors in the presence of a suitable acceptor chromophore embedded in the COF pore10 or at an adjacent linker in the framework,11,12 facilitating charge separation. Furthermore, because 2D COFs with chromophore linkers are highly ordered materials with extensive π conjugation and relatively short interlayer stacking distances, significant charge mobility can be achieved.13–15 These investigations establish electroactive COFs as a promising platform for the development of organic solar cell materials, but the unique ordering and electronic properties of COFs suggest a further advantage that has yet to be explored.
Among the most highly desirable electronic processes to introduce into a candidate solar cell material is the phenomenon of singlet fission (SF), the creation of two triplet excitons from one singlet exciton.16,17 In SF, a photoexcited organic chromophore, typically in the lowest singlet excited state S1, transfers a portion of its excitation energy to an adjacent chromophore in the ground electronic state S0, yielding a correlated pair of triplet excitons (Fig. 1). The SF rate depends sensitively on an energy-matching condition between the singlet exciton and the correlated pair of triplet excitons, ΔE(S1) ≈ 2ΔE(T1). By creating up to two electron–hole pairs per absorbed photon, SF materials permit quantum efficiencies exceeding 100%.18
 | ||
| Fig. 1 Molecular orbital representation of electronic states involved in the direct mechanism for singlet fission. (a) Pair of chromophores in the S0 ground state. (b) Absorption of a photon excites one chromophore to the S1 excited state. (c) Energy transfer from the chromophore in the S1 state to the second chromophore in the ground state produces two chromophores in the T1 state. | ||
Recent progress in the development of novel intramolecular SF materials19,20 has been complemented by the construction of predictive theoretical models of SF.21–23 Many of these developments have been recently reviewed.24,25 The energy transfer in SF typically proceeds from an exciton described by a localized S1 state either directly26–28 or through an intermediate charge-transfer (CT) state23,29,30 to a pair of triplet excitons (TT state). SF has long been known to occur in a limited number of organic crystals including tetracene, pentacene, and certain of their derivatives, but the number of organic materials, and small organic dyads in particular, known to support SF has swelled in recent years.25,31
This study is motivated by the notion that COFs offer several potential advantages in the search for cheap, easily processible organic materials that could support SF. First, the assembly of 2D COFs results in stacked, closely spaced layers which could facilitate SF between adjacent organic linkers embedded in the framework. It is well documented that intermolecular packing can significantly modulate SF rates in crystalline tetracene, pentacene, and their derivatives.23,32 SF in pentacene and tetracene depends sensitively on molecular packing in the crystal, and in particular on the relative orientation of neighboring molecules. While herringbone and slip-stacked pentacene configurations exhibit significant SF rates, orthogonal orientations drastically hinder SF.33
Another potential advantage of COF architectures in the search for SF materials lies in the vast but largely unexplored chemical space accessible through combinatorial matching of COF linker and core units. Together, the degree of control over intermolecular interactions and the large chemical space suggest to us that COFs constructed from chromophore linkers already implicated in SF could be engineered not only to support but to enhance SF rates.
Here we employ quantum mechanical simulations to investigate the effects of linker orientation and interlayer packing on the energies and electronic properties of the series of acene-linked 2D COFs in Fig. 2. The investigated model COFs are derived from experimentally achieved materials containing acene linkers to ensure that our findings are directly applicable to synthetically accessible COFs.34 In particular, the anthracene-linked COF (n = 3) was recently synthesized and shown to respond to UV irradiation through the same heat-reversible photodimerization as its acene parent molecule.35 Our focus here is on the response of acene-COFs to visible and near-IR radiation, presupposing that a suitable method of filtering UV wavelengths from the incident spectrum can be achieved for applications where the photodimerization process would be detrimental to device performance.
 | ||
| Fig. 2 Chemical structure of the unit cell for the acene-linked COF monolayers. | ||
The energy level condition ΔE(S1) ≈ 2ΔE(T1) is necessary but not sufficient to achieve a highly efficient SF material. Once triplet excitons are produced, the generation of a photocurrent requires charge separation and transport through the COF. Because the relaxation from T1 to S0 is spin forbidden, the probability of radiative recombination from T1 is relatively small, and the corresponding exciton diffusion length is relatively long.29 To address these downstream processes computationally, we also probe the relationship between acene length and electron mobility through the framework.
Energy level spacings, electronic couplings, and charge mobility must be considered concurrently: modifications to the COF structure to tune one of these properties can easily influence the others. If modifications that favor one property prove detrimental to another, trade-offs may be required to optimize the COF structures' overall internal quantum efficiency.
In the next section, we describe the details of our computational methods and simulation strategy. Results concerning the geometry and interlayer stacking of acene COFs are the focus of Section 3.1. In Section 3.2 we identify several important features of the acene-COF structures revealed by finite-temperature molecular dynamics (MD) which are not evident in the geometry-optimized structures. Section 3.3 discusses the energy levels of acene-COFs and their implications for SF, and electron mobility through the materials is characterized in Section 3.4. We conclude with an assessment of the strengths and weaknesses of these acene-COFs as candidate SF materials and offer guidance for the future design of electroactive COFs for SF.
2 Computational details
2.1 Geometry optimization and molecular dynamics
Unit cells for the acene-COFs in Fig. 2 were constructed in Avogadro.36 For each structure, four commonly observed stacking patterns (AA, inclined AA, serrated AA, and AB)37 were optimized using self-consistent charge density functional tight-binding (SCC-DFTB) with empirical dispersion correction38,39 as implemented in the DFTB+ program package.40 The Slater–Koster parameter set of Heine and co-workers was used for all DFTB calculations.41,42 To obtain optimized structures for each stacking pattern, two-layer unit cells with periodic boundary conditions were used. The binding energy for each stacked arrangement was determined from the difference between the energy of the optimized structure and twice the energy of an isolated monolayer. For DFTB geometry optimizations, an SCC tolerance of 10−6 hartree was used together with a maximum force component of 10−4 hartree per bohr.MD simulations were performed with the same SCC-DFTB method used for geometry optimizations due to the prohibitive computational cost of MD based on full density functional theory (DFT) applied to these extended COF systems. Our DFTB-MD simulations employ a periodically repeated four-layer unit cell of the optimized serrated-AA stacking geometries in order to reduce correlation between the motions of adjacent acenes due to periodic images. After geometry optimization and equilibration, production simulations were performed in the NVE ensemble with initial velocities corresponding to temperatures of 200 K, 300 K, and 600 K. A 1 fs time step was employed, and snapshots were collected at 40 fs intervals. The frequency distribution of linker orientations was constructed from all snapshots obtained from the four-layer simulations with the exception of the first 1 ps of MD, which was discarded in all cases.
2.2 Electronically excited states
We evaluate the S0, S1, TT, and CT state energies of DFTB-MD snapshots using DFT calculations with the PBE0 functional and 6-311G* basis set.43 The S1 state was calculated by the ΔSCF approach44 together with the maximum-overlap method (MOM) to ensure convergence to the target state.45 ΔSCF has been benchmarked against TDDFT and experiment for the S1 state of organic dyes, including anthracene and pentacene, where ΔPBE0 without range separation successfully reproduces the S1 excitation energy.46 We employed constrained DFT (CDFT) to calculate energies and self-consistent electronic densities for the CT states.47,48 CDFT has been successfully used in this context to predict singlet fission rates in tetracene and pentacene crystals.23 Potential energy curves for the CT and TT states were computed by averaging over the relevant microstates; a molecular orbital scheme indicating the corresponding electron spin and charge constraints can be found in the ESI.† All DFT calculations were performed in the Q-Chem 4.3 software package.492.3 Charge mobility analysis
Charge mobility through COF layers was estimated by extracting all neighboring chromophore dimer pairs from each optimized COF model and computing the electronic coupling between one chromophore and the anion of the other within constrained DFT-based configuration interaction (CDFT-CI).48 While the electronic couplings obtained from CDFT-CI should be considered qualitative, their success in predicting experimental SF rates in molecular crystals23 and their best-in-class performance in recent benchmarks50 support their application in our study. To retain charge balance, acene linker units extracted from the COF model were capped with hydrogens.The computed couplings were incorporated into an adjacency matrix to evaluate the Kirchoff transport index (KT), a recently introduced metric which ranks the relative electron transport ability of related molecular structures.51 Our approach to the evaluation of KT closely follows the algorithm of ref. 51.
To break the high translational symmetry of the optimized COF structures and explore the effect of disorder on charge mobility, we applied the coupling estimation scheme above to snapshots from the DFTB-MD simulations. Electronic couplings from 100 snapshots per structure were aggregated to find average couplings between acene units for the Kirchoff transport index analysis.
3 Results and discussion
3.1 Stacking energies and patterns
X-ray diffraction (XRD) patterns of many 2D COFs have revealed a general preference for AA or slipped-AA stacking over AB stacking.52 The typically broad nature of the XRD peaks indicate that lower-symmetry slipped stacking orientations are most prevalent.42,53 Variations in the COF stacking pattern affect the relative orientations of the embedded acenes, thereby influencing the energies, electronic couplings, and ultimately the rate of SF. To account for this orientation dependence, the four stacking patterns commonly observed in 2D COFs were investigated to determine the preferred stacking of these acene-linked COFs. Optimized structures for the stacking patterns considered in this study were calculated using SCC-DFTB41 and are depicted in Fig. 3. | ||
| Fig. 3 Molecular representation of the stacking patterns investigated in this study: (A) AB (B) serrated AA (C) inclined AA (D) perfect AA stacking. | ||
The binding energy for each stacking configuration was determined by subtracting twice the total energy of an isolated monolayer from that of a periodically repeated two-layer unit cell. The binding energies (Table 1) show that serrated AA stacking is slightly preferred over inclined and AA stacking, all of which are more stable than AB stacking. The order of stacking preference is mirrored in the optimized interlayer distances (ESI Table S1†), with the lowest energy structures corresponding to the smallest interlayer distances.
| Acene unit | AB | Serrated AA | Inclined AA | AA |
|---|---|---|---|---|
| Benzene | −40.00 | −100.55 | −98.64 | −96.68 |
| Naphthalene | −42.12 | −116.72 | −115.81 | −112.06 |
| Anthracene | −43.85 | −133.07 | −132.26 | −127.54 |
| Tetracene | −46.01 | −148.53 | −147.99 | −142.80 |
| Pentacene | −48.73 | −164.42 | −163.76 | −158.12 |
Our finding that slipped stacking is preferred to cofacial AA stacking is consistent with recent nonadiabatic fewest-switches surface hopping (FSSH) simulations of pentacene dimers.33 Furthermore, investigations of SF in perfluoropentacene have shown that SF is enhanced by slip stacking, whereas a perfectly eclipsed orientation suppresses the mechanism.32 Thus the apparent preference for slip stacking in acene-linked COFs supports their potential as candidate frameworks for materials supporting SF.
3.2 Framework flexibility at room temperature
To account for the possibility that flexibility in the COF structure might affect electronic energies and couplings at device operating temperatures, i.e. at room temperature, we used molecular dynamics (MD) simulations to sample commonly adopted COF configurations. In these multilayer room temperature MD simulations, the acene units within the COF adopt an unexpectedly wide distribution of orientations relative to the COF plane. Surprisingly, the observed distribution of angles was effectively independent of acene length (Fig. 4). | ||
| Fig. 4 Acene unit angle distribution observed in molecular dynamics simulations at 300 K. | ||
At 300 K, angles between 5 and 10° were most common, and the angular distributions show only a small skew toward larger angles. At 200 K, the distributions are narrower and favor orientations closer, on average, to planar (0°). At 600 K, the increased thermal energy enhances fluctuations in the orientation of the acene unit relative to the COF plane. This creates a much broader distribution with the most frequent angle occurring at 10° and with this key angle regularly exceeding 20°. Vertically adjacent acene units remain mostly co-planar, but herringbone-like orientations occur as well. This fluctuation in the relative angle of the embedded acene units can impact the energy and coupling between adjacent acenes; the rate of SF is highly sensitive to both of these factors.
3.3 Electronic energies relative to SF criteria
As the orientations of nearby acenes in adjacent layers fluctuate, the relative energies of the ground and low-lying excited states of the acene-COFs may also vary. To determine which configurations are most favorable for SF, we calculated the energies of the first singlet excited state (S1), the lowest charge-transfer (CT) states, and the triplet-pair state (TT) from Fig. 1 along two orientational degrees of freedom. The first degree of freedom θ is an out-of-plane rotation of one acene unit relative to the other, connecting co-planar and herringbone conformations (Fig. 5a). The second degree of freedom ϕ is a collective rotation of both acenes relative to the rest of the COF (Fig. 5b). The acenes remain co-planar along the second degree of freedom, but the effective distance and degree of slip-stacking between the acene varies. These calculations are performed on borate-capped acenes extracted from the full COF structures for computational tractability with CDFT-CI. Excitation energies from S0 to each state are shown in Fig. 6 for tetracene and pentacene; analogous results for smaller acenes are available in the ESI.† | ||
| Fig. 5 Rotation of capped acene segments from a slip-stacked planar position (middle). (a) Rotation of a pair of segments to a herringbone orientation. The angle θ is the internal angle the segments make with one another. (b) Rotation of a pair of segments, which remain co-planar, relative to the COF plane. The angle ϕ is the angle either acene makes with the COF plane. | ||
 | ||
| Fig. 6 Excitation energies from S0 to the S1, CT, and TT states for capped acene dimers along rotational degrees of freedom described by the angles θ and ϕ introduced in Fig. 5. | ||
Up to 40° from co-planarity, we observe only small changes in the S0-CT and S0-TT energy gaps with respect to θ (Fig. 6a and b) and a modest decrease in the S0–S1 energy gap. Along ϕ, there is a steeper decrease in all three energy gaps between 30° and 40°, especially for the S1 state (Fig. 6c and d). As ϕ increases, the effective distance between the acene units decreases, giving rise to steric effects that may partially explain the trend as a destabilization of S0 relative to the excited states. At the same time, the relative phase of the acene frontier orbitals changes with ϕ; such phase shifts can significantly alter electronic energies and couplings.54 However, such large values of ϕ are not commonly observed at room temperature (Fig. 4). The orbital phase change should have the greatest impact on the CT state because the dominant donor and acceptor orbitals for this state are on different acenes. Given that the observed decrease is independent of the particular acene or electronic state in question, we attribute the change in S1 excitation energy along ϕ primarily to the changing effective distance between adjacent acenes.
Excitation energies for the S1, CT, and TT states all decrease with increasing acene length, in keeping with the known properties of their isolated chromophore equivalents and with the particle-in-a-box model of electronic excitations in quasi-1D systems. Although the energies of all states considered here decrease with increasing acene length, the decrease in the S0-TT energy gap as a function of acene length is more drastic than that of the S0–S1 energy gap. Critically, the energy difference between the S1 state and the TT state falls from 3.7 eV in the benzene-COF to less than 0.4 eV in the pentacene-COF. The energy level criterion for SF is approximately met when these two levels become nearly degenerate. These results indicate that the pentacene-embedded COF comes close to meeting a key condition for SF in its optimized geometric structure and has the highest likelihood, among the structures investigated here, of adopting configurations favorable to SF.
3.4 Electron mobility along slip-stacked COF layers
For acene-COFs to be viable candidates for the active layer of a bulk heterojunction solar cell, it is not only enough to generate excitons and electron–hole pairs in sufficient quantity. These COFs must also demonstrate sufficient electron and hole mobility to deliver charge carriers to the electrodes. In the case of ordered 2D COFs, in order to utilize the energy captured by the embedded chromophores, electrons must be readily transported across the COF layers, as illustrated schematically in Fig. 7. | ||
| Fig. 7 Schematic of charge mobility through acene-COFs. (a) Vertical mobility depicting the transfer of an electron one segment away (purple to green) and two segments away (purple to red). (b) Horizontal mobility depicting the transfer of an electron one segment away (purple to green). | ||
It has been demonstrated that certain electroactive COFs can achieve high electron mobilities.37 Tetrathiafulvalene-based COFs can achieve conductivity on the order of 10−5 to 10−6 Ω−1 cm−1 due to their highly ordered π-stacking structure.14 To estimate the relative mobility of our acene-COFs, we implemented and applied a recently developed measurement tool called the Kirchoff transport index.51 This approach to the characterization of electron mobility in organic materials treats the conduction of electrons among available sites by analogy to classical resistor network theory.
The Kirchoff transport index is not an absolute measure of electron mobility in cm2 V−1 s−1; instead, it provides a relative measure of transport that can be used to compare the electron mobility of related materials. Nevertheless, for a rough estimate of the electron mobility in these acene-COFs, one can compare the KT values obtained in this study against the values of approx. 100 meV obtained for PC60BM in ref. 51, which correspond to an electron mobility of 5 × 10−2 cm2 V−1 s−1.55 By constructing an adjacency matrix from electronic couplings between adjacent neutral and anion electronic states via CDFT-CI, we evaluated the Kirchoff transport index KT for vertical and horizontal electron transport in each of the acene-COFs investigated here.
Relative charge mobilities for the acene-COFs are reported in Table 2. Charge mobility was assessed between adjacent COF planes (vertical) as well as across acenes within the same plane (horizontal). Consistent with the interrupted nature of the π conjugation between adjacent acenes within the same plane, the horizontal mobility is predicted to be smaller than the vertical mobility between planes. This preferential mobility along the stacking direction is consistent with other investigations of conductive COFs.14 While the coupling is strongest between adjacent acenes, a small interaction is also seen between acenes two layers away, with the effect dropping below any meaningful magnitude beyond two layers. The deviation from the trend of decreasing KT with increasing acene length observed in naphthalene was traced back to a slightly larger-than-expected optimized interlayer spacing, reflecting the fact that electronic couplings between distant molecular fragments can be exponentially sensitive to distance.
| Acene unit | Vertical KT | Horizontal KT |
|---|---|---|
| Benzene | 101.401 | 32.932 |
| Naphthalene | 14.170 | 17.688 |
| Anthracene | 81.480 | 7.746 |
| Tetracene | 74.022 | 4.740 |
| Pentacene | 43.903 | 4.150 |
Snapshots from the previously detailed MD simulations were used to gain insight into the fluctuations of KT with structure at room temperature. The observed distributions of KT are illustrated in Fig. 8. In contrast to the optimized slip-stacked geometries at 0 K, the value of KT increases with acene length at finite temperature. With an average value of 74 meV for the benzene containing COF and 139 meV for the pentacene containing COF, charge mobility for the pentacene-linked COF is estimated to be almost double that of the benzene-linked COF at operating temperatures. The sharply contrasting trends between the predicted electron mobilities in the static versus dynamic analyses illustrates the high sensitivity of electronic coupling to molecular orientation in organic semiconductors and the importance of configurational sampling in predicting electronic properties like the charge carrier mobility.
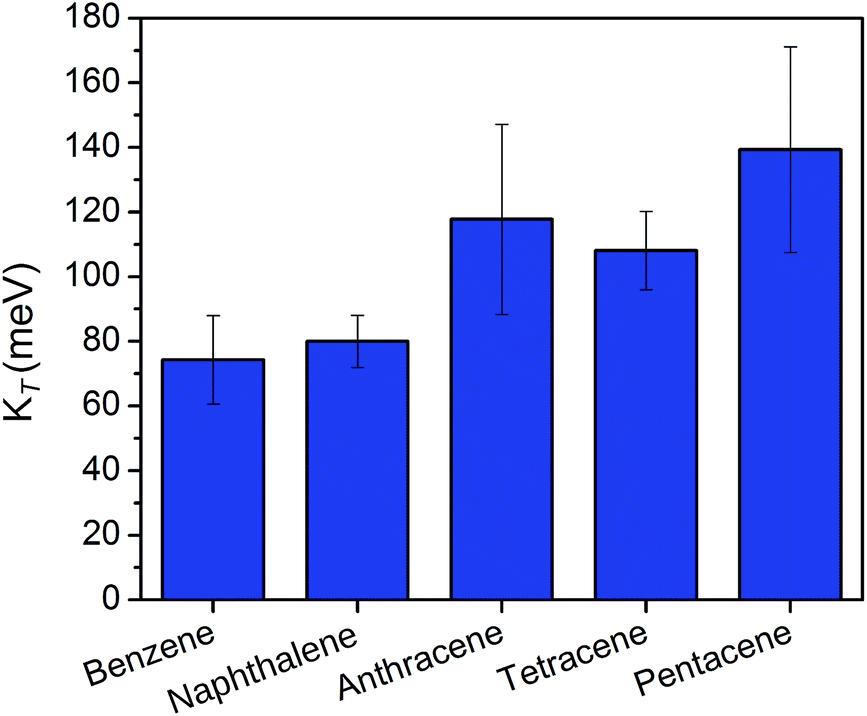 | ||
| Fig. 8 Kirchoff index (KT) values in meV calculated for various acene-COFs at 300 K. Error bars indicate standard deviations in KT across MD snapshots. | ||
4 Conclusions
In summary, we have investigated the electronic properties of experimentally realizable 2D COFs with embedded acene linkers to determine their capacity to support singlet fission. We found that the embedding of acenes within the COF does not significantly perturb the energy levels of the S1 and triplet-pair (TT) excited states of the embedded acenes. This finding offers preliminary evidence that monomer properties are indeed meaningful for the design of COFs capable of supporting SF. Consequently, our study suggests that the search for COFs capable of SF can rely heavily on the underlying electronic properties of their embedded organic chromophore dimers, greatly simplifying both computational and experimental screening efforts.The acene-COF materials examined here prefer to adopt a slip-stacked orientation which is known to be beneficial for SF. Molecular dynamics simulations revealed that the acene units can adopt a distribution of angles that appears to be independent of acene length but that broadens with increased temperature. From the construction of potential energy curves along two key orientational degrees of freedom, we showed that pentacene-embedded COFs can satisfy the energetic requirements for SF at orientations achieved at room temperature. This finding is especially ripe for future experimental validation or refutation.
From analysis of the charge mobility by the Kirchoff transport index, we found that increasing the length of the embedded acene unit from benzene to pentacene significantly increases mobility along the stacking direction. By embedding a longer acene into the COF, the energy requirements for SF are more closely met with the added benefit of increased charge mobility when compared to those with smaller embedded acenes.
This investigation suggests several avenues for future work. Given that the pentacene-embedded COFs best matched the energy level criteria for SF, one may consider whether even longer embedded acenes may offer better energetics, provided that a viable synthetic route to such COFs could be identified. This study considered one specific COF-linker chemistry, but there exist a number of alternative strategies which would result in analogous acene-COFs with different stacking patterns and energetics. Thus there already exist several known routes to closely related structures which may be even better-suited to SF than the family of acene-COFs studied here. Beyond these alternative linker chemistries, it is interesting to consider the wide range of options available for fine-tuning the embedded chromophore's electronic structure for SF;56 for example, one may adjust the interlayer stacking distances or even restrict the chromophore's orientational degrees of freedom through bulky substituents or by silicon substitution within the chromophore.57 This investigation lays the foundation for computational screening towards the experimental realization of electroactive COFs as SF materials.
Acknowledgements
We thank Shane R. Yost for valuable discussions. T. K. gratefully acknowledges start-up support from Western Washington University.References
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- M. E. Davis, Nature, 2012, 417, 813–821 CrossRef PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- N. K. Jensen, T. E. Rufford, G. Watson, D. K. Zhang, K. I. Chan and E. F. May, J. Chem. Eng. Data, 2012, 57, 106–113 CrossRef CAS.
- J. Dong, Y. Liu and Y. Cui, Chem. Commun, 2014, 50, 14949–14952 RSC.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- A. Primo and H. Garcia, Chem. Soc. Rev., 2014, 43, 7548–7561 RSC.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- J. L. Mendoza-Cortes, W. A. Goddard, H. Furukawa and O. M. J. Yaghi, J. Phys. Chem. Lett., 2012, 3, 2671–2675 CrossRef CAS PubMed.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920–2924 CrossRef CAS PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- S. Wan, F. Gandara, H. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- S. Jin, T. Sakurai, T. Kowalczyk, S. Dalapati, F. Xu, H. Wei, X. Chen, J. Gao, S. Seki, S. Irle and D. Jiang, Chem.–Eur. J., 2014, 20, 14608–14613 CrossRef CAS PubMed.
- H. Ding, Y. Li, H. Hu, Y. Sun, J. Wang, C. Wang, C. Wang, G. Zhang, B. Wang, W. Xu and D. Zhang, Chem.–Eur. J., 2014, 20, 14614–14618 CrossRef CAS PubMed.
- C. E. Swenberg and W. T. Stacy, Chem. Phys. Lett., 1968, 2, 327–328 CrossRef CAS.
- R. Merrifield, P. Avakian and R. P. Groff, Chem. Phys. Lett., 1969, 3, 386–388 CrossRef CAS.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- Y. Zhai, C. Sheng and Z. V. Vardeny, Philos. Trans. R. Soc., A, 2015, 373, 20140327 CrossRef PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X.-Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- P. E. Teichen and J. D. Eaves, J. Phys. Chem. B, 2012, 116, 11473–11481 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114102 CrossRef PubMed.
- S. R. Yost, J. Lee, M. W. B. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sfeir, M. G. Bawendi, T. M. Swager, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- P. M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, J. Am. Chem. Soc., 2011, 133, 19944–19952 CrossRef CAS PubMed.
- W.-L. Chan, M. Ligges, A. Jailaubekov, L. Kaake, L. Miaja-Avila and X.-Y. Zhu, Science, 2011, 334, 1541–1545 CrossRef CAS PubMed.
- F. Mirjani, N. Renaud, N. Gorczak and F. C. Grozema, J. Phys. Chem. C, 2014, 118, 14192–14199 CrossRef CAS.
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef.
- W.-L. Chan, T. C. Berkelbach, M. R. Provorse, N. R. Monahan, J. R. Tritsch, M. S. Hybertsen, D. R. Reichman, J. Gao and X.-Y. Zhu, Acc. Chem. Res., 2013, 46, 1321–1329 CrossRef CAS PubMed.
- S. T. Roberts, R. E. McAnally, J. N. Mastron, D. H. Webber, M. T. Whited, R. L. Brutchey, M. E. Thompson and S. E. Bradforth, J. Am. Chem. Soc., 2012, 134, 6388–6400 CrossRef CAS PubMed.
- K. Kolata, T. Breuer, G. Witte and S. Chatterjee, Beilstein J. Nanotechnol., 2014, 8, 7377–7383 CAS.
- L. Wang, Y. Olivier, O. V. Prezhdo and D. Beljonne, J. Phys. Chem. Lett., 2014, 5, 3345–3353 CrossRef CAS PubMed.
- M. Dogru, A. Sonnauer, A. Gavryushin, P. Knochel and T. Bein, Chem. Commun, 2011, 47, 1707–1709 RSC.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8704–8707 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. Dogru and T. Bein, Chem. Commun, 2014, 50, 5531–5546 RSC.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- M. Elstner, P. Hobza, T. Frauenheim, S. Suhai and E. Kaxiras, J. Chem. Phys., 2001, 114, 5149 CrossRef CAS.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim and S. Suhai, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- B. Lukose, A. Kuc, J. Frenzel and T. Heine, Beilstein J. Nanotechnol., 2010, 13, 60–70 CrossRef PubMed.
- C. Adamo and B. Vincenzo, J. Chem. Phys., 1999, 13, 6158–6170 CrossRef.
- T. Ziegler, A. Rauk and E. J. Baerends, Theor. Chim. Acta, 1977, 43, 261–271 CrossRef CAS.
- A. Gilbert, N. Besley and P. Gill, J. Phys. Chem., 2008, 112, 13164–13171 CrossRef CAS PubMed.
- T. Kowalczyk, S. R. Yost and T. Van Voorhis, J. Chem. Phys., 2011, 134, 054128 CrossRef PubMed.
- Q. Wu, C.-L. Cheng and T. Van Voorhis, J. Chem. Phys., 2007, 127, 164119 CrossRef PubMed.
- B. Kaduk, T. Kowalczyk and T. Van Voorhis, Chem. Rev., 2012, 112, 321–370 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kus, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stueck, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- A. Kubas, F. Gajdos, A. Heck, H. Oberhofer, M. Elstner and J. Blumberger, Phys. Chem. Chem. Phys., 2014, 17, 14342–14354 RSC.
- N. E. Jackson, B. M. Savoie, L. X. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2015, 6, 1018–1021 CrossRef CAS PubMed.
- M. A. Addicoat and M. Tsotsalas, Front. Magn. Mater., 2015, 2, 7 Search PubMed.
- B. Lukose, A. Kue and T. Heine, Chem.–Eur. J., 2011, 17, 2388–2392 CrossRef CAS PubMed.
- L. J. Wang, Q. K. Li and Z. Shuai, J. Chem. Phys., 2008, 128, 194706 CrossRef CAS PubMed.
- F. Steiner, S. Foster, A. Losquin, J. Labram, T. D. Anthopoulos, J. M. Frost and J. Nelson, Mater. Horiz., 2015, 2, 113–119 RSC.
- C. Quarti, D. Fazzi and M. Del Zoppo, Phys. Chem. Chem. Phys., 2011, 13, 18615–18625 RSC.
- K. Bhattacharyya, S. M. Pratik and A. Datta, J. Phys. Chem. C, 2015, 119, 25696–25702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic configurations for CDFT-CI calculations; optimized interlayer distances of COF structures; calculation of core-linker angular distortions; optimized geometries. See DOI: 10.1039/c6ta03708a |
| This journal is © The Royal Society of Chemistry 2016 |