
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly insensitive and thermostable energetic coordination nanomaterials based on functionalized graphene oxides†
Qi-Long
Yan
a,
Adva
Cohen
a,
Natan
Petrutik
a,
Avital
Shlomovich
a,
Larisa
Burstein
b,
Si-Ping
Pang
c and
Michael
Gozin
*a
aSchool of Chemistry, Faculty of Exact Science, Tel Aviv University, Tel Aviv, 69978, Israel. E-mail: cogozin@gmail.com (M. Gozin); terry.well@163.com (Q.-L. Yan); Tel: +972-36405878
bWolfson Applied Materials Research Center, Tel Aviv University, Tel Aviv, 69978, Israel
cSchool of Material Science & Engineering, Beijing Institute of Technology, Beijing, 100081, China
First published on 1st June 2016
Abstract
In this research, a group of new energetic coordination nanomaterials (CNMs) based on functionalized graphene oxide sheets (FGS) have been designed and characterized. GO was first functionalized with N-rich energetic ligands such as triaminoguanidine (TAG), and then the resulting FGS was coordinated with metal ions to prepare energetic CNMs with high thermostability and insensitivity to mechanical stimuli. The density of GO–TAG–Cu(II)/Cu(I) is as high as 3.14 g cm−3, while it has a Tp of 495 °C and VoD of 7723 m s−1 by using 40 wt% ammonium perchlorate as the oxidant. These insensitive (Im > 81 J) and highly thermostable energetic CNMs in combination with oxidizers are good candidate ingredients of low-vulnerability solid propellants and charges of deep-well perforating guns.
1 Introduction
The reduced sensitivity and improved thermostability of high performance energetic materials (EMs) are essential requirements for their practical applications.1 The EMs are still under fast development in terms of new energetic molecules, novel energetic compositions, advanced energetic composites, as well as unique energetic systems.2 There has been a significant amount of EMs being developed during the past decades, including new energetic polymers,3 novel nitrocompounds,4 energetic salts,5 nitrogen rich compounds, based on azole and azine rings,6 energetic ionic liquids,7 nanothermites,8 energetic supermolecules (host/guest complexes, cocrystals)9 and polynitrogen compounds.10 However, the newly developed energetic compounds, in most cases, are still not able to completely replace the currently used high EMs due to various problems including chemical incompatibility, instability, high sensitivity, and high cost. As one of the important research topics, the highly thermostable EMs are becoming more and more attractive, since the oil/gas industry is searching for more thermostable explosive charges that could be used in perforator guns for deeper well exploration (Fig. S1†).11 The currently used thermostable explosives including octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) with a melting point (m.p.) of 291 °C (ref. 12) and 1,3,5-triamino-2,4,6-trinitrobenzene (TATB, Scheme 1) with a m.p. of 350 °C are not sufficiently thermostable to fulfil the upcoming requirements.13 There are several strategies for the development of highly thermostable energetic compounds. The most common one is the formation of salts, where the potassium salt of 3,3′-diamino-2,2′,4,4′,6,6′-hexanitrodiphenylamine (HNDPA, m.p. = 334 °C) is a typical example. The second methodology is the introduction of amino groups into the structure of aromatic explosophore. For instance, the introduction of –NH2 groups into a nitrobenzene ring results in more thermostable energetic compounds such as 1,3-diamino-2,4,6-trinitro benzene (DATB, m.p. = 286 °C)14 and TATB. The third strategy is to form conjugations in the molecular structure, where the best example is hexanitrostilbene (HNS),15 whose heat resistance can be further enhanced by the introduction of amino groups.16 The fourth way is condensation with triazole ring/s, such as picryl-substituted and picrylamino-substituted 1,2,4-triazoles by condensing 1,2,4-triazole or amino-1,2,4-triazole with picryl.17 The insensitive energetic compounds 3-picrylamino-1,2,4-triazole (PATO) and 1,3-bis(1′,2′,4′-triazol-3′-ylamino)-2,4,6-trinitrobenzene (BTATNB) with a m.p. of 310 and 320 °C, respectively, are good representatives of this approach. By this strategy, very stable energetic compounds such as 4,4′-bis(1,2,4-triazol-3-ylamino)-2,2′,3,3′,5,5′,6,6′-octanitroazobenzene (BTDAONAB) and 5,5′-bis(2,4,6-trinitrophenyl)-2,2′-bi(1,3,4-oxadiazole) (TKX-55) were prepared (Scheme 1).18 These explosive molecules are symmetric and contain conjugation. BTDAONAB has the unique distinction of being the most thermostable explosive reported so far (with a broad exothermic peak at 550 °C),18a while TKX-55 normally starts to decompose at 335 °C.18b More recently, another thermostable energetic guanidine salt of 3-nitro-1-(2H-tetrazol-5-yl)-1H-1,2,4-triazol-5-amine (HANTT) was prepared, which has a decomposition peak temperature of 321 °C, with density (1.65 g cm−3) even lower than that of RDX.19 However, most of the abovementioned thermostable EMs have not been applied in the perforating guns due to difficult multi-step preparation and low synthetic yields. Recently, alternative superior ways to stabilize the energetic molecules have been developed, including the construction of energetic super structures such as co-crystals and coordination polymers.20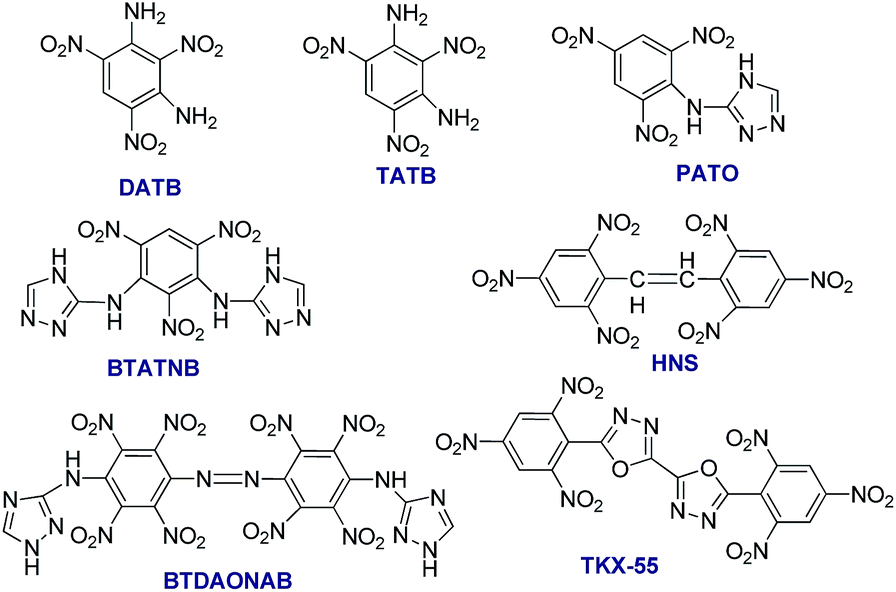 | ||
| Scheme 1 Typical examples of highly thermostable energetic compounds and their molecular structures. | ||
It has been also reported that graphene oxide (GO) could stabilize HMX and some other energetic materials, resulting in reduced sensitivity and improved mechanical strength.21 In addition to mechanical mixtures, GO was modified into several novel energetic supermolecules, because GO itself is energetic and can readily undergo fast exothermic decomposition.22
Typically, GO can be covalently functionalized with selected small molecules or polymers through activation and amidation/esterification reactions of either the carboxyl or hydroxyl functional groups on the GO surface.23 The GO carboxylic acid groups could be first activated by thionyl chloride (SOCl2) or by a mixture of 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS), followed by the coupling of amines, and the resulting functionalized products were found to be soluble in common organic solvents.24 The epoxy groups in GO nanomaterials can also be covalently functionalized with amines.25 Alternatively, the noncovalent functionalization of GO can also be accomplished via π–π stacking, cation–π, van der Waals interactions, or hydrogen bonding.26 In addition, GO has also shown strong oxidizing properties and could be used as an effective oxidant in a broad range of reactions.27
Many N-containing compounds are very good ligands in metal coordination chemistry. Among them, diaminoguanidine (DAG) and triaminoguanidine (TAG) are promising ligands, in protonated, deprotonated and neutral forms depending on their pH value, binding Cu(I) or Cu(II) as the typical metal centers.28 The complexes Cu(DAG)(NO3)2, Cu(DAG)2(NO3)2, and Cu(DAG)2(NO3)2·HNO3 have been reported, where the DAG ligand is coordinated in the first two compounds. For the latter complex, both the DAG molecule and the DAGH+ cation are coordinated,29 and it was also the case in the [Cu(DAGH)Cl3] complex.30 The reaction between Cu(NO3)2 and TAG in aqueous solution results in the formation of the Cu(TAG)(NO3)2 complex.29 The coordinated compounds containing protonated TAGH+ were prepared by the reaction of CuCl2 and TAG in concentrated HCl.31 However, the energetic properties of all the reported metal complexes of DAG and TAG were not explored.
In this work, we report the preparation of novel energetic coordination nanomaterials (CNMs) based on DAG and TAG functionalized GO, using the abovementioned strategies for GO functionalization. In particular, GO–TAG–Cu(I) and GO–TAG–Cu(II) CNMs were synthesized by the amidation process, followed by coordination reactions with CuCl or Cu(NO3)2. This method is facile and avoids the issues of multi-step preparation processes, typical to many other highly thermostable energetic molecules listed in Scheme 1. The first key process is to chemically graft DAG or TAG moieties onto GO flakes by EDC/NHS coupling. The second process is reacting the functionalized GO flakes (GO–TAG and GO–DAG) with metal ions (Fig. 1), where the resulting structures are supported by characterization of the mononuclear 4-coordinated TAG–Cu(II) complexes.
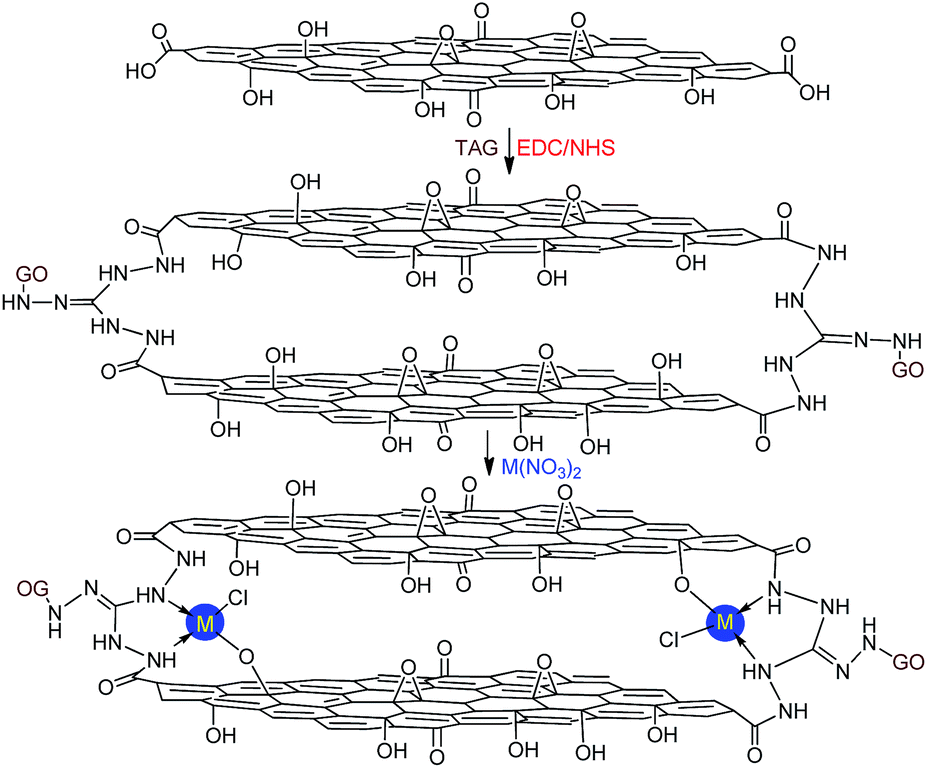 | ||
| Fig. 1 The GO–TAG–M (M = Cu2+, Ni2+, Co2+ and Fe2+) coordination nanomaterials prepared by the reaction of ammonized GO with corresponding metal nitrates: mononuclear coordination nanomaterials (CNMs) were formed based on the triaminoguanidine (TAG) ligand (ESI†). | ||
2 Experimental section
2.1. Materials
The commercially available graphene oxide (GO, 1–5 layers) was used as received from the market with an oxygen content of over 42%. Guanidine (99%), diaminoguanidine (98%), copper nitrate trihydrate (99%), nickel nitrate (99%), cuprous chloride (98%), cobalt nitrate (98%), iron(II) chloride tetrahydrate (99%) and hydrazine monohydrate (98%, N2H4 64–65%) were obtained from Sigma-Aldrich and stored under nitrogen to prevent oxidation. Ultrapure deionized water (resistivity > 18 MΩ) was obtained from a Milli-Q Biocel system. Whatman Anodisc™ membranes (0.2 μm pore size, 47 mm diameter) were used during filtration for support of fabricated papers.2.2. Preparation of GO–TAG
40 mg GO was dispersed in 60 mL H2O by high power ultra-sonication in a glass reactor. 10 mL aqueous mixed solution of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 8 mg) and N-hydroxysuccinimide (NHS, 6 mg) was added to the dispersed GO, and stirred for 30 min. A dark grey agglomeration was formed. Then 400 mg of triaminoguanidine hydrochloride (TAG·HCl) was added to the obtained mixture in the glass reactor, which should be kept in a water bath at a controlled temperature of 75–80 °C for four hours with magnetic stirring. Black flocculent precipitates were gradually formed, which were filtered and washed with distilled water three times. The filtered liquid has a pH value of 1.5–2.0 due to the presence of HCl molecules. The as-prepared product (231 mg) is the TAG functionalized GO via a bridge of –CO–NH–(GO–TAG).2.3. Preparation of GO–TAG-metal coordination nanomaterials
120 mg GO–TAG was dispersed in 30 mL H2O, and then 10 mL aqueous solution of a certain amount of metal salts can be added to the GO–TAG solution. The metal salts can be 200 mg of Cu(NO3)2, 199 mg of nickel acetate, 238 mg of Ni(NO3)2, 163 mg of FeCl2, 239 mg of Co(NO3)2, 121 mg of CuCl or a mixture of 100 mg Cu(NO3)2 with 50 mg CuCl. The mixture was reacted at a temperature of 75 °C for 5 h. The black precipitates were filtered and washed with H2O. The obtained filtered liquids have different pH values: 3.1 for Cu(NO3)2, 4.5 for nickel acetate, 3.8 for Ni(NO3)2, 1.6 for FeCl2, 3.7 for Co(NO3)2, 3.6 for CuCl and 3.5 for mixture of Cu(NO3)2 with CuCl. The as-prepared products are coordination nanomaterials: GO–TAG–Cu(II), GO–TAG–Ni(II), GO–TAG–Co(II), GO–TAG–Fe(II), GO–TAG–Cu(I) and GO–TAG–Cu(I)/Cu(II). Only the GO–TAG–Cu(II), GO–TAG–Cu(I) and GO–TAG–Cu(I)/Cu(II) are found to be highly thermostable EMs.3. Results and discussion
3.1. Chemical structure of the ECPs
Successful TAG functionalization was first confirmed by elemental analysis (EA) and SEM/EDS analysis (Fig. 2), with the compositions and formula listed in Table S1 in the ESI.† As fabricated, GO has a CGA/O ratio of 2.0, with a water content of less than 3.0 wt% after drying under vacuum for 48 h. In contrast, the water content for the GO–TAG was slightly increased to 5.3 wt%, while the CGA/O ratio was significantly increased to 4.6 due to the loss of H2O during the amidation of carboxyl groups by the TAG moiety. The newly functionalized GO–TAG has a NTAG/CGA ratio of 0.293. GO–TAG was further coordinated with copper ions.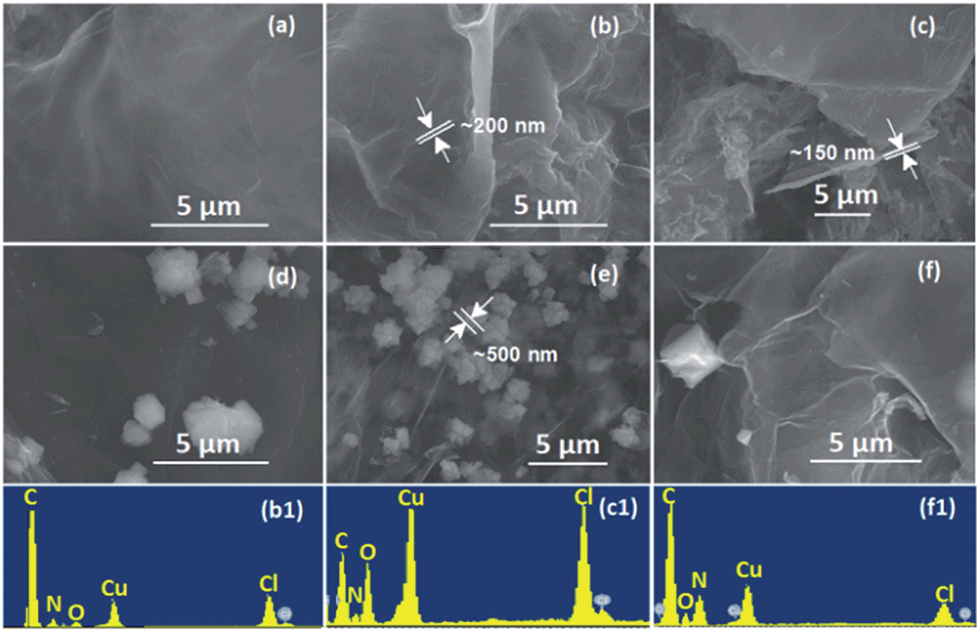 | ||
Fig. 2 The SEM images of energetic CNMs: (a) GO–TAG, (b) GO–TAG–Cu(II), (c) GO–TAG–Cu(I) and the GO–complex clusters (d) TAG–Cu(I), (e) TAG–Cu(II), (f) GO–TAG–Cu(II)/Cu(I), prepared by simultaneous dispersion of GO–TAG and precipitation of TAG with Cu(NO3)2, CuCl and their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 aqueous solution (molar ratio), respectively. The EDS surface elemental analyses of the CNMs (b, c and f) are also included (b1, c1 and f1), which reveal that GO–TAG–Cu(II) has an atomic C/O ratio of 4.6:1, and Cl/Cu ratio of about 6:1. It is due to the significantly reduced oxygen content, as compared to pristine GO that has an atomic C/O ratio of 2:1 (Fig. S1, ESI†). The atomic O/N ratio is about 3:4, where the N is due to the functionalization of TAG. The molar ratio of Cl/Cu in both GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I) materials is 1:1. :1 aqueous solution (molar ratio), respectively. The EDS surface elemental analyses of the CNMs (b, c and f) are also included (b1, c1 and f1), which reveal that GO–TAG–Cu(II) has an atomic C/O ratio of 4.6:1, and Cl/Cu ratio of about 6:1. It is due to the significantly reduced oxygen content, as compared to pristine GO that has an atomic C/O ratio of 2:1 (Fig. S1, ESI†). The atomic O/N ratio is about 3:4, where the N is due to the functionalization of TAG. The molar ratio of Cl/Cu in both GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I) materials is 1:1. | ||
It has been demonstrated that TAG·HCl readily reacts with copper dichloride in concentrated HCl, forming an energetic complex, whose crystal structure is composed of [Cu3(GO–TAGH)Cl3]+ cations, NO3− anions and water molecules.31 Our materials exhibit a similar coordination chemistry. By examining the morphologies of our CNMs by SEM and EDS (Fig. 2), it is clear that GO–TAG, GO–TAG–Cu(II) and GO–TAG–Cu(I) CNMs are homogeneous materials containing N and Cu atoms, indicating successful functionalization and coordination. The molar ratio of C/O/Cu is found to be very different for GO–TAG–Cu(II) and GO–TAG–Cu(I)/Cu(II) CNMs, while the ratio of Cu/Cl was maintained to be the same (1:1), which was also confirmed by XPS analysis of these materials. During functionalization, the GO flakes were linked together by the TAG moiety bridge, resulting in a much higher thickness (150–200 nm) as compared to 2–5 nm of stacked GO in 1–5 layers.24,25 The coordination of Cu(II) did not change the thickness of GO–TAG due to simple doping in the voids between the layers.
The crystal clusters of the TAG–Cu(I) and TAG–Cu(II) complexes were formed on the GO–TAG surfaces when extra TAG·HCl was added during the complexation reaction. The formed TAG–Cu(I) complex has smaller crystal sizes (<500 nm) as compared to TAG–Cu(II) complex crystals, with 1–4 μm in size. Interestingly, when the Cu(I) and Cu(II) ions reacted with GO–TAG in aqueous solution, several crystals were formed on the surface or between the layers of GO–TAG even without using extra TAG·HCl during complexation. It could be assumed that the reduction of GO–TAG occurred in the presence of Cu(I) ions as a reducing agent, which was then converted to Cu(II), as confirmed by XPS analysis (Fig. S3†). During the reduction, the oxygen functionalities on the surface of GO were almost completely removed and some of the TAG ligands were detached and coordinated with Cu(II), forming the crystals on the surface. To study the functionalization and the resulting disorder on the GO flakes for energetic GO–TAG–M(II) CNMs, we employed Raman spectroscopy (Fig. 3), and the corresponding peak parameters are summarized in Table S1.†
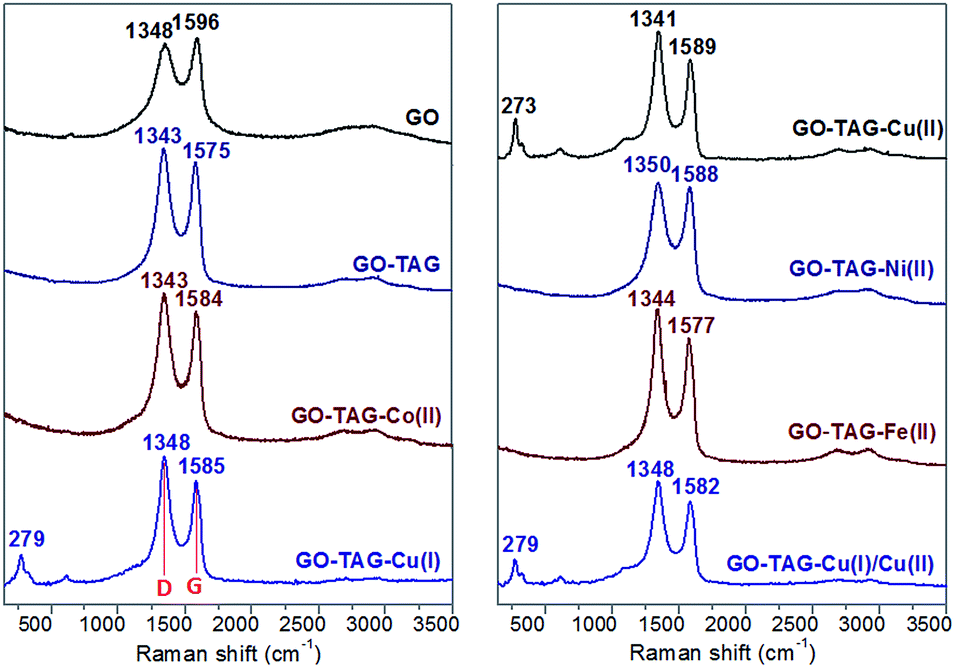 | ||
| Fig. 3 Raman spectra (using 3.0 mW laser) with D, G, and 2D peaks. The decreased D peak intensity of GO compared to TAG functionalized GO (GO–TAG) and its coordinated nanomaterials, indicates more structural defect sites on GO after functionalization and complexation. | ||
In general, a higher disorder in graphite leads to a broader G band, as well as to a broad D band, with relatively higher intensity as compared to the G band.32 The functionalization of GO with TAG and the corresponding metal ion coordination process slightly shift the peak position and largely change the intensity of both D and G bands (Table S1†). All the CNMs based on GO–TAG exhibit a similar couple of Raman-active bands, with the D band inbetween 1343–1350 cm−1, corresponding to a breathing mode of k-point photons of A1g symmetry and the D band at 1575–1589 cm−1, related to the first order scattering of the E2g phonon of sp2 in C atoms.32 The G band of natural graphite could be attributed to the in-phase vibration of the graphite lattice.33 After oxidation, the intensity of the D band is increased, and the G band of GO becomes much broader, owing to the presence of isolated double bonds that resonate at higher frequencies than that of graphite.32,33 Two bands could be observed in the samples, in a comparable D/G intensity ratio (ID/IG) to that of GO, suggesting that the skeleton structure of GO remains in GO–TAG after functionalization. With the evaporation of the solvent, abundant TAG groups bound to the edges of GO flakes could make GO–TAG nanosheets aggregate together as multi-layered platelets with significantly increased thickness (Fig. 2). The peaks at 273, 279 cm−1 and shoulder peaks correspond to Ag and Bg modes of Cu–O bonds, while the small bands at around 620 cm−1 are probably due to coordination bonds between N atoms in the TAG ligand and copper. However, there are no strong peaks observed for nickel(II), cobalt(II) and iron(II) CNMs, which is also the case for glycine functionalized GO during water purification.34 One could also notice that the ID/IG values for Ni- and Co-based nanomaterials (Table S1†) are very close to that of GO–TAG, due to partial van der Waals interactions between the metal ions and the TAG-grafted GO surfaces.
To further address the binding chemistry of GO–TAG with metal ions, in order to generate energetic CNMs, XPS was used. Fig. 4 presents C 1s and N 1s curve fitted spectra of GO before and after its functionalization. It shows that the functionalization of GO with the TAG moiety was successful, showing additional peaks for N-bonding (Fig. S3†). The C 1s and N 1s curve fitting also shows the presence of HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –HN–CO bonding components (288.9 and 400.9 eV, respectively). It enables direct quantification of the degree of GO functionalization with TAG.35 The GO surface oxygen content was reduced during the TAG functionalization and the following coordination processes, causing the disappearance/decrease of the C–O (or/and C–O–(C) peak, with simultaneous appearance of C–N, CN, –HN–CO and C–O–Cu peaks (Fig. 4e and f). Although C–O (or C–O–C) and CN peaks could be in a close proximity to each other, it is difficult to resolve the observed shift of peak binding energy in the C 1s structure from 286.6 eV (C–O or/and C–O–(C)) to 286.4 (CN). This finding is supported by the observation of the appearance of CN in N 1s spectra. All these data allow us to deduce that the covalent bonding of TAG–NH2 to GO–COOH, and the coordination reaction occurred for both GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I). Concurrently, four new peaks corresponding to C–N, CN, NHCO and C–O–Cu bonds appear in the C 1s XPS spectrum for GO–TAG–Cu(II) (Fig. 4b). These chemical bonding types are further confirmed by FTIR analysis (Fig. S4 and Table S3†).
O and –HN–CO bonding components (288.9 and 400.9 eV, respectively). It enables direct quantification of the degree of GO functionalization with TAG.35 The GO surface oxygen content was reduced during the TAG functionalization and the following coordination processes, causing the disappearance/decrease of the C–O (or/and C–O–(C) peak, with simultaneous appearance of C–N, CN, –HN–CO and C–O–Cu peaks (Fig. 4e and f). Although C–O (or C–O–C) and CN peaks could be in a close proximity to each other, it is difficult to resolve the observed shift of peak binding energy in the C 1s structure from 286.6 eV (C–O or/and C–O–(C)) to 286.4 (CN). This finding is supported by the observation of the appearance of CN in N 1s spectra. All these data allow us to deduce that the covalent bonding of TAG–NH2 to GO–COOH, and the coordination reaction occurred for both GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I). Concurrently, four new peaks corresponding to C–N, CN, NHCO and C–O–Cu bonds appear in the C 1s XPS spectrum for GO–TAG–Cu(II) (Fig. 4b). These chemical bonding types are further confirmed by FTIR analysis (Fig. S4 and Table S3†).
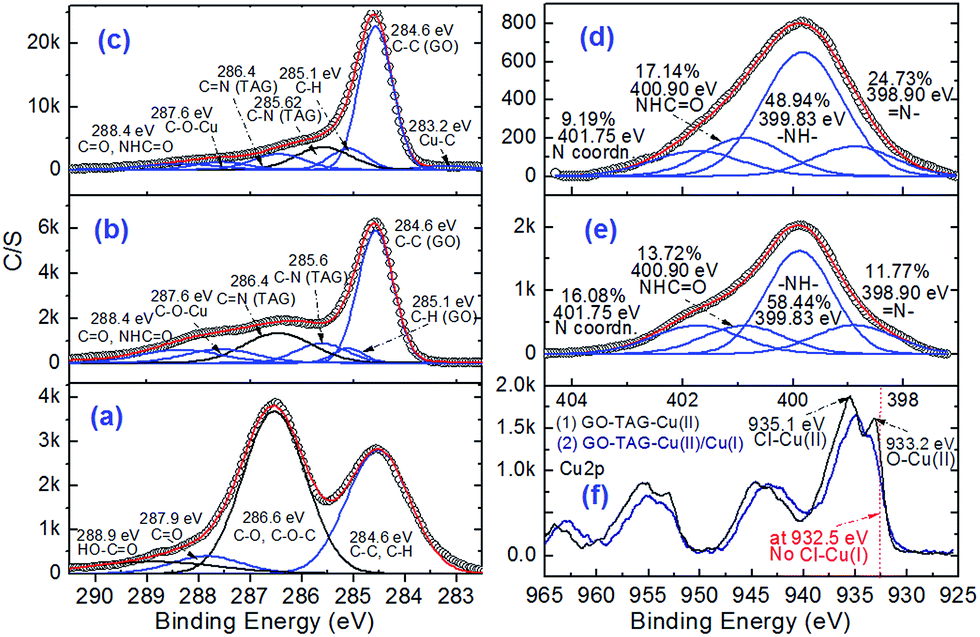 | ||
| Fig. 4 Deconvolution of the peaks in the XPS spectra for: C 1s peaks of (a) GO, (b) GO–TAG–Cu(II), and (c) GO–TAG–Cu(II)/Cu(I) and N 1s peaks of (d) GO–TAG–Cu(II) and (e) GO–TAG–Cu(II)/Cu(I), as well as the peaks of (f) Cu 2p for the latter two coordination nanomaterials. | ||
Fig. 5 shows XRD patterns of GO, GO–TAG based ECP materials, and the TAG–Cu(I) complex. The Miller indexing and corresponding plane distance data of our ECP materials are summarized in Table S5† (determined from PXRD spectra by using Jade software). As shown in Fig. 5a, the peak at 10.63° displays the characteristic peak of GO, corresponding to the Miller plane of 100 with a distance of 8.21 Å. The corresponding plane distances for the ECPs are smaller than GO, resulting in higher density (Table 1). The peaks at about 43.5° represent the peak of the graphite phase, with the atom Miller planes indexed as 202 for GO, 131 and 061 for GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I), respectively. If we compare the XRD patterns of GO-based ECP materials with pristine GO, the difference in the GO/graphite peak ratio is significant, indicating loss of oxygen-containing groups in GO planes during TAG functionalization, in agreement with the finding that there are more graphitic fractions of GO after the emulsion process.36 In the meantime, the peaks representing the GO planes in GO–TAG ECP materials shifted to higher angles. The second and third peaks in the curves (b) and (c) correspond to the Cu–Cl complexation structure in these ECPs, which are much different from those of the pure TAG–Cu(I) complex crystal. The latter complex has a space group of P21/c with cell parameters of a = 6.8581(4) Å; b = 3.6173(1) Å; c = 8.3926(2) Å; β = 98.765(5)°; α = γ = 90°; V = 189.42(3) Å3 with Z = 4. The XRD results further confirmed the successful synthesis of a new structure of GO–TAG ECPs, instead of a mixture of GO with the TAG–Cu(I) complex.
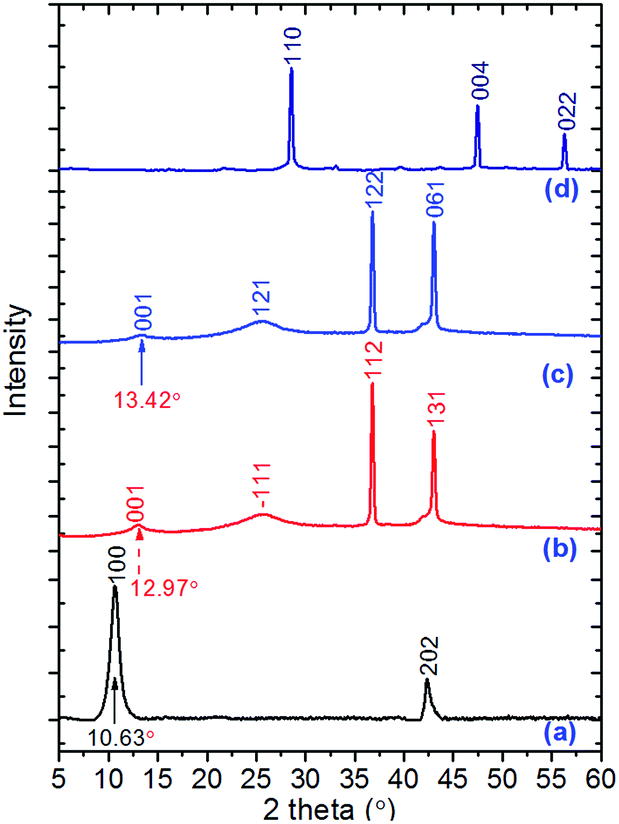 | ||
| Fig. 5 Experimental powder X-ray spectra of all TAG–Cu(I) and their GO-based ECPs: (a) pristine GO; (b) GO–TAG–Cu(II) ECP; (c) GO–TAG–Cu(II)/Cu(I) ECP; (d) TAG–Cu(I) complex. | ||
| Parameters | GO | TAG·HCl | TAG–Cu(I) | CNM(1) | CNM(2) | TNT | RDX |
|---|---|---|---|---|---|---|---|
| a Nitrogen content. b Decomposition peak temperature from DSC (β = 10 °C min−1). c The density was measured by Helium-Pycnometer, 25 °C. d Oxygen balance (for CaHbNcOd compounds, Ω = 1600 × (a − 2b − 0.5d)/Mw). e Calculated enthalpy of formation. f Detonation pressure. g Detonation velocity. h The pure GO is not detonable, and the detonation parameters were calculated for a mixture of 80 wt% of GO with 20 wt% of aluminum. i The detonation performances is compositions containing 40 wt% AP and 60 wt% of the CNMs; CNM(1) and CNM(1) represent GO–TAG–Cu(II) with formula of C41H16O9(C2H10N12)Cl1.5Cu1.5 and GO–TAG–Cu(II)/Cu(I) with formula of C42H13O8(CH5N6)ClCu, respectively. TAG–Cu(I) is the complex prepared in aqueous solution of copper nitrate with TAG·HCl under room temperature with a formulae of Cu2(CN6H4)Cl2·H2O, where Cu(II) ions in the starting materials was reduced to Cu(I). | |||||||
| M w [g mol−1] | 1027.7 | 140.6 | 332.1 | 1003.2 | 845.6 | 227.1 | 222.1 |
| % Na [wt%] | 0.91% | 59.78% | 25.31% | 16.80% | 10.08% | 18.50% | 37.84% |
| T p [°C] | 196.6 | 243.5 | 167.8 | 451.5 | 495.5 | 295.1 | 242.6 |
| ρ [g cm−1] | 1.91 | 1.55 | 3.01 | 2.59 | 3.12 | 1.65 | 1.80 |
| ΩCO2d [%] | −137.8% | −9.1% | −4.8% | −68.6% | −80.6% | −74% | −21.6% |
| ΔHfe [kJ mol−1] | −1869 | +200.2 | −604.2 | −2890.7 | −4486.5 | −59.4 | +83.8 |
| Impact [J] | >98 | 25.0 | 36.0 | >98.0 | 81.3 | 15.0 | 7.5 |
| Friction [N] | > 360 | — | >360 | >360 | >360 | >360 | 120 |
| P C–J [kbar] | 375h | 178 | 133 | 267i | 232i | 213 | 349 |
| VoDg [m s−1] | 3836h | 7198 | 3783 | 7815i | 7723i | 7304 | 8795 |
3.2. Thermal reactivity and stability
After coordination with metal ions, the hygroscopicity was substantially decreased, especially in the case of GO–TAG–Cu(II)/Cu(I) CNM due to the loss of oxygen functional groups of GO. There is a negligible mass loss for these two CNMs below 140 °C (Fig. 6c), which was likely due to the increase of their hydrophobicity caused by a decrease in the polar functionality on GO flakes.37 The observed rapid mass losses between 180–200 °C for GO and GO–TAG are due to the pyrolysis of the oxygen-containing functional groups. This feature accounts for 25% of the initial mass of GO, whereas it is about 21% for GO–TAG, resulting in a slightly higher CGO/O ratio, which is in good agreement with XPS and EDS elemental analysis. The TAG curve of GO–TAG also shows a slow mass-loss process in the temperature range of 310–430 °C in contrast to pristine GO, which is attributed to the detachment and following thermolysis of the TAG moiety, accompanied by a broad exothermic process (Fig. 6b).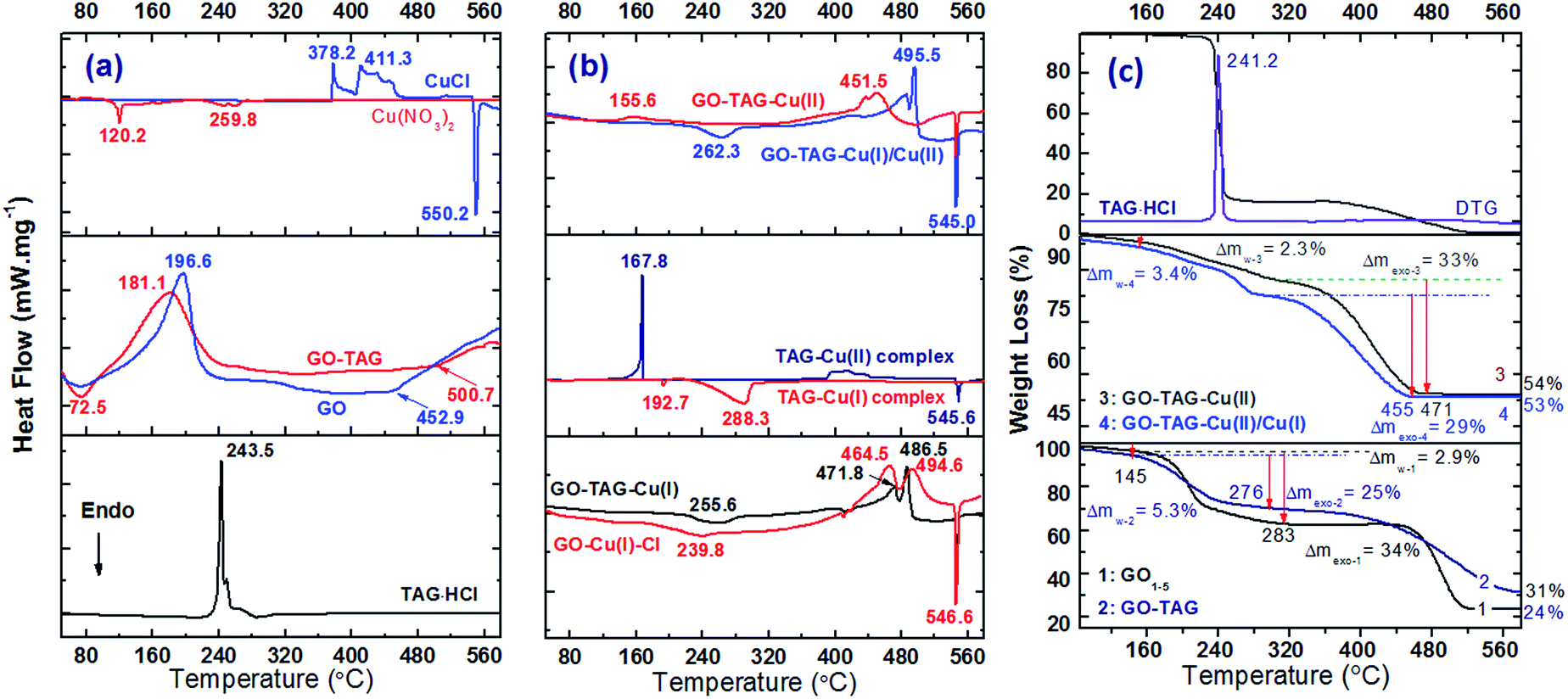 | ||
| Fig. 6 DSC (a, b) and TGA curves (c) of GO, GO–DAG, GO–TAG and their coordination complexes at a heating rate of 10 °C min−1. The TAG–Cu(II) complex was prepared at room temperature with the 1:1 molar ratio of the starting materials according to the literature31 (Tp = 167.8 °C), while GO–TAG–Cu(II) CNMs are much more stable with the exothermic peak temperature above 450 °C. The TGA experiments are in good agreement with the DSC findings, and the residues of the CNMs are over 53% in contrast to 24% for pristine GO, confirming the presence of copper in the GO–TAG–based CNMs. | ||
TAG·HCl decomposes at about 240 °C with a heat release of 2310 J g−1. For the GO–TAG–Cu(I) and GO–TAG–Cu(II)/Cu(I) CNMs, the endothermic peaks are observed at 255.6 and 262.3 °C, corresponding to the loss of water molecules upon reduction of GO. These H2O molecules contribute a mass loss of 4.1%, as shown in the TGA curve (Fig. 6c). The large exothermic peaks were covered with a heat release of 1792, 1192, and 847 J g−1 for GO–TAG–Cu(II), GO–TAG–Cu(I) and GO–TAG–Cu(II)/Cu(I), respectively. The lower heat releases of the latter two CNMs again correspond to the slow reduction of the oxygen functional groups by Cu(I). In comparison, the decomposition of nickel(II)-, cobalt(II)- and iron(II)-based materials show almost the same decomposition peak as GO–TAG in the temperature range of 170–200 °C (Fig. S5†), which further supports the partial physisorption of these metal ions on the GO–TAG surface. However, there is a weak exothermic peak for iron(II)-based materials, probably due to coordination with the TAG ligand. There is a sharp endothermic peak in all cases at about 545–547 °C, due to the phase transition of the CuAl2 alloy (Fig. S6†), which is also the case for the solid-state reaction of CuCl with metallic Al upon heating (Fig. 6a). We assume that CuCl and CuO are the main decomposition products of the described CNMs. These products could be further reduced in the presence of metallic Al to metallic copper upon heating. In the case of functionalized GO with DAG, further coordination was not so successful for all metals, although DAG could readily react with Cu(II) ions, forming an energetic complex with two exothermic peaks at above 250 °C (Fig. S5†).
3.3. Detonation performances and mechanical sensitivity
In order to predict the detonation performance of our new energetic CNMs, the EXPLO-5 software was used based on the experimental heat of formation (ΔHf) and density (ρ) data (Table 1). As shown in Table 1, pristine GO is not detonable due to low energy output and low achievable detonation temperature. When 20 wt% of high energy fuel Al was mixed with GO, the detonation velocity (VoD) was calculated to be 3836 m s−1, close to that of a blasting agent. EXPLO-5 could not calculate VoDs of our CNMs in a single phase due to their very high oxygen deficiency (<−68%), indicating that our CNMs are fuel-rich EMs, which need to be combined with oxidants such as ammonium perchlorate (AP). In a subsequent calculation, when 40 wt% of AP was added to our CNMs, the calculated VoDs for GO–TAG–Cu(II) and GO–TAG–Cu(II) are 7815 and 7723 m s−1, respectively. These VoD parameters are lower than that of 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX, 8750 m s−1 at a density of 1.76 g cm−3). The density of GO–TAG–Cu(II)/Cu(I) (3.14 g cm−3) is even higher than that of its parent TAG–Cu(II) complex. It is also significantly higher than that of pristine GO and GO–TAG–Cu(II), due to the largely reduced oxygen functional groups on the surface of GO. More importantly, as highly thermostable EMs, our CNMs have much higher VoDs than the most commonly used highly thermostable insensitive EMs, including HNS (7000 m s−1) and TATB (7350 m s−1). Our CNMs are also different in their sensitivity to impact due to the change in the amount of oxygen functional groups. GO–TAG–Cu(II)/Cu(I) is more sensitive than GO–TAG–Cu(II), but both of them could be clearly classified as insensitive EMs with excellent impact sensitivity, which is much lower than that of TAG·HCl and the TAG–Cu(I) complex (Table 1). We believe that our CNMs have a potential to be used as energetic catalysts for solid rocket propellants due to the presence of copper. Moreover, because GO could be fully consumed by oxidation during high temperature combustion of propellants with improved oxygen balance, our CNMs can contribute to the fuel energy density. In fact, the functionalized GO was found to be a promising catalyst for the combustion of nitromethane,38 and the presence of nucleophilic oxygen-containing functionalities on the surfaces and edges of our CNMs could be also chemically active sites with catalytic behavior for the combustion of solid propellants.394 Conclusions
The EDC/NHS coupling agent can be used for the functionalization of GO with the TAG moiety, and further coordination reaction with metal ions leads to the formation of highly thermostable CNMs. These CNMs are very oxygen deficient (<−68%) and should be combined with oxidizers for applications such as explosive charges and as combustion catalysts for solid propellants. Our CNMs GO–TAG–Cu(II) and GO–TAG–Cu(II)/Cu(I) have VoDs of 7815 and 7723 m s−1, respectively, when 40% of AP was added as the oxidant. More importantly, our CNMs are very insensitive (Im > 81 J) and thermostable up to more than 450 °C, which is much higher than that of TATB and HNS.Conflict of interest
The authors declare no competing financial interests.Abbreviations
| VoD | Detonation velocity. |
| EMs | Energetic materials. |
| CNMs | Coordination nanomaterials. |
| GO | Graphene oxide. |
Acknowledgements
The financial support from the Planning and Budgeting Committee (PBC) of the Council for Higher Education in Israeli government is greatly appreciated. This work was also partially supported by the Center for Nanoscience and Nanotechnology and by the Faculty of Exact Sciences in Tel Aviv University.References
- J. P. Agrawal, High Energy Materials, Propellants Explos. Pyrotech, Wiley-VCH, Germany, 2010 Search PubMed.
- M. B. Talawar, S. K. Jangid, T. Nath, R. K. Sinha and S. N. Asthana, J. Hazard. Mater., 2015, 300, 307–321 CrossRef CAS PubMed.
- (a) B. Gaur, B. Lochab, V. Choudhary and I. K. Varma, J. Macromol. Sci., Polym. Rev., 2003, 43, 505–545 CrossRef; (b) Q.-L. Yan, S. Zeman and A. Elbeih, Thermochim. Acta, 2012, 537, 1–12 CrossRef CAS.
- (a) J. Zhang, Q. Zhang, T. T. Vo, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 1697–1704 CrossRef CAS PubMed; (b) J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437–4445 CrossRef CAS PubMed; (c) P. Yin, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 4778–4786 CrossRef CAS PubMed.
- (a) H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377 CrossRef CAS PubMed; (b) J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 10532–10535 CrossRef CAS PubMed; (c) Y.-L. Wang, F.-Q. Zhao, Y.-P. Ji, Q.-L. Yan, J.-H. Yi and S.-Y. Xu, et al. , J. Anal. Appl. Pyrolysis, 2014, 105, 295–300 CrossRef CAS.
- (a) Y. Li, J. Hao, H. Liu, S. Lu and J. S. Tse, Phys. Rev. Lett., 2015, 115, 105502 CrossRef PubMed; (b) A. A. Dippold and T. M. Klapötke, J. Am. Chem. Soc., 2013, 135, 9931–9938 CrossRef CAS PubMed; (c) T. M. Klapötke and T. G. Witkowski, Propellants, Explos., Pyrotech., 2015, 40, 366–373 CrossRef.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584–3601 CrossRef CAS PubMed.
- M. Comet, G. Vidick, F. Schnell, Y. Suma, B. Baps and D. Spitzer, Angew. Chem., Int. Ed., 2015, 54, 4458–4462 CrossRef CAS PubMed.
- (a) K. Liu, G. Zhang, J. Luan, Z. Chen, P. Su and Y. Shu, J. Mol. Struct., 2016, 1110, 91–96 CrossRef CAS; (b) J. C. Bennion, A. McBain, S. F. Son and A. J. Matzger, Cryst. Growth Des., 2015, 15, 2545 CrossRef CAS.
- (a) B. Tan, M. Huang, X. Long, J. Li, X. Yuan and R. Xu, Int. J. Quantum Chem., 2015, 115, 84–89 CrossRef CAS; (b) C. Knapp and J. Passmore, Angew. Chem., Int. Ed., 2004, 43, 4834–4836 CrossRef CAS PubMed; (c) M. T. Nguyen, Coord. Chem. Rev., 2003, 244, 93–113 CrossRef CAS.
- G. Sun, X. Wang, F. Sun and Y. Liang, Explos. Mater., 2002, 31, 4 CAS.
- T. R. Gibbs and A. Popolato, LASL Explosive Property Data, University of California Press, Los Angeles, 1980, vol. 38, p. 157 Search PubMed.
- A. J. Bellamy, S. J. Ward and P. Golding, Propellants, Explos., Pyrotech., 2002, 27, 49–58 CrossRef CAS.
- S. Zeman, Thermochim. Acta, 1993, 216, 157–168 CrossRef CAS.
- (a) C. X. Lu, Kogyo Kayaku, 1990, 51, 275–279 CAS; (b) K. G. Shipp, P. Golding and G. F. Hayes, Propellants Explos., 1979, 4, 115–120 CrossRef.
- J. P. Agrawal, Cent. Eur. J. Energ. Mater., 2012, 9, 273–290 CAS.
- (a) J. Li, B. Chen and Z. Ou, Proc. 17th Int. Pyrotech. Seminar, Beijing lnst. Tech. Press, Beijing, vol. 1, 1991, pp. 196–199 Search PubMed; (b) J. P. Agrawal, M. U. S. Prasad and R. N. Surve, New J. Chem., 2000, 24, 583–585 RSC.
- (a) S. N. Mehilal, A. K. Sikder and J. P. Agrawal, Indian J. Eng. Mater. Sci., 2004, 11, 516–520 Search PubMed; (b) T. M. Klapötke and T. Witkowski, ChemPlusChem, 2016, 81, 357–360 CrossRef.
- C. Bian, M. Zhang, C. Li and Z. Zhou, J. Mater. Chem. A, 2015, 3, 163–169 CAS.
- J. P. Agrawal, M. R. N. Surve, and V. K. Bapat, et.al., HEMRL Report No. HEMRL/99/6, 1999 Search PubMed.
- (a) R. Li, J. Wang, J. P. Shen, C. Hua and G. C. Yang, Propellants, Explos., Pyrotech., 2013, 38, 798–804 CrossRef CAS; (b) P. D. McCrary, P. A. Beasley, S. A. Alaniz, C. S. Griggs, R. M. Frazier and R. D. Rogers, Angew. Chem., Int. Ed., 2012, 51, 9784–9787 CrossRef CAS PubMed; (c) X. Zhang, W. M. Hikal, Y. Zhang, S. K. Bhattacharia, L. Li and S. Panditrao, et al. , Appl. Phys. Lett., 2013, 102, 141905 CrossRef; (d) Y. Zhang, Z. Shao, K. Gao, X. Wu and Y. Liu, Integr. Ferroelectr., 2014, 154, 147–153 CrossRef CAS.
- (a) Q.-L. Yan, M. Gozin, F.-Q. Zhao, A. Cohen and S.-P. Pang, Nanoscale, 2016, 8, 4799–4851 RSC; (b) V. Alain-Rizzo, L. Galmiche, P. Audebert, F. Miomandre, G. Louarn and Bozlar, et al. , Chem. Mater., 2015, 27, 4298–4310 CrossRef; (c) A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- (a) S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS PubMed; (b) J. Azevedo, L. Fillaud, C. Bourdillon, J.-M. Noël and F. Kanoufi, et al. , J. Am. Chem. Soc., 2014, 136, 4833–4836 CrossRef CAS PubMed.
- (a) B. Wang, B. Luo, M. Liang, A. Wang, J. Wang and Y. Fang, et al. , Nanoscale, 2011, 3, 5059–5066 RSC; (b) J. Hwang, T. Yoon, S. H. Jin, J. Lee, T.-S. Kim, S. H. Hong and S. Jeon, Adv. Mater., 2013, 25, 6724–6729 CrossRef CAS PubMed; (c) O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892–896 CrossRef CAS PubMed.
- H. F. Yang, C. S. Shan, F. H. Li, D. X. Han, Q. X. Zhang and L. Niu, Chem. Commun., 2009, 3880 RSC.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785 CrossRef CAS PubMed.
- H. P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011, 67, 4431 CrossRef CAS.
- G. R. Andrade, J. Kunsminskas, L. Pizzuti, A. Dos Anjos, S. D. Inglez, B. Tirloni and P. H. Suegama, Inorg. Chem. Commun., 2015, 61, 210–213 CrossRef CAS.
- Z. A. Saveleva, S. V. Larionov and V. N. Ikorskii, Izvestiya SO RAN SSSR, Ser. Khim. Nauk., 1982, 2, 89 Search PubMed.
- Z. A. Saveleva, S. V. Larionov, G. V. Romanenko, N. V. Podberezskaya, O. V. Shishkin and Y. T. Struchkov, Zh. Strukt. Khim., 1995, 36, 936 CAS.
- A. Z. Saveleva, V. G. Romanenko, A. L. Sheludyakova and V. S. Larionov, Polyhedron, 2000, 19, 1737–1740 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prudhomme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- D. D. Zhang, S. Z. Zu and B. H. Han, Carbon, 2009, 47, 2993–3000 CrossRef CAS.
- (a) F. Najafi, O. Moradi, M. Rajabi, M. Asif, I. Tyagi, S. Agarwal and V. K. Gupta, J. Mol. Liq., 2015, 208, 106–113 CrossRef CAS; (b) F. Fang, L. Kong, J. Huang, S. Wu, K. Zhang and J. Wang, et al. , J. Hazard. Mater., 2014, 270, 1–10 CrossRef CAS PubMed.
- (a) N. L. V. Carreño, M. T. Escote, A. Valentini, L. McCafferty, V. Stolojan and Beliatis, et al. , Nanoscale, 2015, 7, 17441–17449 RSC; (b) A. Y. S. Eng, C. K. Chua and M. Pumera, Nanoscale, 2015, 7, 20256–20266 RSC.
- H. V. Kumar, S. J. Woltornist and D. H. Adamson, Carbon, 2016, 98, 491–495 CrossRef CAS.
- (a) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes and Y. Jia, et al. , Carbon, 2007, 45, 1558–1565 CrossRef CAS; (b) O. C. Compton, B. Jain, D. A. Dikin, A. Abouimrane, K. Amine and S. T. Nguyen, ACS Nano, 2011, 5, 4380–4391 CrossRef CAS PubMed.
- J. L. Sabourin, D. M. Dabbs, R. A. Yetter, F. L. Dryer and I. A. Aksay, ACS Nano, 2009, 3, 3945–3954 CrossRef CAS PubMed.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301–306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Descriptions of the experimental details, characterization techniques; figures on the current status of oil extraction perforator charges, XPS spectra and the curves for Cu 2p peaks, SEM images of GO and corresponding EDS results, FTIR of the explored materials, DSC curves of physical mixtures of GO–TAG/Fe(II), GO–TAG/Co(II) and GO–TAG/Ni(II), and a Al–Cu phase diagram; tables on the summary of Raman peak parameters, elemental analysis, FTIR and peak assignments and XPS bonding information. See DOI: 10.1039/c6ta03510h |
| This journal is © The Royal Society of Chemistry 2016 |