
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of Co-adsorbates on CO2 induced phase transition in functionalized pillared-layered metal–organic frameworks†
Andreas
Schneemann
a,
Yukiko
Takahashi
b,
Robin
Rudolf
a,
Shin-ichiro
Noro
*b and
Roland A.
Fischer
*cd
aChair of Inorganic Chemistry II, Ruhr-University Bochum, Universitätsstr. 150, 44801 Bochum, Germany
bResearch Institute for Electronic Science, Hokkaido University, Sapporo 001-0020, Japan. E-mail: noro@es.hokudai.ac.jp
cChair of Inorganic and Metal-Organic Chemistry, Department of Chemistry, Technical University Munich, Lichtenbergstraße 4, D-85748 Garching, Germany. E-mail: Roland.Fischer@tum.de
dCatalysis Research Centre, Technical University Munich, Ernst-Otto-Fischer Straße 1, 85748 Garching, Germany
First published on 25th July 2016
Abstract
Most studies on flexible MOFs which suggest good separation properties of the framework are solely based on single component isotherms and conclusions drawn from these. However, many factors are not considered, particularly the change of the pore space after the opening of the framework can have a distinct effect on the adsorption of a second gas present in a real separation problem that does not induce the phase transition of the material. Within this study, we focus on a series of flexible pillared-layered MOFs of the type [Zn2(fu-bdc)2(dabco)]n bearing flexible side chains and analyze the gas adsorption behavior when the material is exposed to mixtures of CO2 with other adsorptives, including N2, CH4, C2H6 and C3H8, to evaluate the influence of the co-adsorbate on the sorption selectivity and phase transition under these conditions.
Introduction
Among the porous materials, metal–organic frameworks are a class that has gathered a lot of attention in recent years, owing to their outstanding properties.1 They are built up by the combination of metal clusters and multitopic organic linkers i.e. aromatic carboxylates or N-donor ligands.2 The reasons for the high interest in this class of materials arise from their ultrahigh surface areas, their high degree of tunability and their structural flexibility. By combining appropriate linker molecules and metal clusters, record holding materials have been prepared with unprecedented pore volumes and surface areas.3 The aspect of tunability of material properties leads to new tailor-made materials and ranges from simple linker functionalization to more complex functionalization and tuning principles such as defect engineering,4 postsynthetic exchange/incorporation of metals/linkers,5 the precise assembly of different building blocks per pore,6 or postsynthetic metalation of metal sites by atomic layer deposition.7 Structural flexibility describes the possibility of certain frameworks to undergo a reversible phase transition between at least two distinct states.8 The transition between these two states can be initiated by different stimuli, including guest ad/desorption,9 temperature,10 light,11 or mechanical pressure.12 Frameworks that can undergo this kind of transition are denoted as flexible MOFs, 3rd generation MOFs or soft porous crystals in the literature.8b,13 The ability to switch depending on an external stimulus between different states makes flexible MOFs amenable for a handful of potential applications, including chemical sensing,14 as dampers,15 drug release16 or gas separation and storage.17In our study we are focusing on several issues regarding gas separation problems in flexible MOFs, using as an example functionalized derivatives of the well-known compound [Zn2(bdc)2(dabco)]n (bdc = 1,4-benzenedicarboxylate, dabco = 1,4-diazabicyclo[2.2.2]octane).18 These so-called fu-MOFs feature alkoxy sidechains (fu) pinned to the bdc linker to yield fu-bdc (2,5-functionalized-1,4-benzenedicarboxylate). These fu-MOFs are built up from Zn2 paddle-wheels which are fourfold coordinated by fu-bdc linkers, thus generating a [Zn2(fu-bdc)2]n 2D sheet (Fig. 1). The paddle wheels are coordinated in the axial position by dabco pillars, connecting in this fashion the adjacent layers and forming a 3D structure. In the parent material with bdc as the linker only small structural changes are observed (i.e. bending of the linker molecules) when the as-synthesized DMF containing material is desolvated. In contrast, for the functionalized derivatives a large pore contraction is found after desolvation.19 This contraction is mostly initiated by interactions of the solvent-like side chains. The solvated state of the material is also referred to as the large pore (lp) form and the contracted state after solvent removal is termed as the narrow pore (np) form. Interestingly, strong sorption selectivities are initiated by the right choice of fu e.g. for C2H2 over C2H4 when the material is present in the np form.20 Sorption selectivity can also be observed in related materials bearing the same fu-bdc linkers but with a different topology.21 Notably, during CO2 adsorption a transition from the np to the lp phase is detected, for the functionalized pillared-layered MOFs.22Fig. 1(e) shows the shape of a CO2 adsorption isotherm typically observed for these kinds of fu-MOFs (pink dotted line). At low pressures the material takes up a small portion of CO2. When a certain threshold pressure is surpassed, the pore opens which is indicated by a drastic increase of the CO2 uptake in the material. The threshold pressure of the phase transition can be modulated by the choice of the fu-bdc linker and can be further tuned by the preparation of mixed linker MOFs.23 Only very little N2 uptake is monitored for these materials and the common N2 isotherm shape is also depicted in Fig. 1(e) (green dotted line). For many flexible and guest responsive MOFs this kind of situation is observed for single component adsorption: pore opening at threshold pressure p for gas A and no pore opening for gas B. After the pore opening by A, a steep increase of the adsorption of A is observed, while the uptake of B remains low over the whole pressure range of the adsorption isotherm. From the data obtained from such single component adsorption experiments, it is usually deduced, that the material can separate a gas mixture consisting of the two components A and B. However, the entire pore surface of the flexible MOF is changed after gas A induced the np → lp phase transition in the material and hence two different scenarios are imaginable (Fig. 1(f)):
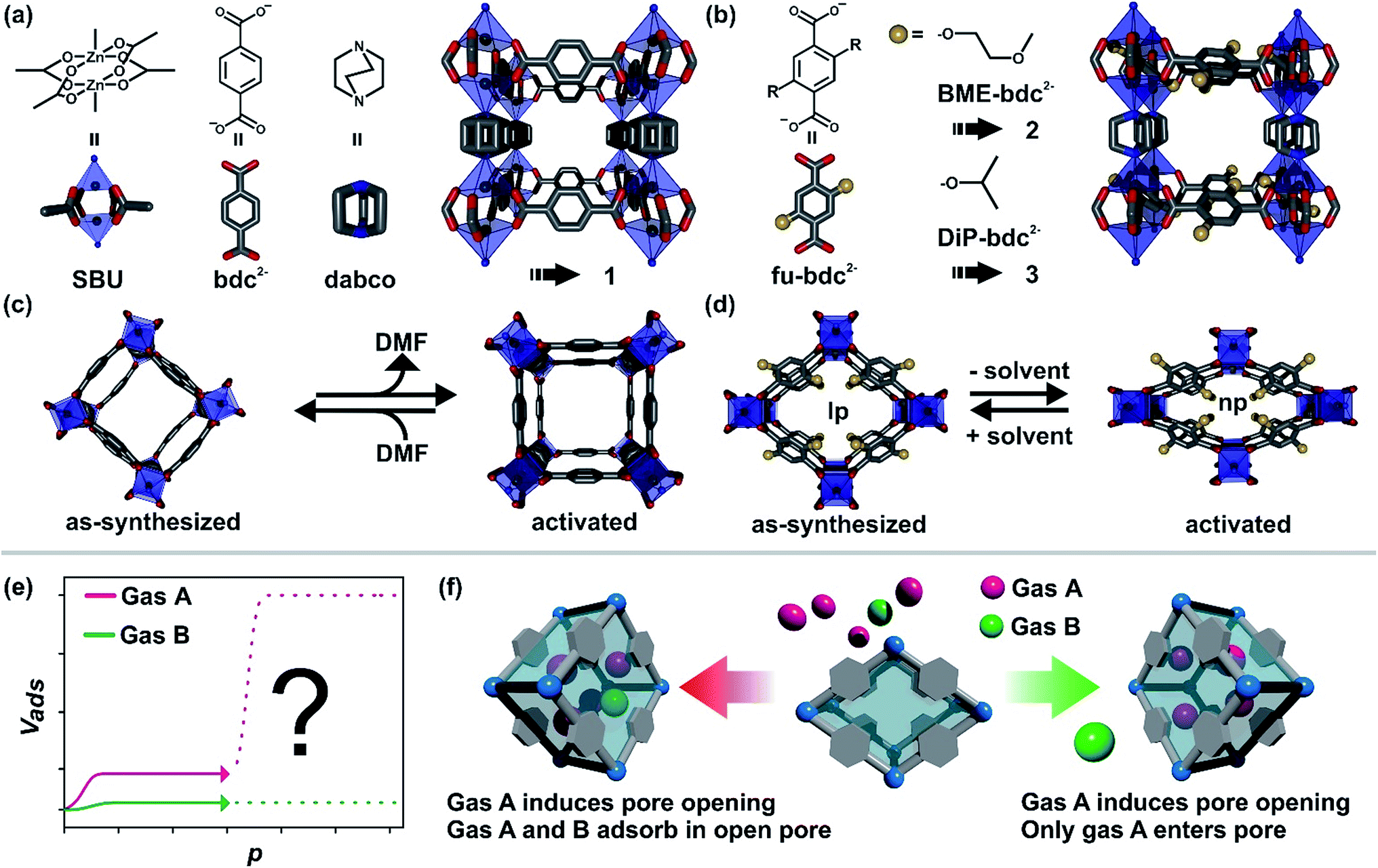 | ||
| Fig. 1 (a) Schematic illustration of the building blocks of the pillared layered framework [Zn2(bdc)2(dabco)]n. (b) Depiction of the functionalized derivatives 2 and 3 of the type [Zn2(fu-bdc)2(dabco)]n. (c) Scheme of the structural flexibility of 1 after solvation/desolvation with DMF and (d) of 2 and 3 after solvation/desolvation. Carbon atoms are depicted in grey, nitrogen in blue and oxygen in red, respectively. Coordination environment around zinc atoms is represented by blue polyhedra and functional groups by yellow spheres. (e) Illustration of the adsorption isotherm when two gases are introduced to a flexible MOF. (f) Illustration of two scenarios that can occur when a mixture of gas A and gas B is introduced to a flexible MOF, whereas gas A induces the pore opening and gas B does not. | ||
(a) Gas A opens the pore and is adsorbed inside the framework and gas B remains outside.
(b) Gas A opens the pore and is adsorbed inside the framework and gas B is co-adsorbed with gas A in the lp.
The first scenario would be necessary for a good separation of two compounds. The other scenario, i.e. the transport of species B into the framework opened by gas A, is less desirable and would reduce the sorption selectivity.
With our study presented herein, we wish to address these issues mentioned above. In particular, we want to pursue the question of what happens if the material is exposed to two different gases that both initiate a phase transition (i.e. np → lp). [Zn2(bdc)2(dabco)]n (1) as the non-functionalized and essentially rigid reference material and the two functionalized and responsive fu-MOFs [Zn2(BME-bdc)2(dabco)]n (H2BME-bdc = 2,5-bis(2-methoxyethoxy)-1,4-benzenedicarboxylic acid) (2) and [Zn2(DiP-bdc)2(dabco)]n (H2DiP-bdc = 2,5-diisopropoxy-1,4-benzenedicarboxylic acid) (3) were employed (Fig. 2). A series of co-adsorption experiments was conducted using CO2 (gas A) and a selection of gases B such as N2, CH4, C2H6 and C3H8 which feature different pore opening properties. The results gathered here should be transferable to other flexible MOF materials showing promising adsorption isotherms for gas separation applications.
 | ||
| Fig. 2 Illustration of the organic linker molecules used for the synthesis of the MOF materials 1–3. | ||
Experimental section
All materials were bought from commercial suppliers, such as Sigma Aldrich, TCI and Alfa Aesar and used without further purification unless otherwise noted.Linker synthesis
The functionalized linkers H2BME-bdc and H2DiP-bdc used for the preparation of fu-MOFs [Zn2(fu-bdc)2(dabco)]n were prepared via Williamson ether synthesis from dimethyl 2,5-dihydroxy-1,4-benzenedicarboxylate. A detailed description of the synthetic route to obtain the organic precursors and the linker molecules can be found elsewhere.19b,20MOF synthesis
Compounds 1–3 were prepared under solvothermal reaction conditions only slightly modified from already reported synthesis methods.18,19b[Zn2(bdc)2(dabco)]n (1), [Zn2(BME-bdc)2(dabco)]n (2), [Zn2(DiP-bdc)2(dabco)]n (3)
Zn(NO3)2·6H2O (1.33 g; 4.5 mmol), H2bdc/H2fu-bdc (4.5 mmol) and dabco (270 mg; 2.4 mmol) were put in a beaker and 100 ml DMF were added. The mixture was sonicated until the reagents were dissolved. Afterwards, the synthesis solution was divided into three equal portions and the portions were put into three individual 50 ml screw top jar. The jars filled with the solutions were then placed in a preheated oven and left for 24 hours at 120 °C. The oven temperature was then reduced to room temperature. A white, microcrystalline precipitate formed in all three cases (off-white in the case of materials 2 and 3). The solvent from the synthesis was replaced by fresh DMF and a stirring bar was added to each vessel. The mixture was vigorously stirred for 30 minutes, in order to assure a good solvent exchange and was afterwards left to settle for 24 hours. The solvent was removed by decantation and exchanged with CHCl3 and again stirred for 30 minutes and left to settle for 24 hours. This process was repeated a second time, also with CHCl3. The three identical batches were combined. Now, the material was filtered and washed three times with 15–20 ml CHCl3, transferred to a Schlenk tube, and dried in vacuo (oil pump) for 24 hours at elevated temperature (120 °C). The purity of the isolated compounds was controlled by measuring 1H-NMR spectra (after sample digestion) and PXRD patterns (see ESI†). Yield (based on H2bdc/H2fu-bdc): 1 – 79% (1.03 g, 1.8 mmol), 2 – 56% (1.09 g, 1.26 mmol), and 3 – 63% (1.15 g, 1.42 mmol).Adsorption measurements
The single component adsorption isotherms and co-adsorption measurements were performed using a Belsorp VC apparatus (MicrotracBEL Corp.) coupled with an Agilent 490 Micro gas chromatographic system equipped with a thermal conductivity detector. For each measurement approximately 1 g of sample was used. The MOF was placed in the sample cell, which was sealed with a metal O-ring and connected to the instrument. The sample cell is located within the isothermal part of the Belsorp VC and the temperature within this area can be precisely controlled. The lowest stable temperature was at 278 K and hence all measurements were performed at this temperature. Prior to each measurement, the samples were evacuated at elevated temperatures (120 °C) overnight (first measurement on sample) or at least for three hours (each additional measurement). The single component adsorption isotherms were measured volumetrically. For the co-adsorption measurements, a pre-defined gas mixture with distinct partial pressures for each component was introduced into the manifold of the instrument. In the manifold the two gases are mixed for 2200 seconds and dosed onto the sample. The gas phase was then allowed to equilibrate until the pressure variation was minimized. Afterwards, the total uptake was determined volumetrically. Subsequently a small amount of the gas phase remaining in the manifold was removed and the composition was analyzed by GC (gas chromatography). From the composition of the gas phase not adsorbed on the sample, the exact adsorbed portions of each component of the mixture on the sample were calculated. The process of sampling the gas above the sample followed by equilibration (until pressure variation is minimal) was repeated five times, thus measuring five data points with relatively similar total gas pressure and uptake.Calculation of selectivity coefficients
The co-adsorption measurements provided experimental values for the molar fraction of both components remaining in the gas phase (yi) above the sample and adsorbed on the sample (xi) and hence the selectivity coefficients were calculated by the equation: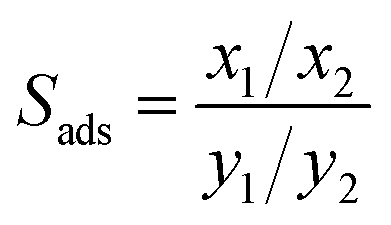
General methods
Additional data including PXRDs of the as-synthesized and activated MOFs, IR spectra, TG and NMR spectra of compounds 1–3 are given in the ESI.†Results and discussion
In the following paragraph, all co-adsorption experiments, measurements and data are represented in a similar manner. Three different data sets were acquired in each experiment. The total uptake was determined volumetrically and is always depicted as green diamonds in the figures. The CO2 uptake (determined by GC as described in the experimental part) is indicated by red diamonds. The co-adsorbate is depicted by a differently colored diamond (N2, CH4, C2H6 and C3H8 are shown in blue, black, turquoise and orange, respectively). Five measurements were undertaken during each co-adsorption experiment with the selected gas mixtures. The black arrows point from the first to the fifth measurement point. In addition, the relevant single component adsorption isotherms are plotted in the same diagrams for better understanding and comparison of the effect of the simultaneous presence of the two components.[Zn2(bdc)2(dabco)]n (1)
The first material under study, in order to test the experimental set-up and measurement conditions, was the parent MOF [Zn2(bdc)2(dabco)]n and the respective single component adsorption isotherms for N2, CO2, CH4, C2H6 C3H8 at 278 K are shown in Fig. 3(a). The measurements were undertaken in the range from 0 to 3200 kPa (C2H6 and C3H8 adsorptions were measured in a smaller range, due to their lower condensation pressures). It is directly visible that the adsorbed amount of CO2 on 1 is much higher than for the other gases. The maximum uptakes are for CO2, 330 cm3(STP) per g (1624 kPa); for N2, 107 cm3(STP) per g (3100 kPa); for CH4, 141 cm3(STP) per g (3050 kPa); for C2H6, 127 cm3(STP) per g (1047 kPa) and for C3H8, 95 cm3(STP) per g (189 kPa). The shapes of the isotherms do not indicate gate opening behaviour, since for all adsorbed gases no steps are visible in the adsorption isotherm. In fact, this is in good accordance with the expectations, since the parent material 1 only undergoes small structural changes upon guest loading. Fig. 1 highlights the slight structural flexibility upon ad/desorption of the solvent DMF inside the pores of 1. | ||
| Fig. 3 Depiction of the excess single component and co-adsorption measurements of material 1. N2, CO2, CH4, C2H6 and C3H8 single component isotherms are blue, red, black, turquoise and orange, respectively. CO2, N2 and CH4 fractions of the co-adsorption experiments are shown in red, blue and black. Total adsorption is shown in green. | ||
Material 1 was chosen as a reference for comparison and will be also used to explain and evaluate the co-adsorption data. Fig. 3(b) shows the co-adsorption of a mixture of N2 and CO2. The applied mixture has a total gas pressure of 925 kPa. The partial pressures of the two components amount to 513 and 421 kPa of N2 and CO2, respectively. At the applied pressures barely any N2 from the mixture is adsorbed in the pore (below detection limit). Moreover, 274 cm3(STP) per g CO2 are taken up, which coincides nicely with the single component adsorption isotherm at the same pressure. During the CO2/CH4 measurement, the measured pressure points are not overlaying with the single component gas adsorption isotherms as nicely as for the CO2/N2 mixture. During the measurement a total gas pressure of 1324 kPa was applied to the sample, containing 640 kPa CO2 and 713 kPa CH4. A total amount of 169.9 cm3(STP) per g of CO2 is adsorbed which is distinctively less than for the single component adsorption isotherm at the same pressure. From the mixture a total of 27.7 cm3(STP) per g CH4 are adsorbed.
[Zn2(BME-bdc)2(dabco)]n (2)
Among the flexible fu-MOF materials that have been introduced by Fischer and co-workers over the recent years, [Zn2(BME-bdc)2(dabco)]n has been the most studied material.10c,19,22 The material undergoes a np → lp phase transition when the desolvated np phase is exposed to suitable guest molecules (Fig. 1). In Fig. 4 the single component gas adsorption isotherms of this material measured at 278 K for CO2 and N2 are displayed. From the isotherms it is directly observable, that the material does not take up any N2 at 278 K. However, the CO2 adsorption isotherm shows the anticipated phase transition known for this material featuring a considerably wide hysteresis loop.22 In the low pressure region of the isotherm, before the np → lp transition occurs, already a decent amount of CO2 is adsorbed (10 cm3(STP) per g). After surpassing the threshold pressure at 1700 kPa, a marked increase of the adsorbed volume of CO2 can be observed. The overall uptake reaches a value of 83 cm3(STP) per g. The desorption of CO2 starts at pressures below 1367 kPa and the initial plateau observed during the adsorption is reached again.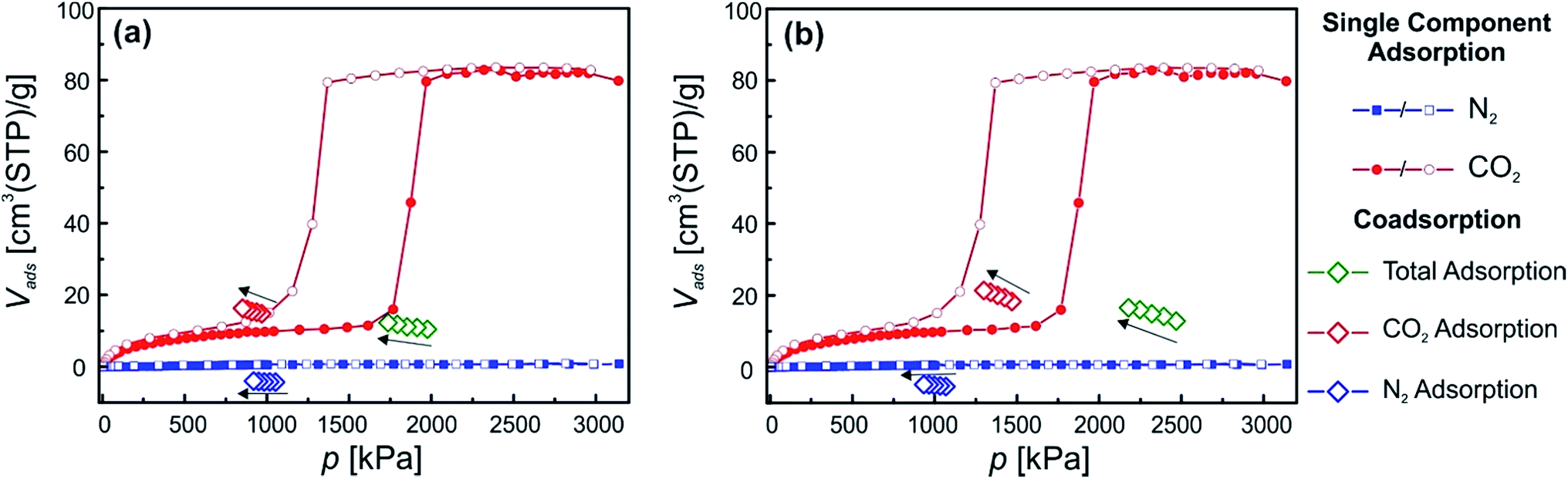 | ||
| Fig. 4 Depiction of the excess single component and co-adsorption measurements of material 2. N2 and CO2 single component isotherms are blue and red, respectively. CO2 and N2, fractions of the co-adsorption experiments are shown in red and blue, respectively. Total adsorption is shown in green. | ||
In order to characterize this gate opening and how it is affected by the presence of N2, co-adsorption experiments were performed using CO2/N2 mixtures containing ratios of 986![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1051 kPa (Fig. 4(a)) and 1469:1068 kPa (Fig. 3(b)). The total pressure (1975 and 2468 kPa, respectively) is higher than the characteristic single-component CO2 phase transition pressure of material 2. However, the partial CO2 pressure is below the phase transition pressure. A total CO2 uptake of 15 cm3(STP) per g is measured at the first equilibration step (Fig. 4(a)) (986 kPa partial CO2 pressure), slightly more than that of the CO2 single component adsorption isotherm. It should be noted, that the N2 uptake during co-adsorption is below the detection limit and thus might have an influence on the correct determination of the CO2 uptake. During the 5 equilibration steps, the CO2 uptake slightly increases, most likely due to the longer exposure of the material to CO2 (higher equilibration time). For the second experiment a substantially higher CO2 content is present in the CO2/N2 mixture, however the partial CO2 pressure is still below the threshold pressure inducing the phase transition. The total CO2 uptake is only slightly higher compared to the first experiment (18 cm3(STP) per g). As for the first experiment, the CO2 uptake increases over the course of the five set equilibrium points. The N2 uptake again is below the detection limit. Unfortunately, it was not possible to increase the pressure of CO2 to surpass the threshold pressure of the phase transition due to limitations of the applicable pressures of the co-adsorption instrument. Nonetheless some valuable conclusions can be drawn. Material 2 will not undergo a np → lp phase transition for CO2/N2 mixtures unless the characteristic CO2 threshold is surpassed, i.e. the pore cannot be pushed open by a great excess of N2 in the presence of CO2, even when the applied partial CO2 pressure in the mixture is only slightly lower than the threshold pressure.
:1051 kPa (Fig. 4(a)) and 1469:1068 kPa (Fig. 3(b)). The total pressure (1975 and 2468 kPa, respectively) is higher than the characteristic single-component CO2 phase transition pressure of material 2. However, the partial CO2 pressure is below the phase transition pressure. A total CO2 uptake of 15 cm3(STP) per g is measured at the first equilibration step (Fig. 4(a)) (986 kPa partial CO2 pressure), slightly more than that of the CO2 single component adsorption isotherm. It should be noted, that the N2 uptake during co-adsorption is below the detection limit and thus might have an influence on the correct determination of the CO2 uptake. During the 5 equilibration steps, the CO2 uptake slightly increases, most likely due to the longer exposure of the material to CO2 (higher equilibration time). For the second experiment a substantially higher CO2 content is present in the CO2/N2 mixture, however the partial CO2 pressure is still below the threshold pressure inducing the phase transition. The total CO2 uptake is only slightly higher compared to the first experiment (18 cm3(STP) per g). As for the first experiment, the CO2 uptake increases over the course of the five set equilibrium points. The N2 uptake again is below the detection limit. Unfortunately, it was not possible to increase the pressure of CO2 to surpass the threshold pressure of the phase transition due to limitations of the applicable pressures of the co-adsorption instrument. Nonetheless some valuable conclusions can be drawn. Material 2 will not undergo a np → lp phase transition for CO2/N2 mixtures unless the characteristic CO2 threshold is surpassed, i.e. the pore cannot be pushed open by a great excess of N2 in the presence of CO2, even when the applied partial CO2 pressure in the mixture is only slightly lower than the threshold pressure.
[Zn2(DiP-bdc)2(dabco)]n (3)
Since material 2 showed some restrictions, due to the relatively high phase transition pressure under the used conditions, we chose the differently functionalized material [Zn2(DiP-bdc)2(dabco)]n (3) because it reveals the lowest np → lp CO2 phase transition pressure among our library of fu-MOFs,19b making this material much more amenable for examining the behaviour when exposed to gas mixtures, in particular when using the specific set up of this study. Fig. 5(a) and (b) shows the single component adsorption isotherms for CO2, N2, CH4, C2H6 and C3H8 recorded at 278 K. Notably, a stepped isotherm with a hysteresis loop, indicating the np → lp phase transition of the activated material is observed for all probe gases except N2 and CH4. The uptake of these two gases is very low. Before the np → lp transition occurs, CO2, C2H6, and C3H8 uptakes of 6.5, 3.1 and 2.6 cm3(STP) per g were recorded. After exceeding the threshold pressures of the np → lp transition at 306 kPa (CO2), 115 kPa (C2H6) and 11 kPa (C3H8) the uptakes were drastically increased, peaking at 92.1 (CO2), 53.2 (C2H6) and 47.2 (C3H8) cm3(STP) per g. For N2 and CH4 only comparatively low values of 2.4 and 4.6 cm3(STP) per g were found. Fig. 5(c) and (d) shows the co-adsorption experiments on 3 for CO2/N2 mixtures. In one case the partial CO2 pressure is kept below the threshold pressure at 290 kPa (Fig. 5(c)) and in the other case the partial CO2 pressure is above the np → lp transition pressure at 621 kPa (Fig. 5(d)). The total pressure of the gas mixture was above the threshold pressure in both cases. The first experiment shows that an excess of N2 (903 kPa) cannot help to induce the np → lp transition (Fig. 5(c)), as was also observed for material 2. When this mixture is applied the framework takes up 8.5 cm3(STP) per g CO2 and barely any N2 (amount below the detection limit). Both values are in line with the values found for the single component isotherms. For the second experiment the threshold pressure of the np → lp transition was surpassed by the applied partial CO2 pressure (621 kPa) (Fig. 5(d)). In comparison to the previous experiment, the CO2 uptake is significantly increased and completely in line with the uptake of 3, determined from the single component adsorption isotherm. Interestingly, the N2 uptake is not increased (compared to the single component isotherm), attesting that the material does not take up any N2, even when being present in the lp. Thus, this material might be feasible for CO2/N2 separations. After these experiments we moved on to a different gas mixture, to assess the ability of 3 to separate CO2 from a more polarizable co-adsorbate as compared to N2 and chose CH4. From the single component adsorption we know that CH4 does not induce a phase transition and the overall adsorbed amount is only slightly higher than for N2 (Fig. 5(a)).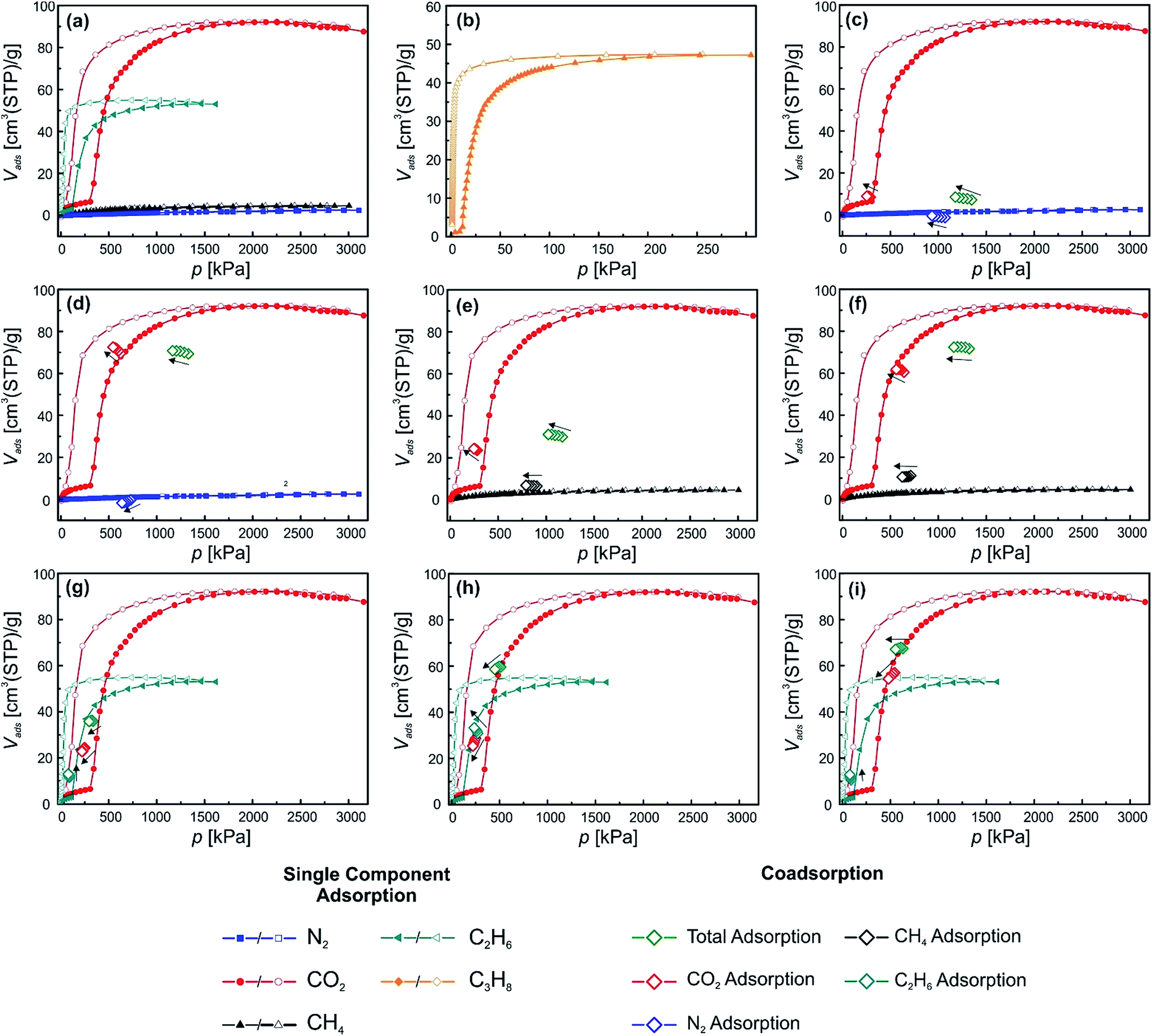 | ||
| Fig. 5 Depiction of the excess single component and co-adsorption measurements of material 3. N2, CO2, CH4, C2H6 and C3H8 single component isotherms are blue, red, black, turquoise and orange, respectively. CO2, N2, CH4 and C2H6 fractions of the co-adsorption experiments are shown in red, blue, black and turquoise, respectively. The total adsorption is shown in green. | ||
Again two experiments were performed. In the first run, the partial CO2 pressure is slightly below the np → lp transition pressure and during the second experiment the partial CO2 pressure was above the np → lp transition pressure. The results are depicted in Fig. 5(e) and (f). From the first experiment (Fig. 5(e)) it can be seen, that the adsorbed amount of CO2 during the co-adsorption experiments is much higher than for the single component adsorption at the same pressure (23.47 cm3(STP) per g compared to 6.5 cm3(STP) per g), while the CH4 uptake is only slightly increased (6.5 cm3(STP) per g compared to 4.6 cm3(STP) per g). We propose a synergy of the CH4 and CO2 co-adsorption that allows for a lower partial CO2 pressure to trigger the np → lp transition. In the second experiment (Fig. 5(f)) it can be nicely seen that the CO2 uptake coincides with the CO2 uptake of the single component experiment at this pressure after the np → lp pore transition. Most interestingly, also the amount of CH4 adsorbed in the framework is now drastically increased when compared to the first CO2/CH4 co-adsorption experiment. We assume that after the CO2 induced opening CH4 is able to co-adsorb in the lp phase. This behaviour is substantially different from the findings obtained from the CO2/N2 co-adsorption experiments and attributed to the much higher polarizability of the CH4 molecule compared to N2 (see ESI† for information on properties of used adsorptives).
In order to see how material 3 behaves when two guests are adsorbed that both initiate a np → lp transition CO2/C2H6 and CO2/C3H8 co-adsorption experiments were conducted (Fig. 5(g)–(i) (C2H6) and ESI† (C3H8)). Three different measurement conditions were chosen for CO2/C2H6 mixtures. In the first case, the partial pressures of both gases were below the respective threshold pressures of the np → lp transition (306 and 115 kPa for CO2 and C2H6, respectively). In the second and third case the partial pressure of one of the components was below the corresponding phase transition pressure and the other one was set above. The applied mixtures contained CO2/C2H6 ratios of 250:89 kPa (Fig. 5(g)), 249:271 kPa (Fig. 5(h)) and 546:91 kPa (Fig. 5(i)), respectively. In the first measurement (Fig. 5(g)) a marked increase of the adsorption was found for both components when compared to the respective single component isotherms. Values of 24.4 cm3(STP) per g adsorbed CO2 and 11.5 cm3(STP) per g of C2H6 are measured in contrast to 6.1 cm3(STP) per g and 2.8 cm3(STP) per g (single component adsorption). This suggests that strong cooperative effects between the two gases allow the gate opening transition to occur at lower partial pressures of each component. In the experiments depicted in Fig. 5(h) and (i) in both cases very similar behavior is observed. In Fig. 5(h), p(CO2) and p(C2H6) amount to 249 kPa and 271 kPa, respectively. The measurement reveals, that the CO2 uptake (28.7 cm3(STP) per g) is significantly increased in comparison to the single component adsorption (6.1 cm3(STP) per g). We conclude that the CO2 is co-adsorbing with the C2H6 that opened the pore. A similar explanation can be drawn for the experiment in Fig. 5(i), where p(CO2) is above (545.7 kPa) and p(C2H6) is below (90.7 kPa) the np → lp transition pressure. The CO2 adsorption at this point nicely coincides with the single component isotherm, while the C2H6 adsorption at 90.7 kPa is notably increased from 2.9 cm3(STP) per g to 10.4 cm3(STP) per g, which is in line with the uptake found in the first co-adsorption experiment of these two species when both partial pressures are below the np → lp transition pressure (Fig. 5(g)).
For the CO2/C3H8 mixtures similar results were found as for CO2/C2H6 and a detailed description can be found in the ESI (Fig. S9†). A summary of the results found for material 3 is shown in Table 1.
| p(CO2) < ppta | p(CO2) > ppt | p(CO2) < ppt | p(CO2) > ppt | ||
|---|---|---|---|---|---|
| a p pt is the phase transition pressure of the respective single component adsorption isotherm. | |||||
| CO2/N2 | No pore opening; only CO2 adsorbed in np | Pore opening; only CO2 adsorption | CO2/CH4 | Pore opening before reaching ppt; enhanced CO2 uptake | Pore opening; CH4 co-adsorbs in lp |
| p(CO2) and p(C2H6) < ppt | p(CO2) < ppt; p(C2H6) > ppt | p(CO2) > ppt; p(C2H6) < ppt | |
|---|---|---|---|
| CO2/C2H6 | Pore opening before reaching ppt; cooperative induction of phase transition (enhanced uptake of both component) | Pore opening; C2H6 transports CO2 into pore (enhanced CO2 uptake) | Pore opening; CO2 transports C2H6 into pore (enhanced C2H6 uptake) |
Calculation of selectivities
For all co-adsorption measurements the selectivity coefficients were also calculated based on the molar fractions adsorbed on the material and left in the gas phase during each co-adsorption measurement. The calculated values for the 1st equilibration point for material 3 are presented in Table 2 (values for the other materials and other equilibration points are listed in the ESI†). First of all, it needs to be mentioned, that the CO2/N2 measurements conducted on all three materials look very promising and a good separation of the two components is directly visible from the co-adsorption experiments shown before. However, the values determined during the N2 adsorption are below the detection limit and a meaningful evaluation was not possible. However, in particular for material 3 some interesting conclusions can be drawn. In the case of the CO2/CH4 co-adsorption experiments a decent selectivity towards CO2 is calculated, for the case that the CO2 pressure is below the threshold pressure of the phase transition (5.85). In this case CH4 helps inducing a phase transition at lower partial pressures as expected. When the CO2 pressure is increased and the material is present in the lp, CH4 is co-adsorbing and the selectivity is drastically increased to 11.57 (the increase in CO2 adsorption is higher than the increase in CH4 adsorption induced by the observed cooperative effects). From the CO2/C2H6 co-adsorption measurements only very low selectivities are determined. The values towards CO2 are in the range of 0.75 to 1.02 – the material seems to slightly favour the adsorption of C2H6 and a separation of these two gases over 3 does not seem feasible. The CO2/C3H8 co-adsorption measurements show a clear trend towards a favoured adsorption of C3H8 in the presence of CO2 (see ESI Table S1† for data). In particular at low overall pressures, when the C3H8 partial pressure is below the threshold pressure of the phase transition determined by the single component isotherm, a good selectivity of C3H8 over CO2 (7.32 and 3.24) results. This result seems a bit odd, as the adsorbed amount of C3H8 is very low compared to the adsorbed CO2. However, the molar ratios of C3H8/CO2 in the gas phase are much lower than for the adsorbed state. Thus C3H8 is relatively enriched over CO2 by adsorption.| Fig. | Material | Gas mixture | p e /kPa | Molar fraction gas phase | Molar fraction adsorbed | Selectivity (CO2) | Selectivity (co-adsorbate) | ||
|---|---|---|---|---|---|---|---|---|---|
| CO2 | Co-adsorbate | CO2 | Co-adsorbate | ||||||
| a Data is from the 1st equilibrium point. b Equilibration pressure of the gas mixture. | |||||||||
| 5(c) | 3 | CO2/N2 | 1328.59 | 0.47 | 0.53 | 1.00 | 0.00 | — | — |
| 5(d) | 3 | CO2/N2 | 1347.56 | 0.22 | 0.78 | 1.18 | −0.18 | — | — |
| 5(e) | 3 | CO2/CH4 | 1323.98 | 0.48 | 0.52 | 0.84 | 0.16 | 5.85 | 0.17 |
| 5(f) | 3 | CO2/CH4 | 1167.08 | 0.24 | 0.76 | 0.79 | 0.21 | 11.57 | 0.09 |
| 5(g) | 3 | CO2/C2H6 | 336.52 | 0.74 | 0.26 | 0.68 | 0.32 | 0.75 | 1.33 |
| 5(h) | 3 | CO2/C2H6 | 511.79 | 0.48 | 0.52 | 0.48 | 0.52 | 1.02 | 0.98 |
| 5(i) | 3 | CO2/C2H6 | 630.01 | 0.86 | 0.14 | 0.84 | 0.16 | 0.87 | 1.15 |
Conclusions
The results leave a lot of room for discussion and further experiments and theoretical modelling. Below we summarize our findings (I–IV) regarding the adsorption of binary gas mixtures on fu-MOFs of the type [Zn2(fu-bdc)2(dabco)]n and suggest some conclusions that might hold true for other flexible MOFs.(I) The presence of N2 is not affecting the CO2 uptake in fu-MOFs. The results found for 2 and 3 show that the phase transition cannot be shifted to lower pressures and N2 does not assist in the pore expansion process.
(II) N2 is not adsorbed in the pore after the CO2 threshold pressure is surpassed and the np → lp phase transition occurred.
(III) More polar guests, with stronger adsorption enthalpies at the adsorption sites and more favourable interactions with the pendant side chains can help inducing a preliminary CO2 induced phase transition. Additionally, CH4 can travel into the pore with the adsorbent and the CH4 uptake is strikingly higher than during single component adsorption when only the np is present.
(IV) Adsorbates that show stepped isotherms (gate opening behaviour) can induce strong cooperative effects with CO2. In the presence of C2H6 and C3H8 the CO2 adsorption is strongly increased below the CO2 phase transition pressure.
We conclude that a separation of two gases that possess a two-step isotherm over a flexible MOF is highly unlikely. The gas with the lower threshold pressure will open the pore and considerable amounts of the second gas will be adsorbed in the pore as well. Nevertheless, if the concentrations/pressures of both components are wisely chosen, our data suggest that a separation can be achieved under some specific conditions.
Finally it needs to be highlighted that the np → lp phase transition is a phenomenon governed by thermodynamics. The energy needed for the phase transition must be compensated by the adsorption enthalpy of the respective gases at lp. From our recent molecular dynamics simulations and NMR studies we know that the favoured adsorption sites for CO2 within this class of fu-MOFs are located in the vicinity of the carboxylate groups connecting to the metal-ion.23a Thus, the interactions of the guest with the side chains need to be taken into account in order to pass through and reach the adsorption site. Furthermore, the attractive side-chain/side-chain and side-chain/linker interactions favour the pore contraction and need to be compensated to initiate the phase transition. In case of co-adsorption, both components (guests) contribute to the over-all energetics of the np → lp gate-opening phenomenon. It is thus quite conceivable that a cooperative gate opening is possible (as we observed) in case of setting the partial pressures of the two components below the individual phase transition pressures (CO2/C2H6 and CO2/C3H8). If a guest is chosen (N2) exhibiting low uptake and adsorption enthalpies for the np (experimental evidence) and also (presumably) in the lp state of the material, it will neither assist in the pore opening nor will it significantly co-adsorb. In the case of CO2/CH4 experiments, we assume that the adsorption enthalpy of CH4 is increased in the lp as compared to the np state (increased accessibility of favourable adsorption sites, less hindrance by the side chains to reach adsorption sites). We are aiming to understand the effects reported in this study more quantitatively by focusing on a comprehensive thermodynamic analysis of the co-adsorption process in combination with molecular dynamics simulations in the future. Nevertheless, we suggest some exciting potential functions for flexible MOFs from the current results. For example, it is imaginable that the one gas A of a mixture can work as a switching gas. The second gas B can undergo a reaction or cause a signal inside the framework, only if a certain partial pressure of gas A is set. The obvious necessity of fine-tuning the chosen flexible framework could be done by choosing a suitable linker-functionalization as it is offered by our library of fu-MOFs including mixed-component (or multivariate) fu-MOFs (with different metal ions, linkers and functionalized side chains). Such systems may be tailored to operate at different temperature and pressure regimes and feature guest-specific and adjustable gate-opening properties.
Acknowledgements
A. S. gratefully acknowledges the Ruhr University Research School PLUS for a travel grant to conduct experiments at Hokkaido University, Japan. Ruhr University Research School PLUS is funded by Germany's Excellence Initiative [DFG-GSC 98/3]. The authors gratefully acknowledge Michael S. Denny Jr. (University of California, San Diego) for proof checking the manuscript and helpful discussion.Notes and references
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed; (b) A. Schneemann, S. Henke, I. Schwedler and R. A. Fischer, ChemPhysChem, 2014, 15, 823 CrossRef CAS PubMed; (c) H. Furukawa, U. Mueller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417 CrossRef CAS PubMed; (d) A. G. Slater and A. I. Cooper, Science, 2015, 348, 988 CrossRef CAS PubMed.
- (a) S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; (b) G. Ferey, Chem. Soc. Rev., 2008, 37, 191 RSC; (c) O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166 CrossRef CAS PubMed.
- (a) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed; (b) J. An, O. K. Farha, J. T. Hupp, E. Pohl, J. I. Yeh and N. L. Rosi, Nat. Commun., 2012, 3, 1618 Search PubMed; (c) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed; (d) T. C. Wang, W. Bury, D. A. Gomez-Gualdron, N. A. Vermeulen, J. E. Mondloch, P. Deria, K. Zhang, P. Z. Moghadam, A. A. Sarjeant, R. Q. Snurr, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 3585 CrossRef CAS PubMed.
- (a) O. Kozachuk, I. Luz, F. X. Llabres i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058 CrossRef CAS PubMed; (b) Z. Fang, J. P. Duerholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Poeppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627 CrossRef CAS PubMed.
- (a) O. Karagiaridi, W. Bury, A. A. Sarjeant, C. L. Stern, O. K. Farha and J. T. Hupp, Chem. Sci., 2012, 3, 3256 RSC; (b) M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082 CrossRef CAS PubMed; (c) H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011 CrossRef CAS PubMed; (d) O. Karagiaridi, W. Bury, E. Tylianakis, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Chem. Mater., 2013, 25, 3499 CrossRef CAS; (e) M. B. Lalonde, J. E. Mondloch, P. Deria, A. A. Sarjeant, S. S. Al-Juaid, O. I. Osman, O. K. Farha and J. T. Hupp, Inorg. Chem., 2015, 54, 7142 CrossRef CAS PubMed; (f) C. J. Stephenson, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 1361 CrossRef CAS PubMed.
- (a) L. Liu, K. Konstas, M. R. Hill and S. G. Telfer, J. Am. Chem. Soc., 2013, 135, 17731 CrossRef CAS PubMed; (b) C. A. Allen and S. M. Cohen, Inorg. Chem., 2014, 53, 7014 CrossRef CAS PubMed; (c) L. Liu and S. G. Telfer, J. Am. Chem. Soc., 2015, 137, 3901 CrossRef CAS PubMed; (d) A. C. H. Sue, R. V. Mannige, H. Deng, D. Cao, C. Wang, F. Gandara, J. F. Stoddart, S. Whitelam and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5591 CrossRef CAS PubMed; (e) S. Yuan, W. Lu, Y.-P. Chen, Q. Zhang, T.-F. Liu, D. Feng, X. Wang, J. Qin and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 3177 CrossRef CAS PubMed; (f) S. Yuan, Y.-P. Chen, J.-S. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 8912–8919 CrossRef CAS PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294 CrossRef CAS PubMed.
- (a) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC; (b) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed; (c) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC; (d) Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432 CrossRef CAS PubMed; (e) F.-X. Coudert, Chem. Mater., 2015, 27, 1905 CrossRef CAS.
- (a) A. Kobayashi, Y. Suzuki, T. Ohba, T. Ogawa, T. Matsumoto, S.-i. Noro, H.-C. Chang and M. Kato, Inorg. Chem., 2015, 54, 2522 CrossRef CAS PubMed; (b) V. Bon, N. Kavoosi, I. Senkovska, P. Mueller, J. Schaber, D. Wallacher, D. M. Toebbens, U. Mueller and S. Kaskel, Dalton Trans., 2016, 45, 4407 RSC; (c) M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022 CrossRef CAS PubMed; (d) S.-m. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920 CrossRef CAS PubMed; (e) S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Toebbens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- (a) Y. Liu, J.-H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813 CrossRef CAS PubMed; (b) L. D. DeVries, P. M. Barron, E. P. Hurley, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 14848 CrossRef CAS PubMed; (c) S. Henke, A. Schneemann and R. A. Fischer, Adv. Funct. Mater., 2013, 23, 5990 CrossRef CAS; (d) Y. Kim, R. Haldar, H. Kim, J. Koo and K. Kim, Dalton Trans., 2015, 45, 4187–4192 RSC; (e) M. T. Wharmby, S. Henke, T. D. Bennett, S. R. Bajpe, I. Schwedler, S. P. Thompson, F. Gozzo, P. Simoncic, C. Mellot-Draznieks, H. Tao, Y. Yue and A. K. Cheetham, Angew. Chem., Int. Ed., 2015, 54, 6447 CrossRef CAS PubMed.
- (a) N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501 CrossRef CAS PubMed; (b) R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695 CrossRef CAS PubMed.
- (a) S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087 CrossRef CAS PubMed; (b) F.-X. Coudert, A. Boutin, A. H. Fuchs and A. V. Neimark, J. Phys. Chem. Lett., 2013, 4, 3198 CrossRef CAS; (c) S. Henke, W. Li and A. K. Cheetham, Chem. Sci., 2014, 5, 2392 RSC; (d) E. C. Spencer, M. S. R. N. Kiran, W. Li, U. Ramamurty, N. L. Ross and A. K. Cheetham, Angew. Chem., Int. Ed., 2014, 53, 5583 CrossRef CAS PubMed; (e) J. Rodriguez, I. Beurroies, T. Loiseau, R. Denoyel and P. L. Llewellyn, Angew. Chem., Int. Ed., 2015, 54, 4626 CrossRef CAS PubMed; (f) P. G. Yot, L. Vanduyfhuys, E. Alvarez, J. Rodriguez, J.-P. Itie, P. Fabry, N. Guillou, T. Devic, I. Beurroies, P. L. Llewellyn, V. Van Speybroeck, C. Serre and G. Maurin, Chem. Sci., 2016, 7, 446 RSC; (g) C. L. Hobday, R. J. Marshall, C. F. Murphie, J. Sotelo, T. Richards, D. R. Allan, T. Dueren, F.-X. Coudert, R. S. Forgan, C. A. Morrison, S. A. Moggach and T. D. Bennett, Angew. Chem., Int. Ed., 2016, 55, 2401 CrossRef CAS PubMed.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739 CrossRef CAS.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787 CrossRef CAS PubMed.
- I. Beurroies, M. Boulhout, P. L. Llewellyn, B. Kuchta, G. Ferey, C. Serre and R. Denoyel, Angew. Chem., Int. Ed., 2010, 49, 7526 CrossRef CAS PubMed.
- (a) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774 CrossRef CAS PubMed; (b) D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaim, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767 CrossRef CAS.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS PubMed.
- (a) S. Henke, R. Schmid, J.-D. Grunwaldt and R. A. Fischer, Chem.–Eur. J., 2010, 16, 14296 CrossRef CAS PubMed; (b) S. Henke, A. Schneemann, A. Wuetscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464 CrossRef CAS PubMed.
- A. Schneemann, E. D. Bloch, S. Henke, P. L. Llewellyn, J. R. Long and R. A. Fischer, Chem.–Eur. J., 2015, 21, 18764 CrossRef CAS PubMed.
- (a) S. Henke and R. A. Fischer, J. Am. Chem. Soc., 2011, 133, 2064 CrossRef CAS PubMed; (b) S. Henke, A. Schneemann, S. Kapoor, R. Winter and R. A. Fischer, J. Mater. Chem., 2012, 22, 909 RSC; (c) Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920 CrossRef CAS PubMed.
- S. Henke, D. C. F. Wieland, M. Meilikhov, M. Paulus, C. Sternemann, K. Yusenko and R. A. Fischer, CrystEngComm, 2011, 13, 6399 RSC.
- (a) V. Bon, J. Pallmann, E. Eisbein, H. C. Hoffmann, I. Senkovska, I. Schwedler, A. Schneemann, S. Henke, D. Wallacher, R. A. Fischer, G. Seifert, E. Brunner and S. Kaskel, Microporous Mesoporous Mater., 2015, 216, 64 CrossRef CAS; (b) I. Schwedler, S. Henke, M. T. Wharmby, S. R. Bajpe, A. K. Cheetham and R. A. Fischer, Dalton Trans., 2016, 45, 4230 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: PXRDs, NMRs, IR spectra, TGs of the compounds, details on the measurement method and additional selectivity coefficients. See DOI: 10.1039/c6ta03266d |
| This journal is © The Royal Society of Chemistry 2016 |