
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical reduction of carbon dioxide coupled with water oxidation using various soft-oxometalate (SOM) based catalytic systems†
Santu
Das
,
Subharanjan
Biswas
,
Tuniki
Balaraju
,
Soumitra
Barman
,
Ramudu
Pochamoni
and
Soumyajit
Roy
*
Eco-Friendly Applied Materials Laboratory (EFAML), Materials Science Centre, Department of Chemical Sciences, Indian Institute of Science Education & Research, Mohanpur Campus, Kolkata, 741246 West Bengal, India. E-mail: s.roy@iiserkol.ac.in
First published on 5th May 2016
Abstract
Simultaneous CO2 reduction and water oxidation as a coupled process is an important challenge in the quest of clean energy production. Herein, we report a metal oxide based heterogeneous catalytic system, which not only couples CO2 reduction with water oxidation, but also provides very high turnover number for CO2 reduction with scalability. Such a catalytic system can simultaneously oxidize water and release the generated electrons for reduction of CO2 with a maximum turnover number and turnover frequency as high as 1.4 × 106 and 610 s−1, respectively, following effective catalyst concentration; whereas, turnover number and turnover frequency is 1366 and 1380 h−1 per mole of catalyst, respectively. The starting materials for this catalytic process are CO2 and water while the end products are oxygen and formic acid and in few cases, formaldehyde. The prospect of using the formic acid generated during our process in fuel cells to generate green energy is also worth mentioning.
Introduction
Nature performs CO2 reduction and couples it with water oxidation regularly and with very high efficacy in photosynthesis. CO2 emissions are ever increasing. Water and light are amply abundant. Fossil fuel reserves are depleting and up-stream C1 feed-stocks demands are steeply increasing. Hence, the immediate question is can CO2 reduction and water oxidation be coupled in a synthetic scalable catalytic system? Herein, we report such a catalytic system based on three different metal oxide based catalysts that can simultaneously oxidize water, and release the generated electrons for the reduction of CO2 with a high TON (turn over number). The starting materials of this catalytic process are CO2 and water while the end products are oxygen and formic acid and in a few cases, formaldehyde. Now we describe briefly the background of our present catalytic process and the development of its design.Our design of the present catalyst is centred on two phenomena that are coupled together, viz., photo-chemical CO2 reduction1–13 and water oxidation.14–22 Photochemical CO2 reduction has attracted enormous interest in recent times. During the 1980s Lehn23 and Sauvage24 pioneered the field of photoelectrochemical CO2 reduction using Re-porphyrins, Co, Ni cyclams and polyporphyrins, respectively. For CO2 reduction, four prime approaches can be identified as follows. (1) CO2 reduction based on macrocycles such as cyclams with Ni2+ and Co2+.24,25 (2) Polypyrridyl based materials with Re+, Ru+ and Mn.26–29 (3) Pd2+ based pincer type complexes reported by Dubois30 and the related complexes of Ir pioneered by Peruzzini.31 (4) Cantat and co-workers have pioneered metal free CO2 reduction using nitrogen containing bases such as amidines and guanidines with hydrosilanes as the reducing agents.32 Molybdenum alone has been shown to be capable of CO2 reduction.33 Likewise, water oxidation by metal complexes has also attracted enormous attention since the seminal concept of artificial photosynthesis proposed by Nocera.2 The use of optical semi-conductors coupled with a metal complex for CO2 reduction has been very recently proposed.33 The possibility of achieving scalable coupled CO2 reduction with water oxidation has also been proposed.2,4 However, to date and to the best of our knowledge, no scalable approach for the realization of this effort has been reported. Some related noteworthy efforts warrant mention. For instance, Carpenter and co-workers have explored the possibility of CO2 reduction without the presence of any external reducing agent.9 With a prototype example of an amine, they have shown this reduction in an indirect fashion.12 More recently, Yang et al.34 have shown the possibility of coupling of CO2 reduction and water oxidation with nanocomposites of tantalum-based pyrochlore nanoparticles and indium hydroxide where pyrochlore nanoparticles catalyze the CO2 reduction reaction or water reduction reaction and indium hydroxide catalyzes water oxidation. Though a very elegant effort, this method also suffers from a negligible turnover number. In another effort, Kudo et al.35 reported a method coupling Ag co-catalyst loaded perovskites with the formula of ALa4TiO4O15 (A = Ca, Sr and Ba). The catalyst, although couples CO2 reduction with water oxidation with simultaneous formation of HCOOH and CO, provides only a negligible yield (maximum CO yield is 22 micromoles per hour with 0.3 g catalyst loading). The single catalysis electrocatalytic CO2 reduction coupled with water oxidation reported by Meyer group36,37 also deserves a mention. They have shown that a Ru(II) polypyridyl carbene complex can catalyze the electrochemical reduction of CO2 in water with added bicarbonate to give synthesis gas (H2 and CO) mixtures at the cathode and water oxidation to O2 at the anode. However, the problems of low turnover numbers and scalability also occur. The issue of scalability has been addressed in the recent work of the Yaghi and Chang group.1 They have shown that cobalt–porphyrin based covalent organic frameworks can be optimized to synthesize the catalytic material, which shows very high efficiency towards the reduction of CO2 to CO. The faradaic efficiency of this reaction was 90% with turnover numbers up to 290![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, with an initial turnover frequency of 9400 h−1.
000, with an initial turnover frequency of 9400 h−1.
Here we take the next step. To perform photochemical CO2 reduction coupled with the water oxidation reaction, we chose a set of various metal oxide clusters. It is already known from earlier studies that metal oxide clusters can act as active and efficient photocatalysts37,38 as well electrocatalysts39,40 in water oxidation and water reduction41,42 reactions. These metal oxide clusters are also cheap and green. To enhance the effective surface area of the catalysts, instead of using the metal oxides in the solid phase, we have prepared self-assembled vesicles with the metal oxides. Such vesicle-like super-structure formation of the metal oxide clusters is well known and studied by our group and we have proposed them to be called soft-oxometalates (SOMs).43–50 Very recently we have exploited various properties of SOMs and thereby, their usability in a large range of applications have been explored. SOMs can undergo topological transformation,51 can be used in patterning52,53 and as active particles.46 The catalytic activity of SOMs has also been explored in various types of reactions viz., in photo-polymerization54–56 and photochemical water oxidation.57 In this article we have shown that SOM type materials based on oxo-molybdate and oxo-tungstate clusters can efficiently catalyse the reduction of CO2 to HCOOH/HCHO by transferring the electrons and protons generated from oxidation of water. The catalysts reported here present a new opportunity as they provide a method that is scalable and fast with a very high turnover number.
Results and discussion
About the catalysts
We applied the design principles in heterogeneous catalysis mode to render the process scalable and rapid. Following the above design principle three catalysts were synthesized (Fig. 1):| {Mo154}x, where x = 1165 | (1) |
| {Mn6P3W24}y, where y = 931 | (2) |
| {Mo132}n@RGO, where n = 1064 | (3) |
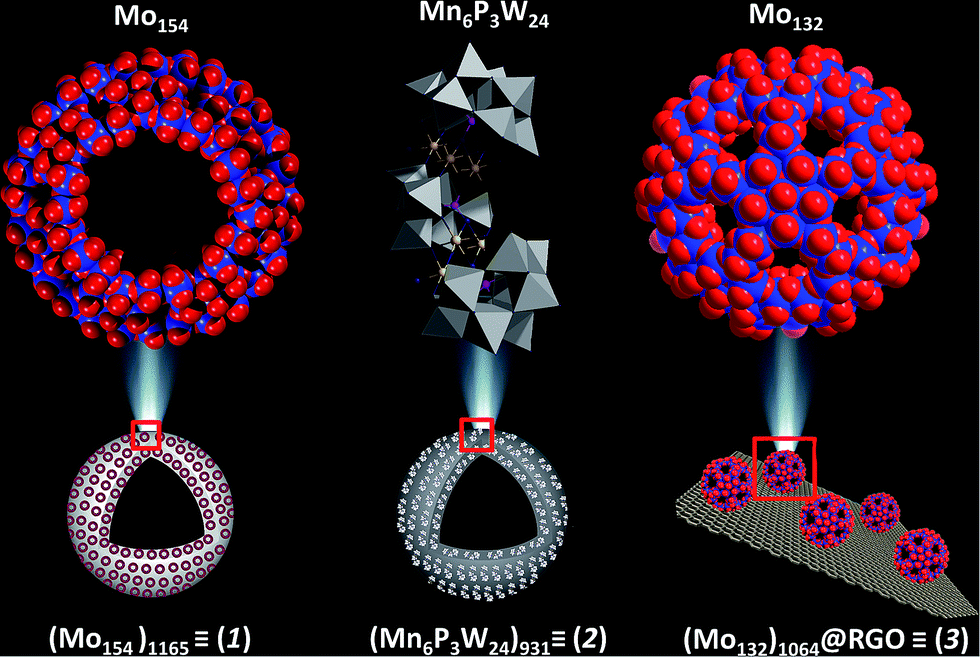 | ||
| Fig. 1 The structures of the three catalysts, {Mn6P3W24} is shown in polyhedral representation while {Mo154} and {Mo132} are shown as a space-filling model. Color code: Mo, red; O, blue; Mn, yellow; W, grey; P, pink. | ||
Before preparing the effective catalysts we first briefly introduce the metal oxide based molecular precursors of the catalysts. We chose three metal oxide based clusters viz. {Mo154}, {Mn6P3W24} and {Mo132}.
Molecular structure of Mo154
{Mo154} is a giant wheel-shaped mixed-valence polyoxomolybdate cluster with the formula {Mo154O462H14(H2O)70}14− comprising 140 {MoO6} octahedra and 14 pentagonal bipyramids of the type {MoO7}.58 It can be described as a tetradecamer comprising 14 {Mo8} units with a central {MoO7} group. This {MoO7} is symmetrically connected to five {MoO6} octahedra by edge sharing. This builds a {(Mo)Mo5} pentagon. Four of the {MoO6} octahedra are linked to further {MoO6} octahedra via corner sharing to form the {Mo8} unit. The two MoO6 octahedra, which are not directly connected to the central MoO7 bipyramid are fused to neighbouring {Mo8} units through corners. Neighbouring {Mo8} groups are additionally fused together by the {Mo2} units, thereby completing the inner-ring parts of the upper and lower half of the ring structure. The complete construction of the ring is achieved by fusing the second half through the 14 {Mo1} groups at the equator after rotating around 360/14° relative to the first. Thus, the ‘giant-wheel’ can be formed in terms of the three different building blocks as [{Mo2}14{Mo8}14{Mo1}14]14−.We have synthesized crystals of these giant-wheel like molybdenum blue compound, {Na15[{Mo126}VI{Mo28}VO462H14(H2O)70]0.5[{Mo124}VI{Mo28}VO457H14(H2O)68]0.5·ca. 400H2O} following a literature procedure.59 {Mo154} has a space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It is characterized by FT-IR (Fig. 2a), resonance Raman spectroscopy (Fig. 2b) and SEM (Fig. 3a). The IR spectrum shows characteristic peaks at 1629 (δH2O), 1409, 1308, 1015, 955 (νMo
. It is characterized by FT-IR (Fig. 2a), resonance Raman spectroscopy (Fig. 2b) and SEM (Fig. 3a). The IR spectrum shows characteristic peaks at 1629 (δH2O), 1409, 1308, 1015, 955 (νMo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 898, 801, 625 and 565 cm−1.59 Using an excitation of the 1064 nm band, a resonance-Raman spectrum is obtained with five bands characteristic for the molybdenum blue species at 802 (νasMo–O), 531, 458, 325 (δO–Mo–O) and 215 (νasMo–O) cm−1.60 The electronic absorption spectrum is dominated by two bands characteristic for molybdenum blue species at 745 and 1076 nm (Fig. 4a). The first maxima is assigned to intervalence (MoV/MoVI) charge transfer transitions (IVCT) and the second maxima corresponds to the abundance of 28 MoV centers.61
O), 898, 801, 625 and 565 cm−1.59 Using an excitation of the 1064 nm band, a resonance-Raman spectrum is obtained with five bands characteristic for the molybdenum blue species at 802 (νasMo–O), 531, 458, 325 (δO–Mo–O) and 215 (νasMo–O) cm−1.60 The electronic absorption spectrum is dominated by two bands characteristic for molybdenum blue species at 745 and 1076 nm (Fig. 4a). The first maxima is assigned to intervalence (MoV/MoVI) charge transfer transitions (IVCT) and the second maxima corresponds to the abundance of 28 MoV centers.61
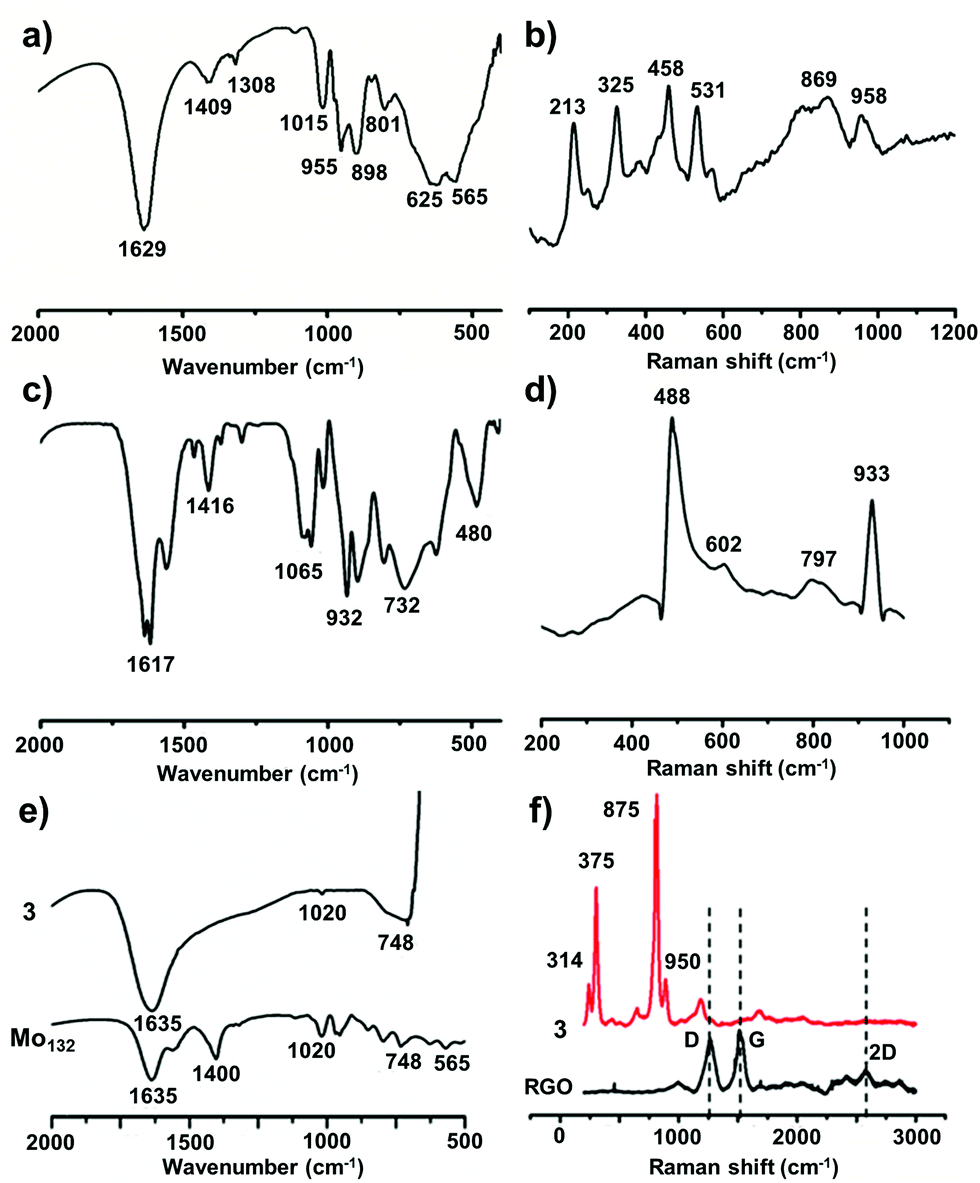 | ||
| Fig. 2 Characterization of all the catalysts by FT-IR and Raman spectroscopy, from the top to bottom row: catalyst 1, 2 and 3. The presence of characteristics bands in the FT-IR (a, c, e) and Raman (b, d, f) spectra are observed for all the catalysts. In the Raman spectrum of catalyst 3 (Mo132@RGO), characteristic D and G bands are shifted somewhat, which might be due to the interaction of Mo132 with the RGO surface. | ||
 | ||
| Fig. 3 (Left column) SEM images of 1, 2 and 3 (a, b & c, respectively). Vesicle like morphology is seen in the case of 1 and 2. (Right column) TEM image of 3 is shown in d–f. Both SEM & TEM images of 3 show embedded Mo132 spheres on the surface of RGO sheet. Curves of RGO sheets are shown by black arrows while white arrows indicate Mo132 spheres. | ||
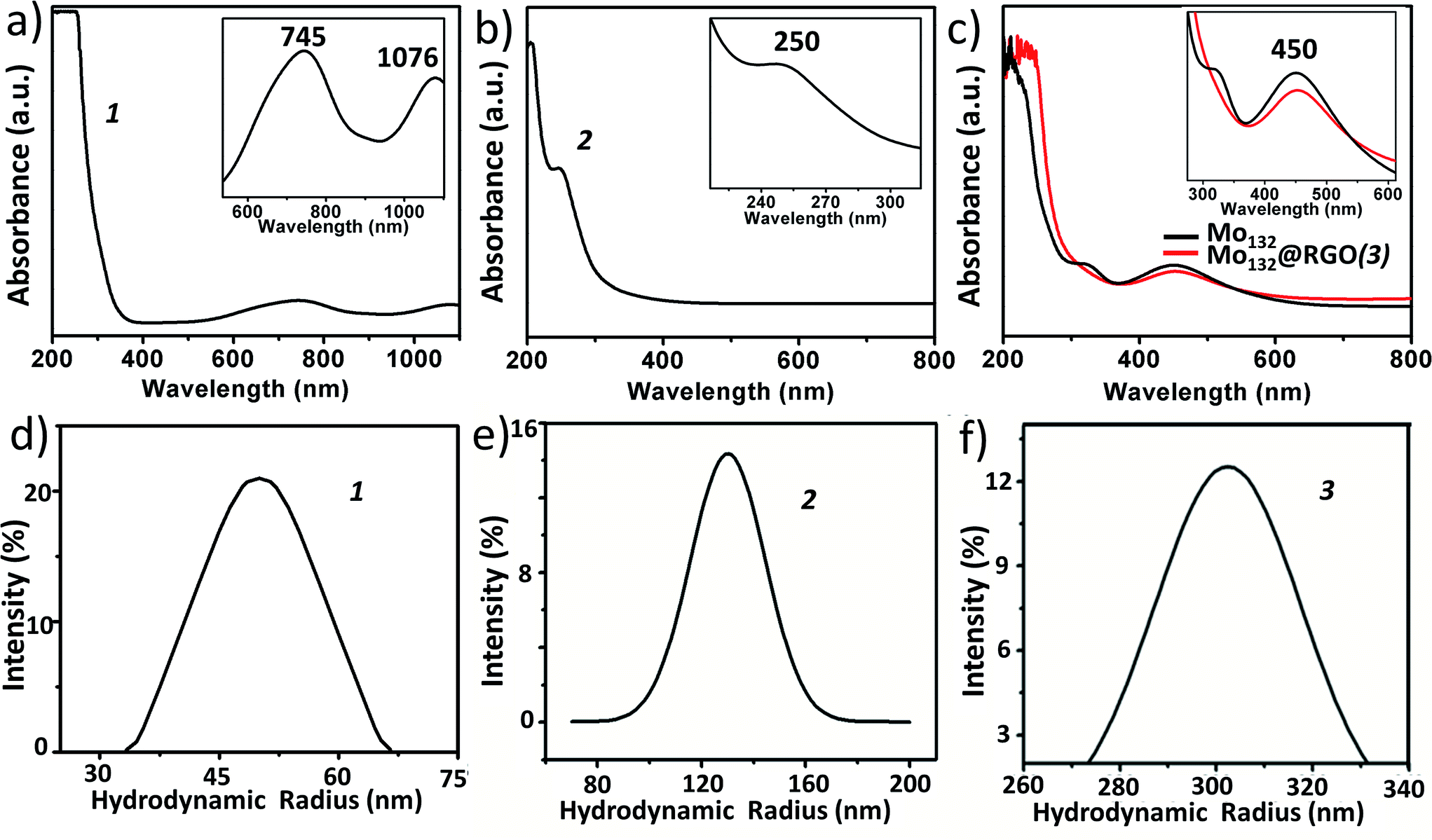 | ||
| Fig. 4 UV-Vis spectroscopy of 1, 2 and 3 (upper panel) shows characteristic bands for {Mo154} (745 and 1076 nm), {Mn6P3W24} (250 nm) and {Mo132} (450 nm). The magnified characteristics bands are shown in the insets. The lower panel represents the hydrodynamic radius obtained from DLS spectroscopy of 1, 2 and 3 with an average of 51, 130 and 300 nm, respectively. | ||
On active catalyst (1)
Molecular structure of {Mn6P3W24}
This polyoxometalate has the formula, {Mn6P3W24O94(H2O)2}17− (Fig. 1). It crystalizes in a monoclinic unit cell, which consists of two Keggin type {Mn3PW9O34} units linked by one {PW6O24} unit. The synthesized POM has C2v symmetry with a banana shape. The molecular structure may also be called a double sandwiched structure that implies the presence of two distinct {Mn3O13} triads formed by octahedral units of {MnO6}. In this structure, all six of the Mn centers are not in similar environment as one of the terminal Mn atoms from each Keggin type cage are coordinated with a water molecule. The Mn–Mn distance in the crystals are not all identical with a minimum distance of 3.30 Å and maximum distance of 3.35 Å. From these Mn–Mn distances we can conclude that there is no Mn–Mn magnetic coupling possible in the molecule. The oxidation state of manganese is +II, which is confirmed from bond valance sum method.63 The characteristic FT-IR peaks (cm−1) are: 1617 (δH2O), 1065 (νP–O), 932 (νWO), 730 (νW–O–W) and 480 (νMn–O) cm−1 (Fig. 2c). The Raman peaks (cm−1) at: 488, 602, 797 and 933 are attributed to (νas,Mn–O), (νWO) and (νP–O), respectively (Fig. 2d). From EAS spectroscopy, the absorption maximum is found to be at 250 nm (Fig. 4b). The structure can be obtained in the CCDC database by quoting the CCDC no. 1057222.
A detailed account of the synthesis of {Mn6P3W24} is given in the experimental section.
On active catalyst (2)
:Mn:W in crystal is 1:2.22:8.3, which is very close to the ratio found in the crystal structure of compound {Mn6P3W24}. The hydrodynamic radius of 2 is measured from the DLS experiment, which shows an average hydrodynamic radius of 130 nm (Fig. 4e). The SEM image also indicates the radius of the vesicles is around 150 nm (Fig. 3b). We used this SOM dispersion of 2 as an active catalyst in our experiments.
Molecular structure of Mo132
{Mo132} of the formula {Mo132O372(CH3COO)30(H2O)72}42− is a type of molybdenum brown “Keplerate” anion with a diameter of 2.9 nm. This spherical cluster contains 132 molybdenum centers of which 72 molybdenum centers are in the +VI oxidation state and the remaining 60 molybdenum centers are in the +V oxidation state. The dark brown color of Mo132 arises due to intervalence charge transfer between the MoV and MoVI centers. Such a Keplerate cluster assembled with acetate as the ligand has an icosahedral point group symmetry. 12 pentagonal {(Mo)Mo5} building blocks are disposed at twelve vertices of an icosahedron. Such disposition creates space for 30 linkers on the icosahedron's surface, which simultaneously link to the prevalent pentagonal building blocks. Omitting the oxygen atoms for the sake of simplicity, the linkers are {Mo2(CH3COO)} in the case of {Mo132}. Hence, the overall cluster formulation can de described as [pentagon]12[linker]30 or {(Mo)Mo5}12{Mo2(acetate)}30 or {Mo132(acetate)30} or {Mo132} or {Mo132O372(CH3COO)30(H2O)72}42−.The IR spectrum (Fig. 2e) shows main peaks at 1635 (δH2O), 1400 (δCH3, νsCOO, δasNH4+), 1020, 748 and 565 cm−1.59 Raman bands (Fig. 2f) are observed at 950 (νMoO), 875, 375 and 314 cm−1 and correspond to the irreducible representations, Hg and A1g (bands with the highest intensity). The electronic absorption spectrum shows an intense band at 450 nm owing to Mo ← O charge transfer (Fig. 4c).
Mo132 was synthesized following a literature procedure.59
On active catalyst (3)
Now we describe the catalytic activity of 1, 2, and 3 in the matter of the photochemical CO2 reduction reaction.
Photocatalytic reduction of CO2
In our present case we used 1, 2 and 3 as photocatalysts for photochemical CO2 reduction. Among the three catalysts, 1 and 2 are one component catalysts while 3 is a two component catalyst. RGO is used for the facile transport of electrons. The principle product of CO2 reduction is formic acid; however, we have also obtained formaldehyde in significant amounts with catalysts 1 and 3. In terms of formic acid production, with respect to every unit of super-structure, 3 gives a maximum yield of formic acid of 205 μmol with a turnover number of ca. 1.4 × 106 in water (while per mole turnover number is 1366). On the other hand, 1 and 2 gives a TON of 0.9 × 106 and 0.25 × 106 (whereas, per mole values are 778 & 270), respectively, at 0.15 μmol loading (for details on the turnover number calculation see later). Here water gets oxidized to produce oxygen, protons and electrons; these protons and electrons are responsible for the reduction of CO2.The formation of formic acid was first characterized by matrix-assisted laser desorption ionization-mass spectroscopy (MALDI-MS) using a α-cyano-4-hydroxycinnamic acid matrix (Fig. 5a) and quantified by high performance liquid chromatography (HPLC) (against an external standard 0.1 M formic acid). Furthermore, the produced formic acid was characterized by coupling it with aniline in the presence of HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate) as a coupling agent to form the amide (Fig. 5b), which was detected by MALDI-MS using an HCCA matrix (Fig. 5c) and 1H NMR spectroscopy (Fig. 5d). The generation of formaldehyde in the reaction mixture was quantified using HPLC (against an external standard of 0.1 M formaldehyde). Formaldehyde was obtained only in the case of 1 and 3; 2 selectively reduced CO2 to formic acid. No other gaseous reduced product was formed during the course of CO2 reduction as confirmed by gas chromatography-mass spectroscopy (GC-MS). Formation of formic acid was further confirmed from the cyclic voltammogram (CV) where we observed a peak around −0.60 V vs. Ag/AgCl electrode, which corresponds to the reduction potential of CO2/HCOOH (Fig. 6a). Simultaneously water oxidation during the course of CO2 reduction was characterized using an YSI Clark type electrode. Further confirmation of water oxidation was obtained from the CV where we observed a sharp increase in current at 1.2 V vs. Ag/AgCl electrode (Fig. 6a), typical of water oxidation. All the reactions were carried out at least five times & the average data for the five sets were taken and analysed accordingly.
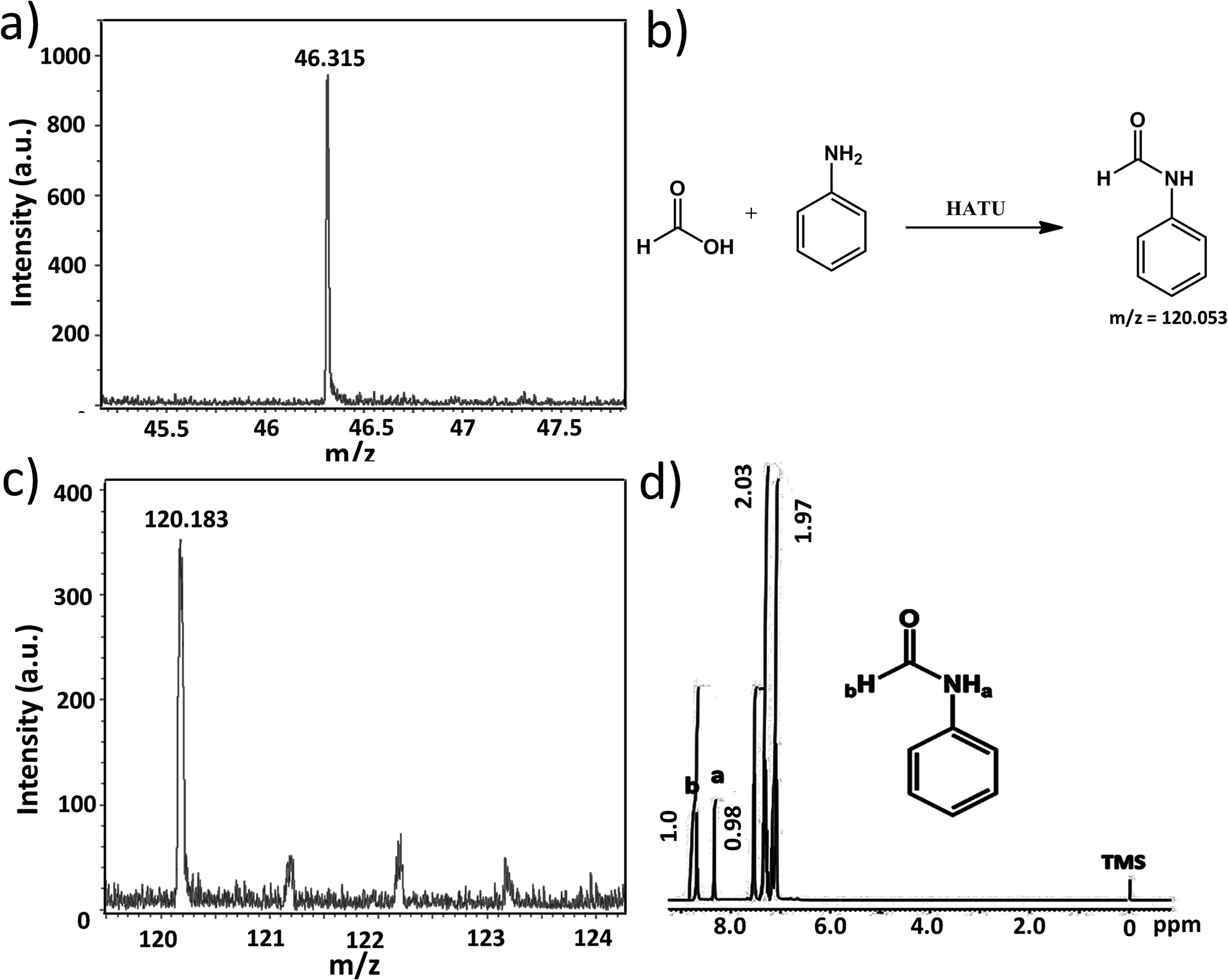 | ||
| Fig. 5 (a) MALDI-MS of the product after CO2 reduction. The formation of formic acid is observed as a peak is seen at 46.31. (b) The reaction of aniline coupling to form amide. (c) MALDI-MS of amide. (d) NMR of amide formed. | ||
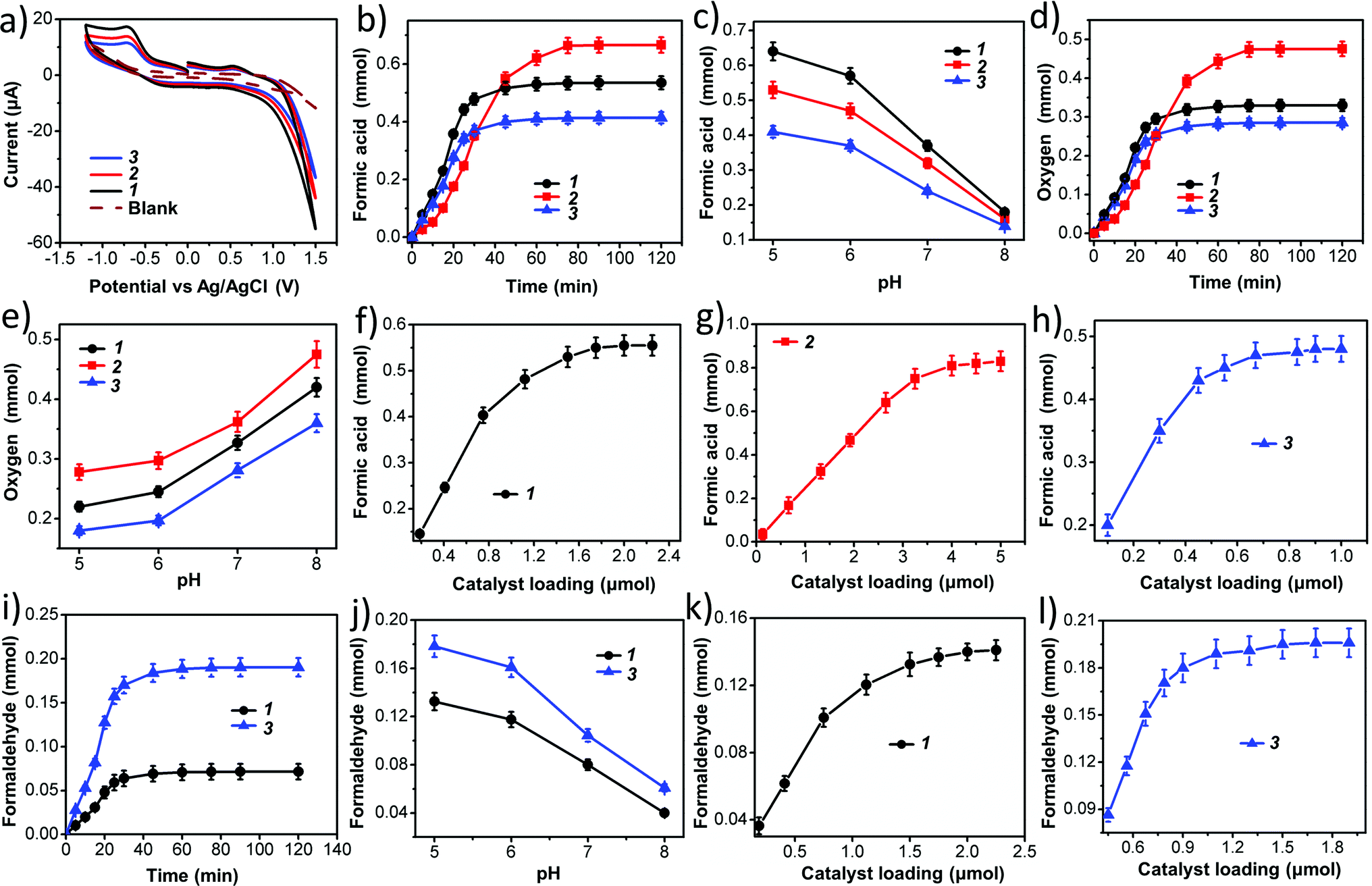 | ||
| Fig. 6 (a) Cyclic voltammetry of the reaction product after CO2 reduction coupled with water oxidation, (b) time-dependent formic acid formation by photochemical carbon dioxide reduction using 1, 2, 3 as the catalyst at pH 5, (c) pH-dependent formic acid formation, (d) time-dependent evolution of oxygen at pH 5, (e) pH-dependent O2 formation, (f, g & h) variation of catalyst 1 – {Mo154}1165, 2 – {Mn6P3W24}931, 3 – {Mo132}1064 on formic acid formation, respectively, (i) time-dependent generation of formaldehyde, catalyst 2 shows no formaldehyde generation, (j) pH variation with catalyst 1 and 3 to get maximum formaldehyde at pH 5 for both cases, and (k) & (l) variation of catalyst loading on formaldehyde formation for 1 and 3, respectively. | ||
Next we performed time-dependent experiments to investigate the kinetics of the reaction. It was observed that for 1 and 3 the reaction is almost completed within 40 minutes (Fig. 6b and i), while 2 takes 75 minutes to complete the reaction (Fig. 6b). These reactions are relatively rapid. This is probably due to the presence of a large number of active catalytic centers in our catalysts.
Then, we investigated the effect of the pH of the reaction medium on the photochemical CO2 reduction reaction as well as the photochemical water oxidation reaction. CO2 reduction increases with decreasing pH (Fig. 6c and j) while water oxidation reaction is hindered (Fig. 6e). This can be explained by considering three different reaction equilibria (eqn (4)–(6)).
| 2H2O → 4H+ + O2 + 4e | (4) |
| CO2 + 2H+ + 2e → HCOOH | (5) |
| CO2 + 4H+ + 4e → 2HCHO | (6) |
As we know that carbon dioxide reduction is a proton coupled reaction and therefore, formation of the reduced product is always favored with increasing proton concentration in the reaction medium i.e. decreasing the pH of the reaction medium. On the other hand, the water oxidation reaction releases protons into the reaction medium and therefore, removal of protons from the reaction medium enhances the rate of water oxidation. Thus, with an increasing pH of the reaction medium, the rate of water oxidation increases.
Catalyst concentration dependent studies on the reactions follow the normal trend of heterogeneous catalyst kinetics.66 Initially the concentration of reduced product increases with the concentration of catalyst up to a certain limit of catalyst loading. Then, the rate of the increment of product formation decreases with catalyst loading and finally, the reaction becomes independent of catalyst concentration. This is due to the fact that such reactions are totally dependent on the active surface area of the catalyst, which does not change after a certain loading of the catalyst. We already mentioned that vesicle-like super-structures of oxometalates in the dispersion are actual active catalysts, therefore the active surface area is generally the surface area of a vesicle, which increases with increasing radius (i.e. hydrodynamic radius) of the vesicle. It is observed that after certain loading of catalyst, the hydrodynamic radius does not increase with the concentration of oxometalate as phase instabilities set in. Thus, after reaching a certain value, the active surface area remains constant, which is reflected in the nature of product concentration profile with catalyst loading (Fig. 6f–h, k and l).
Calculation of the active catalyst concentration and thus, the TON of the reaction
In the case of 1, 2, and 3, 116.7 μmol, 40.6 μmol and 205 μmol formic acid was obtained, respectively with a loading of 0.15 μmol catalyst in each case. The turnover numbers of the reaction per mole of the catalysts are 778, 270 and 1366 for 1, 2, and 3, respectively. We have already mentioned that these oxometalates undergo self-assembly in a dispersion to form a vesicle-like SOM super-structure.43,44,62,67 Therefore, the active catalyst in reaction medium is different to that found for the solid catalyst we have taken in the reaction. Also, due to the self-assembly active surface area of all the catalysts increasing when compared to the surface area of solid catalysts. We have calculated the active concentration of the catalysts, i.e., the number of catalyst molecules forming the SOM vesicle in the dispersion (Table 1).| Catalyst | R (nm) | σ (Å) | Number of metal oxide clusters present per vesicle of SOM |
|---|---|---|---|
| Mo132@RGO | 302 | 29.92 | 1064 |
| Mn6P3W24 | 130 | 13.78 | 931 |
Considering the effective concentration of the catalysts in the reaction, we calculated the effective turnover number of the catalysts. The results are summarized and tabulated in Table 2.
| Catalyst | Amount of solid catalyst (p) (μmol) | Number of catalyst molecule formed catalyst vesicle (m) | Concentration of catalyst vesicle in dispersion (q = p/m) (nmol) | Formic acid formed in reaction (r) (μmol) | TON per mole catalyst (s = r/p) | TON with respect to concentration of catalyst SOM vesicle (t = r/q) | TOF (s−1) |
|---|---|---|---|---|---|---|---|
| 1 | 0.15 | 1165 | 0.129 | 116.7 | 778 | 0.9 × 106 | 377 |
| 2 | 0.15 | 931 | 0.161 | 40.6 | 270 | 0.25 × 106 | 56 |
| 3 | 0.15 | 1064 | 0.140 | 205 | 1366 | 1.4 × 106 | 610 |
In the case of Mo154, the number of metal oxide cluster units present in a single SOM vesicle is 1165.62 Thus, we have calculated the number of metal oxide clusters present per SOM vesicle using eqn (7).
| No. of metal oxide cluster unit = (4πR2)/(72.006σ2) × 60 | (7) |
We can now calculate the actual turnover number of this reaction, which is obtained dividing the concentration of product by the concentration of active catalyst vesicles in the dispersion (Table 2). We have performed each experiment five times, so an average yield out of those sets of experiments has been considered to calculate the turnover numbers. In this way the turnover numbers were found to be 0.9 × 106, 0.25 × 106 and 1.4 × 106 for 1, 2, and 3, respectively.
The water oxidation kinetics for all three catalysts are identical to the carbon dioxide reduction kinetics (Fig. 6d). This indicates that the water oxidation and carbon dioxide reduction reactions take place simultaneously. This also indicates an interesting fact that carbon dioxide reduction in this reaction is coupled with the water oxidation reaction. We will now check whether these two processes are indeed coupled.
On the coupling of the redox processes: the dependency of CO2 reduction on water oxidation
To check whether CO2 reduction depends on the water oxidation reaction, we have performed the photochemical CO2 reduction reaction in dry dimethyl formamide (DMF), keeping the other conditions unchanged. In this aprotic solvent, we observed that there is no reduction of CO2 (Fig. 7a and c) indicating that photochemical CO2 reduction may depend on the photochemical water oxidation reaction. To further prove the dependency, we then performed the reaction using different ratios of water and DMF. Upon gradually increasing the amount of water in the reaction mixture, we noticed a gradual increase in the amount of formic acid and evolved oxygen in the reaction system (Fig. 7b). We have also shown the increase in the generation of HCOOH and O2 by cyclic voltammetry (Fig. 7a). In the CV, we see no peak at −0.6 V in the absence of water, which indicates the absence of any reduced products of CO2 like HCOOH in the reaction medium. Upon gradually increasing the amount of water, there is an increase in the peak at −0.6 V along with a simultaneous increase in the current at 1.2 V, which indicates water oxidation and the production of O2. A similar trend is seen in the amount of HCOOH and O2 formed. For instance, the production of HCOOH increases upon increasing the amount of water. The above results confirm that photochemical CO2 reduction depends on photochemical water oxidation and that these processes are coupled.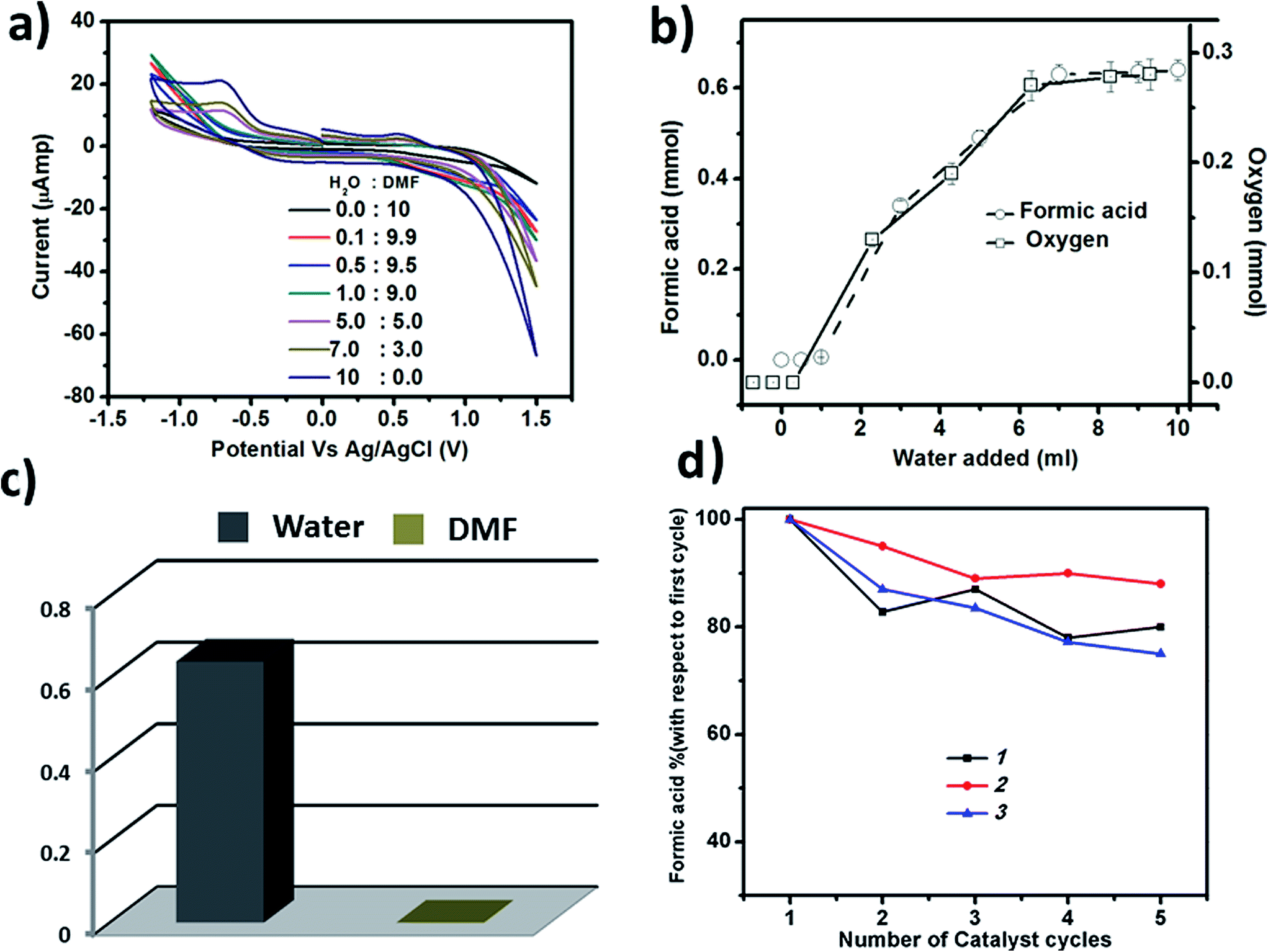 | ||
| Fig. 7 (a) The CV of the solution after the reaction with a gradual increase in the water amount, (b) the increase in the amount of formic acid and oxygen with an increase in the water amount in reaction system, (c) the amount of formic acid formation in 10 mL of water and 10 mL of DMF, showing no formic acid formation in DMF, and (d) the reusability of all three catalysts shown up to 5 cycles of the reaction in terms of the yield of formic acid. | ||
On the reaction pathway
Now, we can delineate the likely reaction pathway. To do so we first identified the essential components of the reaction: 1. Light: without light the reaction does not take place and 2. Water: we have already seen that without water there is no CO2 reduction. All these demonstrate that the catalytic reaction has three crucial components: light, water and CO2.We now sketch a pathway for the reaction. We speculate that the initial clusters go to their excited state upon irradiation with UV-light. Clusters in the excited state generate excitons that in turn create holes (h+) and electrons (e−) in pairs. Now, the generated holes (h+s) oxidize H2O to produce oxygen and release electrons and protons in the reaction.68 Proton coupled reduction of CO2 to form HCOOH/HCHO takes place by subsequent transfer of the electrons and protons released during water oxidation. Thus, in this photochemical CO2 reduction coupled with water oxidation water acts as the only electron source in the reduction reaction. After each cycle the catalyst goes back to its initial state and gets re-excited by light again and the catalytic cycle continues.
Now, to show the actual catalyst in the reaction medium, we performed the photochemical CO2 reduction, keeping the other conditions the same using precursor constituents of 1, 2 and 3. No reaction was observed with those precursor constituents (Table S1 in the ESI†). It implies that the vesicle-like SOM superstructures composed of giant metal oxide clusters are only responsible for the photochemical CO2 reduction reaction in water. All these results are tabulated in Table S2.†
The stability of catalysts 1, 2 and 3
The catalysts are stable up to many cycles of the reaction. This is shown by Raman and infrared spectroscopy (shown in Fig. 8 & S13–S15†). For instance, we have reused the catalysts for five cycles and have seen a maximum 25% decrease in the yield for the production of HCOOH, in the case of catalyst 3. In the case of 1, the decrease is 20% while 2 exhibits the maximum reusability with a decrease in yield of formic acid of only 12% after five cycles (Fig. 7d). Thus, all the catalysts can be reused further to catalyse the same reaction.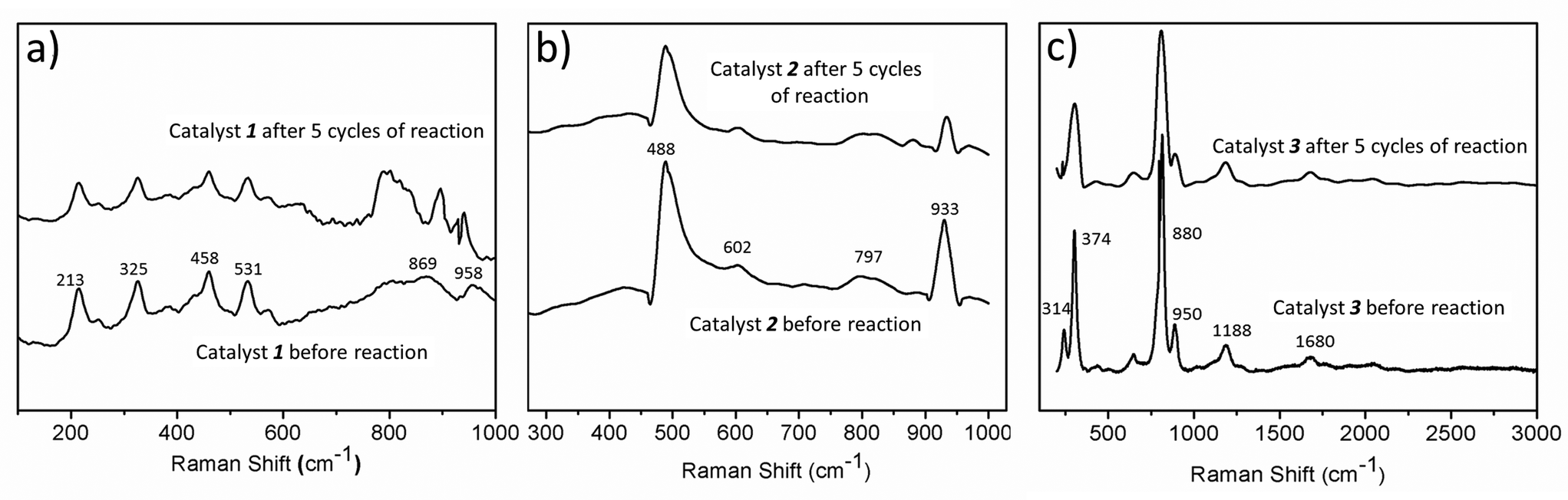 | ||
| Fig. 8 The stability of all three catalysts after the reaction, as shown by Raman spectroscopy. | ||
On the photochemical activity of the catalysts
To show that the catalysts are photochemically active under the reaction condition, we have performed dye degradation studies using 1, 2 and 3. We have used methyl orange for 1, methylene blue for 2, while perylene tetracarboxylate was used in the case of 3, to avoid the absorption maxima overlapping with the catalysts. Methyl orange shows an absorption maxima at 455 nm, methylene blue shows at 560 nm and perylene tetracarboxylate has three bands at 385, 410 and 415 nm. The UV spectra of the dye with the catalysts show that all of the catalysts can degrade their corresponding dyes upon irradiation as evident from the decreasing intensity of the UV-Vis maxima of the dyes with time (Fig. 9).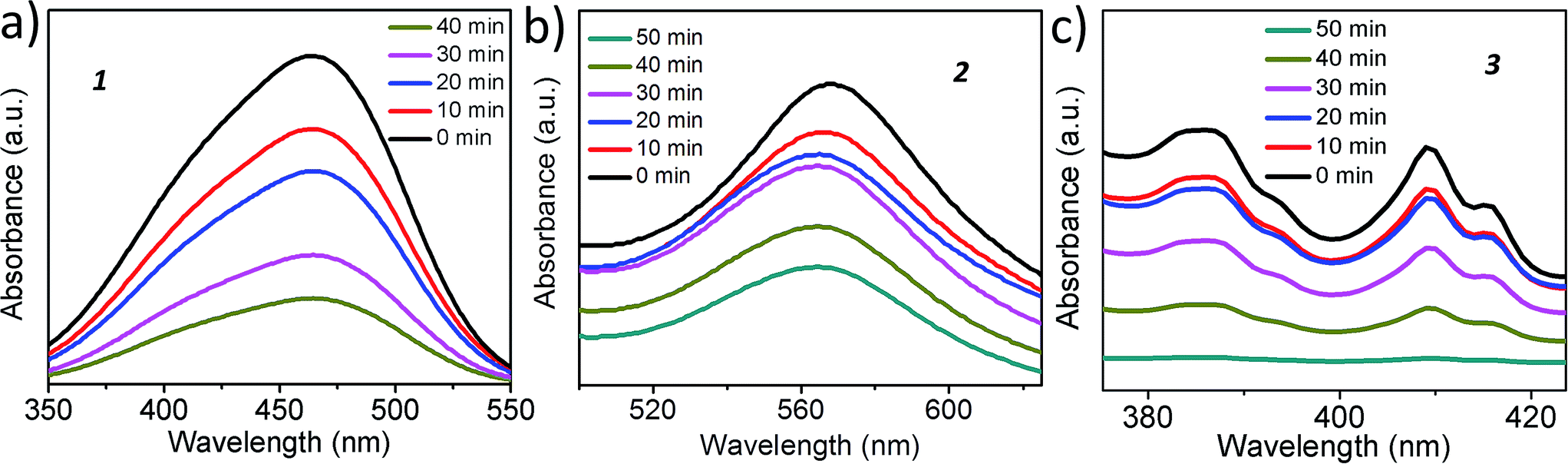 | ||
| Fig. 9 Dye degradation studies of catalyst 1 using methyl orange, 2 using methyl blue and 3 using perylene tetracarboxylate. A gradual decrease in the intensity of the dyes with the irradiated catalysts proves the photoactivity of the catalysts. | ||
Here, it is worth mentioning that the SOM-type catalysts provide better catalytic activity than the conventional catalysts owing to the simultaneous excitation of millions of photoactive clusters in the catalyst vesicles under photochemical conditions due to the high number density of the molecules in the vesicles, which is usually not the case in conventional semiconductors used as photocatalysts.
Experimental section
Materials and reagents
All the materials were purchased from commercially available sources and used without further purification. All glassware was first boiled in an acid bath then washed with water and finally cleaned with acetone. They were dried in hot air oven overnight. We used doubly distilled deionized water in all the experiments.(Na15[MoVI126MoV28O462H14(H2O)70]0.5[MoVI124MoV28O457H14(H2O)68]0.5·hydrate). To a solution of Na2MoO4·2H2O (3.0 g, 12.4 mmol) in 10 mL of water, freshly powdered Na2S2O4 (0.2 g, 1.15 mmol) was added (light yellow coloration). Immediately afterwards, under continuous stirring, 30 mL of hydrochloric acid (1 M) was rushed into the solution (color changes to deep blue). The solution was stirred in an open 100 mL Erlenmeyer flask for further 10 min and then stored undisturbed in a closed flask at 20 °C for 3 days. The precipitated blue crystals were removed by filtration, washed quickly with a small amount of cold water and dried at room temperature over CaCl2. Yield: 0.7 g (13.18% based on Mo).
Synthesis of Mo132. The cluster was prepared according to a literature procedure.69 N2H4·H2SO4 (0.8 g, 6.1 mmol) was added to a solution of (NH4)6Mo7O24·4H2O (5.6 g, 4.5 mmol) and CH3COONH4 (12.5 g, 162.2 mmol) in H2O (250 mL). The solution was then stirred for 10 min (color change to blue green) and 50% CH3COOH (83.0 mL) was subsequently added. The reaction solution, now green, was stored in an open 500 mL Erlenmeyer flask at 20 °C without further stirring (fume hood; slow color change to dark brown). After 4 days, the precipitated red-brown crystals of were filtered off over a glass fritt (D2), washed with 90% ethanol, ethanol and diethyl ether, and finally dried in air. Yield: 3.3 g (52% based on molybdate). Raman spectrocopy confirmed the structure of the prepared Mo132.
Synthesis of RGO. Firstly, graphene oxide (GO) was prepared via the modified Hummers' method.70 Typically, 50 g of H2SO4 (98%) and 2 g of graphite were placed in a reactor cooled to 0 °C using an ice-water bath. After stirring the suspension for 30 min, 0.3 g of KMnO4 was added in small portions keeping the temperature in the reactor less than 10 °C using an ice-water bath. After stirring the mixture for 30 minutes, 6 g of KMnO4 was further added to the suspension gradually. Then, the reactor was heated for 30 minutes to keep the temperature constant at approximately 35 °C. As the reaction progressed, the suspension became pasty and brown in color. At the end of this 30 min period, 90 mL of water was slowly stirred into the paste to prevent violent effervescence, causing an increase in temperature from (90 to 95) °C. The diluted suspension, now brown in color, was maintained at this temperature for 15 min. The suspension was then further treated with a mixture of 7 mL of hydrogen peroxide (30%) and 53 mL of water to reduce the residual permanganate and MnO2 to soluble MnSO4. The suspension was filtered and washed with distilled water three times and the solids were collected by filtration and dried at 45 °C. Graphene was prepared via a hydrothermal process using a basic solution. Generally, 2 g of GO was mixed in 80 mL of ultrapure water and the mixture was stirred for 4 h. The suspension was then ultrasonically treated for 1 h. A concentrated ammonia solution (37 wt%) was used to adjust the suspension to pH 10. The suspension was kept stirred overnight with a cover. Then, the mixture was transferred into a Teflon-lined autoclave (120 mL) and treated at 453 K for 18 h. The precipitate was washed three times with ultrapure water/ethanol. After filtration, the chemically reduced graphene oxide was dried at 353 K.
Preparation protocol and stability of the Mo132@RGO composite. We have added a fixed ratio of RGO and Mo132 and kept it for 1 week in the dark after sonication for 4 h. We have seen that only a RGO/Mo132 ratio of 1.2
:1 provides a stable composite for which a dispersion is stable, even for months. So, this ratio was taken for characterization and the photochemical experiments.
Characterization techniques
969 unique reflections (Rint = 0.0505, 2θmax = 50.06) 18947 were considered to be observed with (I > 2σ(I)). The final cycle of refinement, including the atomic coordinates, anisotropic thermal parameters, (W, P, and Ni atoms), and isotropic thermal parameters (Na and O atoms), converged at R = 0.0869 and Rw = 0.221 (I > 2σ(I)) In the final difference map the highest peak was 7.83 e Å−3 and the deepest hole −10.784 e Å−3. The unique crystal data number is CCDC 1057222.
Next we performed CV experiments with the reaction mass using KCl as an electrolyte within a potential range of +1.5 V to −1.2 V with respect to the Ag/AgCl reference electrode in a standard 3-electrode system. We obtained a peak around −0.6 V, which is indicative of the formation of formic acid from carbon dioxide. To further prove the formation of formic acid in the reaction medium we performed a coupling reaction with the resulting reaction solution. An excess solution of aniline (100 μL) in 2 mL of acetonitrile was added to the reaction mixture and 20 mg HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate) was added as a coupling agent. The reaction mixture was stirred for 1 h at room temperature resulting in the formation of the amide in the reaction medium. The organic components were extracted with EtOAc (3 × 10 mL) and then, the EtOAc was removed in vacuo. Then, the solid mass was further dissolved in acetonitrile to perform GC-MS and MALDI-MS following the above mentioned procedure. After purification we further characterized the amide using 1H NMR spectroscopy.
Conclusions
We have shown a scalable pathway for photochemical carbon dioxide reduction coupled with water oxidation using three different SOM type heterogeneous mixed valent metal oxide based clusters with the highest turnover numbers recorded till date (ca. 1366 per mole, effectively 1.4 × 106). All these metal oxide based clusters are photoactive and therefore, there is no need for any external photosensitizer in the reaction medium. Here water serves as an electron source and when coupled with carbon dioxide reduction, water oxidation obviates the need of any external sacrificial electron donor. Such a successful coupling enabled us to execute the simultaneous reduction of CO2 to HCOOH/HCHO and oxidation of water to oxygen achieving an effective turnover number as high as 1.4 × 106. Thus, in short we have developed a fast, scalable and efficient carbon dioxide reduction model system where water oxidation provides electrons to reduce carbon dioxide. The possibility of adding a visible light sensitizer to make this system operate in sunlight does not escape our attention. The end product formic acid can be used directly in fuel cells providing green and clean energy.Author's contribution
SD, SB and TB contributed equally to this work. SR proposed, initiated and coordinated the project, as well as wrote the paper.Acknowledgements
SR gratefully acknowledges the grants obtained from IISER-Kolkata, India, DST-fast track and BRNS-DAE. SD and TB acknowledge IISER-K; SB and SB (Barman) acknowledge UGC and RP acknowledges DST for fellowship. This paper is dedicated to Prof. Achim Müller.Notes and references
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- J. Collin and J. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- R. D. Richardson, E. J. Holland and B. K. Carpenter, Nat. Chem., 2011, 3, 301–303 CrossRef CAS PubMed.
- Y. Oh and X. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- J. Michl, Nat. Chem., 2011, 3, 268–269 CrossRef CAS PubMed.
- E. de Smit, F. M. de Groot, R. Blume, M. Hävecker, A. Knop-Gericke and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 667–680 RSC.
- J. K. Hurst, Science, 2010, 328, 315–316 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–36 CrossRef CAS PubMed.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- H. J. Lewerenz and L. Peter, Photoelectrochemical Water Splitting: Materials, Processes and Architectures, Royal Society of Chemistry, 2013 Search PubMed.
- M. Bourrez, M. Orio, F. Molton, H. Vezin, C. Duboc, A. Deronzier and S. Chardon-Noblat, Angew. Chem., Int. Ed., 2014, 53, 240–243 CrossRef CAS PubMed.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- C. L. Anfuso, R. C. Snoeberger, A. M. Ricks, W. Liu, D. Xiao, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2011, 133, 6922–6925 CrossRef CAS PubMed.
- B. D. Steffey, A. Miedaner, M. L. Maciejewski-Farmer, P. R. Bernatis, A. M. Herring, V. S. Allured, V. Carperos and D. L. Dubois, Organometallics, 1994, 13, 4844–4855 CrossRef CAS.
- M. Erlandsson, V. R. Landaeta, L. Gonsalvi, M. Peruzzini, A. D. Phillips, P. J. Dyson and G. Laurenczy, Eur. J. Inorg. Chem., 2008, 2008, 620–627 CrossRef.
- C. Das Neves Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Chem.–Eur. J., 2014, 20, 7098–7106 CrossRef CAS PubMed.
- J. Zhao, X. Wang, Z. Xu and J. S. Loo, J. Mater. Chem. A, 2014, 2, 15228–15233 CAS.
- M. C. Hsieh, G. C. Wu, W. G. Liu, W. A. Goddard and C. M. Yang, Angew. Chem., 2014, 126, 14440–14444 CrossRef.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- P. Kang, Z. Chen, A. Nayak, S. Zhang and T. J. Meyer, Energy Environ. Sci., 2014, 7, 4007–4012 CAS.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CAS.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 CAS.
- N. A. Vante, W. Jaegermann, H. Tributsch, W. Hoenle and K. Yvon, J. Am. Chem. Soc., 1987, 109, 3251–3257 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- S. Roy, CrystEngComm, 2014, 16, 4667–4676 RSC.
- S. Roy, Comments Inorg. Chem., 2011, 32, 113–126 CrossRef CAS.
- S. Das, S. Kumar, A. Mallick and S. Roy, J. Mol. Eng. Mater., 2015, 03, 1540008 CrossRef CAS.
- A. Mallick, D. Lai and S. Roy, New J. Chem., 2016, 40, 1057–1062 RSC.
- S. Roy, H. J. D. Meeldijk, A. V. Petukhov, M. Versluijs and F. Soulimani, Dalton Trans., 2008, 2861–2865, 10.1039/b800680f.
- S. Roy, L. C. A. M. Bossers, H. J. D. Meeldijk, B. W. M. Kuipers and W. K. Kegel, Langmuir, 2008, 24, 666–669 CrossRef CAS PubMed.
- A. Müller and S. Roy, Eur. J. Inorg. Chem., 2005, 2005, 3561–3570 CrossRef.
- S. Biswas and S. Roy, Journal of Materials NanoScience, 2014, 1, 6 Search PubMed.
- S. Das, P. Thomas and S. Roy, Eur. J. Inorg. Chem., 2014, 2014, 4551–4557 CrossRef CAS.
- P. Thomas, C. Pei, B. Roy, S. Ghosh, S. Das, A. Banerjee, T. Ben, S. Qiu and S. Roy, J. Mater. Chem. A, 2015, 3, 1431–1441 CAS.
- B. Roy, M. Arya, P. Thomas, J. K. Jürgschat, K. Venkata Rao, A. Banerjee, C. Malla Reddy and S. Roy, Langmuir, 2013, 29, 14733–14742 CrossRef CAS PubMed.
- K. Das and S. Roy, Chem.–Asian J., 2015, 10, 1884–1891 CrossRef CAS PubMed.
- D. Chen, A. Sahasrabudhe, P. Wang, A. Dasgupta, R. Yuan and S. Roy, Dalton Trans., 2013, 42, 10587–10596 RSC.
- A. Sahasrabudhe and S. Roy, J. Mol. Eng. Mater., 2014, 2, 1440002 CrossRef.
- S. Das, A. Misra and S. Roy, New J. Chem., 2016, 40, 994–1003 RSC.
- A. Müller, E. Krickemeyer, J. Meyer, H. Bögge, F. Peters, W. Plass, E. Diemann, S. Dillinger, F. Nonnenbruch and M. Randerath, Angew. Chem., Int. Ed., 1995, 34, 2122–2124 CrossRef.
- A. Muller, K. Das, E. Krickemyer and C. Kuhlmann, Inorg. Synth., 2004, 34, 191–200 Search PubMed.
- A. Müller, S. K. Das, V. P. Fedin, E. Krickemeyer, C. Beugholt, H. Bögge, M. Schmidtmann and B. Hauptfleisch, Z. Anorg. Allg. Chem., 1999, 625, 1187–1192 CrossRef.
- A. Müller and C. Serain, Acc. Chem. Res., 2000, 33, 2–10 CrossRef.
- T. Liu, E. Diemann, H. Li, A. W. Dress and A. Müller, Nature, 2003, 426, 59–62 CrossRef CAS PubMed.
- I. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- T. Liu, J. Am. Chem. Soc., 2002, 124, 10942–10943 CrossRef CAS PubMed.
- M. S. A. Sher Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 CAS.
- M. Boudart and G. Djéga-Mariadassou, Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press, 2014 Search PubMed.
- A. Orlov, A. Roy, M. Lehmann, M. Driess and S. Polarz, J. Am. Chem. Soc., 2007, 129, 371–375 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 CAS.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3359–3363 CrossRef.
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. O. Tadé and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466–5471 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization and stability of the catalysts during the reactions and product quantification table. CCDC 1057222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ta02825j |
| This journal is © The Royal Society of Chemistry 2016 |