
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward highly efficient in situ dry reforming of H2S contaminated methane in solid oxide fuel cells via incorporating a coke/sulfur resistant bimetallic catalyst layer†
Bin
Hua
a,
Ning
Yan
b,
Meng
Li
c,
Yi-Fei
Sun
a,
Jian
Chen
d,
Ya-Qian
Zhang
a,
Jian
Li
c,
Thomas
Etsell
a,
Partha
Sarkar
e and
Jing-Li
Luo
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: jingli.luo@ualberta.ca; Fax: +1 780 492 2881; Tel: +1 780 492 2232
bVan't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Amsterdam, 1098XH, The Netherlands
cCenter for Fuel Cell Innovation, School of Materials Science and Engineering, State Key Laboratory of Material Processing and Die & Mould Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
dNational Institute for Nanotechnology, Edmonton, Alberta T6G 2M9, Canada
eEnvironment & Carbon Management Division, Alberta Innovates-Technology Futures, Edmonton, Alberta T6N 1E4, Canada
First published on 24th May 2016
Abstract
The escalating global warming effects are a reason for immediate measures to reduce the level of greenhouse gases. In this context, dry reforming of methane (DRM), an old yet both scientifically and industrially important process, is making a comeback in contributing to the utilization of CO2. However, catalyst deactivation (sulfur poisoning and coke formation) and the associated high energy consumption remain technological hurdles to its practical implementation. Here we demonstrated that dry reforming of H2S-containing CH4 can be efficiently conducted in conventional solid oxide fuel cells via incorporating a coke/sulfur resistant catalyst layer. The add-on layer, composed of tailored Ce0.8Zr0.2O2 supported NiCu nanoclusters, demonstrated outstanding in situ reforming activity while possessing reasonable coke/sulfur resistance. At 800 °C and in a 50 ppm H2S containing CH4–CO2 mixture, the cell had a maximum power density of 1.05 W cm−2, a value high enough for practical application. Through H2 selective oxidation, the energy required for DRM was partially compensated for and the produced water greatly suppressed the carbon deposition. This study offers a new dimension in cogenerating CO2-derived synthesis gas and electrical power in the context of increasing interests in efficient utilization of H2S-containing CH4 and CO2.
1. Introduction
Synthesis gas or briefly, syngas, is a mixture of carbon monoxide and hydrogen, which is normally referred to as a “chemical intermediate” that could be readily transformed into numerous value-added chemicals or fuels by Fischer–Tropsch processes.1–3 For over a century, much attention from both academia and industries has been paid to syngas production towards more efficient, sustainable, and environmentally benign conversion of fossil fuel feedstocks.4–8 Due to the large reserves of natural gas worldwide along with the recent boom of shale gas/biogas, methane becomes an ideal “precursor” of syngas not just for today but also for the foreseeable future.9–11 There are typically three routes for methane-syngas conversion to date, namely, partial oxidation of methane (POM), steam reforming of methane (SRM) and dry reforming of methane (DRM). Notwithstanding the fact that SRM prevails over the rest in industry, DRM shows advantages over SRM, combining syngas production and greenhouse gas (GHG) utilization.Unfortunately, the practical implementation of DRM still faces several technical challenges:12–22 the first one is coke formation which deactivates almost every type of commercial catalyst for DRM (e.g., Ni).13–17 DRM is thermodynamically more prone to coking than other reforming reactions, albeit abundant achievements have been made regarding the development of coke-resistant catalysts, e.g., Ni and its alloys, during the past decades.14–17,23,24 This is logical since if excess CO2 is introduced to alleviate carbon deposition, it will remain as the main impurity in the effluent (excess H2O in SRM can be removed easily), decreasing the efficiency of the entire process. Secondly, the DRM reaction is extremely endothermic, requiring high temperatures to attain a reasonable yield of syngas. This huge energy input is also a matter of concern for the commercialization of DRM. The third challenge is associated with sulfur poisoning, because H2S is one of the components of methane sources. Once the commonly known H2S poison is present in the raw feedstock, the reforming efficiency will decrease substantially as a result of sulfur deactivation.21,22 Fortunately, much research activity has been dedicated to developing sulfur tolerant DRM catalysts which show promising performances.12,25–27
Solid oxide fuel cells (SOFCs), which typically operate at higher temperatures (above 500 °C) compared to other types of fuel cells, have received particular attention in recent years not only for power generation28–30 but also for the potential of performing simultaneous DRM reactions in their anode compartments.31–33 A high operation temperature is favored by DRM, and at the same time, the heat released during the electro-oxidation process can partially compensate the energy required for DRM. Nonetheless, the conventional Ni–Y2O3-stabilized-ZrO2 (YSZ) electro-catalyst shows neither good in situ dry reforming activity nor an excellent electrochemical performance in a CH4–CO2 mixture,34–36 not to mention that sulfur impurities always significantly deactivate its reforming capability. On one hand, the recent advancement of coke-sulfur resistant DRM catalysts is not fully utilized in SOFC research due to many practical barriers, e.g., the >1300 °C sintering temperature of SOFC fabrication. On the other hand, a typical Ni–YSZ supported cell usually shows a much decreased performance when being directly fed with equal amounts of CH4 and CO2,33 although performance can be improved by introducing noble metal catalysts37,38 or using novel fabrication methods.39,40 To combine the advantages of both hands, we herein design a novel SOFC reactor equipped with a highly coke/sulfur resistant triple-layer anode (TA-SOFC). The so-called electrochemical dry reforming (EDRM) process efficiently converts the H2S-containing (sour) CH4–CO2 into syngas and electricity.
2. Experimental
2.1. Material preparation and cell fabrication
Ni0.8M0.2 (NiM: M = none, Co, Cu, Fe)–Ce0.8Zr0.2O2 (ZDC) catalysts were prepared using a glycine–nitrate combustion process (GNP). The button cell (ϕ 13 × 1 mm) of the conventional SOFC (C-SOFC) is composed of a Ni–YSZ support layer, Ni–YSZ functional layer, YSZ electrolyte, Ce0.9Gd0.1O1.9 (GDC) buffer-layer and a 0.5 cm2 La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF)-GDC cathode. The triple anode layer SOFC (TA-SOFC) was constructed by coating the Ni0.8Cu0.2O (NiCuO)–ZDC slurry onto the surface of the Ni–YSZ support, followed by calcination at 900 °C for 2 h in air. Fabrication details of the cell can be found in the ESI.†2.2. Evaluation of catalyst performance in DRM process
Catalytic activity measurements for the DRM reaction were performed at atmospheric pressure from 550 to 800 °C using a fixed bed reactor (details are described in the ESI†). With regard to the assessments of sulfur tolerance, we performed an accelerated poisoning test via aging the catalysts in a H2S-containing feed stream (H2-500 ppm H2S) at 850 °C for 5 h. During the DRM, the gas mixtures of sweet (H2S-free) CH4–CO2 (mole ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or sour (H2S-containing) CH4–CO2 (mole ratio = 1:1, with 50 ppm H2S) were fed into the reactor at a flow rate of 20 ml min−1. Compositional analysis of the effluent gases was performed with a gas chromatograph (GC, Hewlett Packard Series two). The percentages of CH4 conversion and CO selectivity were calculated according to eqn (1) and (2), respectively.
:1) or sour (H2S-containing) CH4–CO2 (mole ratio = 1:1, with 50 ppm H2S) were fed into the reactor at a flow rate of 20 ml min−1. Compositional analysis of the effluent gases was performed with a gas chromatograph (GC, Hewlett Packard Series two). The percentages of CH4 conversion and CO selectivity were calculated according to eqn (1) and (2), respectively. | (1) |
 | (2) |
2.3. EDRM process measurements
A detailed description and explanation of EDRM are presented in Section 3.1 (vide infra). In the experiment, an Au paste current collecting layer was painted on both sides of the electrode surface and baked in air at 800 °C for 2 h. Pt mesh and wire were used as the current collector and the measuring lead, respectively. Button cells were sealed on an alumina tube using a Ceramabond® glass sealant (Type 552) and reduced in pure H2 atmosphere at 800 °C for 2 h. Prior to electrochemical evaluation, the anode was polarized in H2 or H2-500 ppm H2S (denoted as H2S treated cell below) at 850 °C and 0.8 V constant voltage for 5 h. After reduction and stabilization, I–V polarization curves and electrochemical impedance spectra (EIS) in H2 (40 ml min−1), CO (40 ml min−1), CO–H2 (mole ratio = 1:1, 40 ml min−1) and sweet CH4–CO2 (20 ml min−1, the gas hourly space velocity was ∼9041 h−1) were collected. The performance stability and corresponding effluent gas composition were also measured under constant current with sweet or sour CH4–CO2 (with 50 ppm H2S) as the feeds, and the flow rates were both 20 ml min−1. The air flow rate in the cathode was fixed at 500 ml min−1.
3. Results and discussions
3.1. Electrochemical dry reforming of methane (EDRM)
Fig. 1 shows a schematic of the electrochemical dry reforming of methane in TA-SOFC. In this design, an additional coke/sulfur resistant layer, composed of ZDC supported bimetallic alloys, was attached to the surface of the anode support to perform dry reforming. Prior to entering the Ni–YSZ anode, the sour CH4–CO2 feedstream is therefore effectively reformed, yielding syngas. Sequentially, the acquired syngas reaches the triple-phase boundaries (TPBs) where H2 is preferentially oxidized with negligible CO2 formation, pertaining to H2 conversion in SOFCs being several fold faster than that of CO (vide infra). This unique behavior became more prominent when sulfur was present, which might be attributable to the sulfur–nickel synergetic effect,41–43 as the sulfur poisoning suppressed the oxidation of CO much more significantly. In addition, the H2O generated from the H2 electrochemical reaction suppresses coke formation, and the heat released during the energy conversion process can partially compensate for that required for DRM.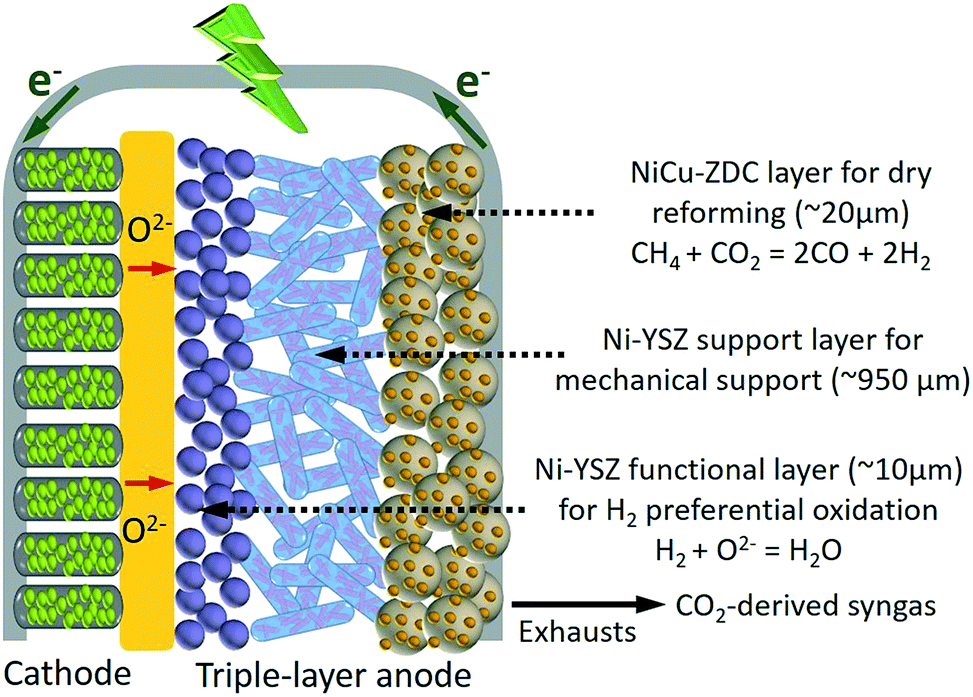 | ||
| Fig. 1 A schematic drawing of the electrochemical dry reforming of methane in SOFC. | ||
3.2. Screening sulfur/coke resistant DRM catalyst
The prepared NiM (M = none, Co, Cu, Fe) bimetallic alloys on ZDC were initially characterized via X-ray diffraction (XRD). The results showed that single metallic phases formed in all of the catalysts (Fig. S2†). For example, Fig. 2a displays the XRD patterns of the as-synthesized and reduced NiCuO–ZDC, confirming that the ZDC crystal remained stable in both the reducing and oxidizing atmospheres, whereas NiCuO was fully reduced to NiCu bimetallic alloy. The broad diffraction peak of NiCu also implied its fine particle size, ensuring high catalytic activity for the DRM reaction. The reduction behaviors of these catalysts were characterized using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) performed in 10% H2 (see Fig. 2b and S3†). As pure ZDC demonstrated good weight stability, we concluded that the two exothermic peaks (250.80 °C and 383.64 °C) of NiCuO–ZDC came from the reduction of NiCuO. Transmission electron microscopy (TEM) analysis was performed to further investigate both the compositional and morphological characteristics of the NiCu–ZDC catalyst. The bright field (BF) image in Fig. 2c shows the good dispersion of the bimetallic nanoparticles (NPs). The energy dispersive X-ray spectroscopy (EDX) elemental mappings further confirmed the formation of the bimetallic NiCu phase (cf. XRD data above). The high resolution TEM (HRTEM) micrographs in Fig. S4 †reveal that the NiCu NPs are spherical, 10 to 25 nm in size, and well dispersed over the cube-like ZDC (Fig. S5†). | ||
| Fig. 2 (a) XRD patterns of the as-synthesized and reduced NiCuO–ZDC; (b) TGA–DSC curves of ZDC and NiCuO–ZDC catalysts in 10% H2; (c) TEM bright field image of NiCu–ZDC and EDX elemental mappings of Ni, Co, Ce and Zr. | ||
The performances of the NiM–ZDC catalysts, regarding their catalytic activity and coke/sulfur resistance, for the DRM reaction were evaluated prior to EDRM application. The state-of-the-art Ni–YSZ electro-catalyst (calcined at 1390 °C to simulate the sintered state of the SOFC anode) was also included as a control. Fig. 3a to e compare the CH4 conversion, CO selectivity and the effluent composition when different catalysts were applied in sweet CH4–CO2 from 550 to 800 °C. Apparently, CH4 conversion and syngas yield increased rapidly with an increase of temperature. NiM–ZDC catalysts showed excellent activities, and >90% methane conversion was recorded at temperatures higher than 750 °C. Conversely, the performance of Ni–YSZ was rather poor, most likely as a result of the sintering effect causing severe agglomerations of Ni particles. In fact, this observation was in accordance with those documented in the literature, showing that the conventional Ni–YSZ anode of SOFCs was not capable of achieving effective internal DRM under SOFC conditions.44–46 Its inferior performances were also reflected in the measured low power density and high polarization resistance.39,44,45
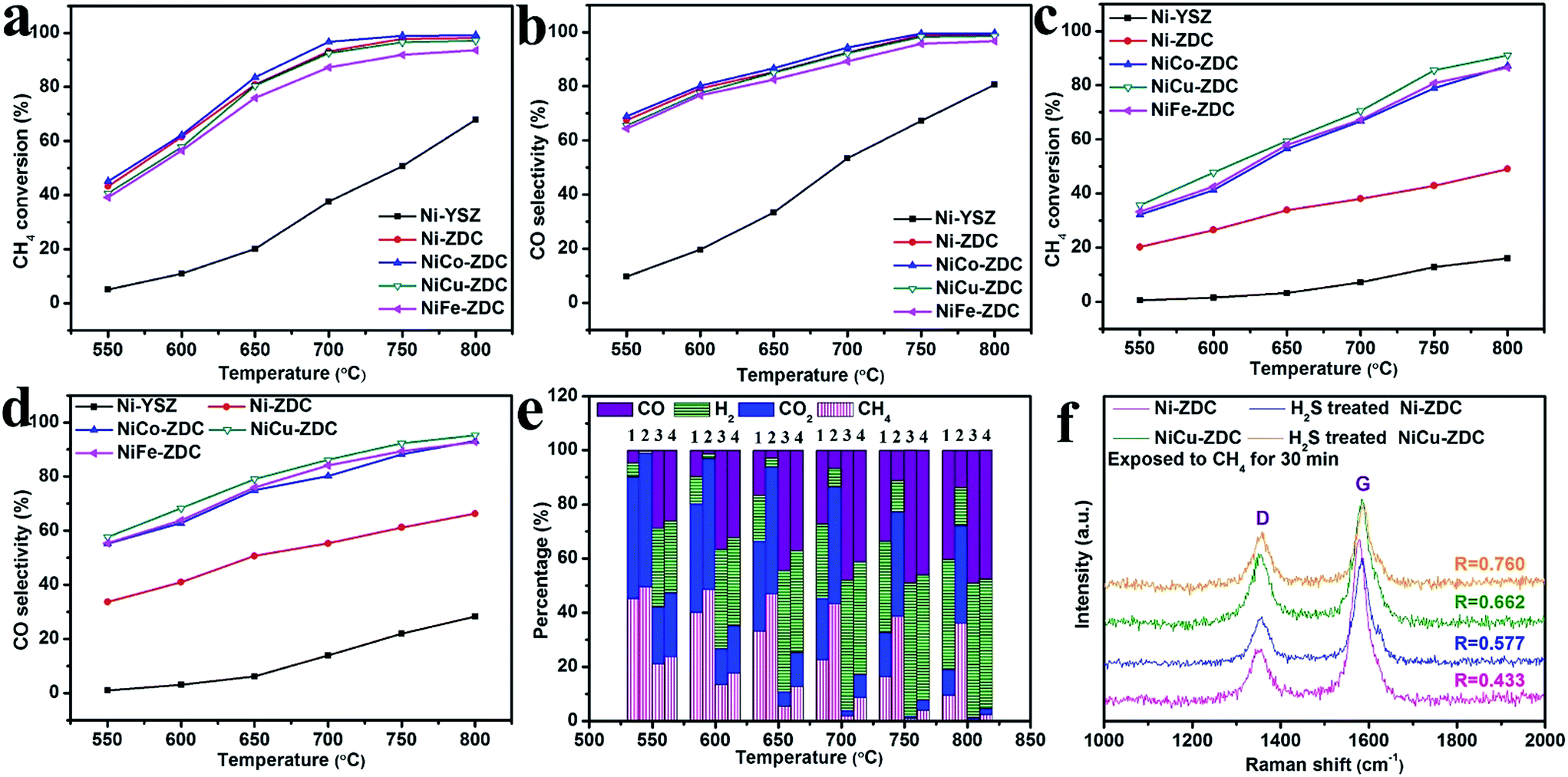 | ||
| Fig. 3 (a and c) CH4 conversion and (b and d) CO selectivity in the DRM process at various temperatures using (a and b) pristine and (c and d) 500 ppm H2S–H2 pretreated catalysts in a fixed bed reactor; (e) the effluent compositions when using (1) Ni–YSZ, (2) NiCu–ZDC, (3) H2S treated Ni–YSZ and (4) H2S treated NiCu–ZDC catalysts; (f) Raman spectra and R values of carbon deposits on the catalysts after being exposed to CH4 at 800 °C for 30 min. | ||
Sulfur tolerance was evaluated by initially exposing the catalyst to H2-500 ppm H2S at 850 °C for 5 h prior to the DRM test. Although the H2S treatment deactivated all the catalysts regarding the DRM reaction, the bimetallic catalyst showed a much enhanced sulfur tolerance.47,48 The methane conversion on H2S treated Ni alloys could reach 91%, approximately 4-fold higher than that of Ni–YSZ (see Fig. 3c). In particular, NiCu seemed to outperform the controls. The poisoned NiCu catalyst showed a roughly identical CH4 conversion and syngas yield in comparison with the pristine at temperatures above 750 °C. This is because the alloying elements, such as Cu and Co, have lower affinities to H2S and are thus thermodynamically more stable in a H2S environment.47,48
Coke resistance is another pivotal parameter to consider when designing DRM catalysts. Therefore, we further evaluated their carbon resistance via exposing them to pure CH4 for 30 min at 800 °C. Such a coking test under extreme conditions (in pure CH4 rather than 50% CO2 + CH4) could accelerate the carbon deposition rate.35,36,39,40 The Raman spectra in Fig. 3f and S6† compare the carbon peak of the Ni–YSZ, pristine and sulfur poisoned NiM–ZDC catalysts. Two intense bands related to the deposited carbon appeared in the spectra, i.e., the D (defect) band associated with the disordered structure of carbon, and the G (graphite) band featuring the graphitic layers and the tangential vibration of carbon atoms. Usually, the amorphous carbon can be easily removed when oxidants are present (e.g., CO2, H2O or O2−).39,40,49 The intensity ratio R(ID/IG) reflects the graphitization degree. A higher value of R here implies that the material might be more coke resistant under DRM conditions (cf. the quantitative coke deposition study via TGA below).35,36,39,40 The NiCu–ZDC catalyst yielded the highest R value (0.662) compared with the other bimetallic catalysts in pure methane. This graphitization degree decreased further on the poisoned catalyst, NiCu–ZDC, yet it exhibited the highest R value (0.760). In fact, copper is commonly documented as an excellent “coke-suppressing” alloying element, as it dilutes the active site on the surface of nickel, restraining carbon–carbon bond formation.12,23,24
After screening a series of DRM catalysts, we concluded that the conventional Ni–YSZ electro-catalyst was not appropriate for in situ dry reforming of methane, particularly due to its low activity and poor sulfur resistance. Alternatively, NiM–ZDC bimetallics exhibited a greatly improved catalytic performance, among which NiCu behaved complementarily in reference to its activity and coke/sulfur resistance. It was thus selected as the catalyst in the EDRM process.
3.3. Designing SOFCs with a triple anode layer for the EDRM process
Despite the excellent performances of the NiCu bimetallic catalyst towards the DRM reaction, applying it to the SOFC anode remains a challenging task. This was simply due to the low melting points of both copper and copper oxide which can sublimate during the co-firing step of SOFC fabrication (>1300 °C). To cope with this difficulty, we designed a triple-layer anode in SOFCs (denoted as TA-SOFC) as shown in Fig. 4a. The extra layer on the anode, containing nanosized NiCu bimetallic catalyst, was fabricated using a 900 °C calcination process. No delamination of this layer from the anode support was observed afterward (Fig. S7†), which could be ascribed to the addition of a Zr doping element that increased the compatibility at the YSZ and ZDC interface (also to the adoption of a Ni based alloy). Meanwhile, this seamless integration led to efficient electron transfers during electrochemical reactions. | ||
| Fig. 4 (a) Cross-section microstructure of the reduced TA-SOFC; (b and c) the I–V and I–P curves of (b) pristine and (c) 500 ppm H2S–H2 pretreated C-SOFC and TA-SOFC in CH4–CO2 and CO–H2 at 800 °C; (d) the current-dependent anode exhaust composition of TA-SOFC fed with sweet CH4–CO2 after H2S pretreatment. | ||
The addition of the bimetallic layer did not deteriorate the SOFC performance, and the TA-SOFC and conventional SOFC (denoted as C-SOFC) demonstrated essentially identical performances in H2, CO and syngas (see Fig. 4 and S8–S10†). For instance, the peak power densities (PPDs) in H2 were ∼1.4 W cm−2 for both C-SOFC and TA-SOFC, whereas they decreased to ∼1.0 W cm−2 in sour atmospheres. We used the cell performance in syngas as a descriptor to evaluate the degree of internal reforming when feeding the cell with CH4–CO2. This is because when methane was completely dry reformed in situ, the actual feed of the SOFC became exactly the same as CO–H2. Intuitively, similar SOFC performances should be therefore recorded. Hence, Fig. 4b compares the I–V and power density profiles in various fuels at 800 °C. Though the PPDs of C-SOFC and TA-SOFC were close in CO–H2 (∼1.2 W cm−2), the PPD of TA-SOFC in CH4–CO2 was roughly 12.5% higher than that obtained in C-SOFC (1.26 W cm−2vs. 1.12 W cm−2). Since TA-SOFC indeed exhibited an almost identical performance in both feedstreams, it became reasonable to speculate that the additional catalyst layer, NiCu–ZDC, fostered the conversion of methane. This was in good agreement with our catalyst screening tests (vide supra). Conversely, the noticeable PPD decrease of C-SOFC in CH4–CO2 in comparison with that in syngas was related to the weak reforming capability of the conventional Ni–YSZ anode.
More importantly, after H2S pretreatment, the TA-SOFC showed much better performances. Albeit both TA-SOFC and C-SOFC demonstrated satisfactory performances in syngas (see Fig. 4c), the PPD of C-SOFC dropped drastically to 0.29 W cm−2 when the feed was switched to CH4–CO2. In contrast, a high PPD, equal to 0.96 W cm−2, was preserved in TA-SOFC. This privileged feature was likely to relate to the excellent sulfur tolerance of NiCu–ZDC regarding the methane reforming reaction.
This instinctive conjecture regarding the reforming activity of the catalyst in SOFCs was then re-assessed quantitatively via analyzing the gas composition of the anode effluent (see Fig. 4d and S11†). Obviously, a high content of both CH4 and CO2 remained in the effluent of the C-SOFC under all current conditions. In the TA-SOFC, the percentages of CO2 and CH4 were both ∼3% at the open-circuit, affirming the previous claim that CO2 and CH4 were almost completely converted to syngas (cf.Fig. 3e). Interestingly, as the applied current rose up, CO concentration increased whereas that of H2 decreased; in the meantime, a negligible amount of CO2 was produced from CO electrochemical conversion when the current was below 1.5 A cm−2. Such a “selective” oxidation of H2 contributed to the power generation, leading to nearly zero GHG emission. This effect is beneficial in terms of GHG control/chemical coproduction, and has been rarely reported in hydrocarbon fueled SOFCs.50–52
In fact, both the C-SOFC and TA-SOFC were employed with oxygen-conducting electrolytes. Theoretically, O2− can readily oxidize both CO and H2 when the SOFC is biased. The selective oxidation of H2 observed above can be explained from two perspectives. On one hand, intrinsically, the electro-oxidation of H2 in SOFCs proceeds more rapidly, and its rate is believed to be several fold faster than that of CO.53,54 Our measurements in Fig. S8† also proved this. On the other hand, such a rate difference became more prominent in the sour feed stream, mainly due to the extremely poor electrochemical performance of the TA-SOFC in CO. Its PPD decreased to merely 0.30 W cm−2 (1.06 W cm−2 in H2, see Fig. S9†). In the electrochemical EIS in Fig. S10,† the polarization resistance (RP) in H2 decreased to 0.17 Ω cm2 when the cell was biased at −0.5 V (relative to OCV), but RP reached 2 Ω cm2 in CO under the same conditions. From the thermodynamic calculation55 and our former XPS data,42 we ensured that the adsorbed sulfur species were formed on Ni in this study, alternating the chemisorption geometry of CO on Ni.43 This might have possibly prevented the diffusion of CO to the TPB, consequently restraining its electrochemical oxidation.
We also performed stability tests on the NiM–ZDC catalyst. Fig. 5a displays the time dependent CH4 conversion of the NiCu–ZDC catalyst at 800 °C under open circuit conditions. It decreased as a function of time in sweet CH4–CO2, presumably due to coke formation.17,18 Such degradation was suppressed on the poisoned catalyst. Under EDRM conditions when TA-SOFC was biased at 1.5 A cm−2 (50 ppm H2S contained CH4–CO2), the output voltage was stable at ∼0.68 V whereas the corresponding power density attained 1.02 W cm−2 during the entire 48 h test (see Fig. 5b). This significantly surpassed most reports in the literature, particularly considering the sour feed conditions of this work (see Table S2† for details). Meanwhile, the effluent gas composition profile shows high conversions of CH4 and CO2 during this period of time yet the produced CO was barely oxidized (see the CO2 concentration under OCV condition in Fig. 4d). This strongly indicates that EDRM in TA-SOFC is a promising process with little CO2 emission.
 | ||
| Fig. 5 (a) Time dependent CH4 conversion of the DRM process at 800 °C; (b) time dependence voltage of H2S-pretreated TA-SOFC in sour CH4–CO2, containing 50 ppm H2S, at 800 °C and at 1.5 A cm−2, and the corresponding variations of anode exhaust composition; (c) TGA signals in air of various fresh and used catalysts, and the weight loss quantitatively reflects the burning of coke; (d) thermodynamic calculation of equilibrium carbon content vs. fuel utilization during the EDRM process. | ||
We thus quantitatively investigated the coke formation in the NiCu–ZDC reforming layer of the TA-SOFC after the 48 h stability test using a temperature programmed oxidation technique (TPO via coupled TGA-MS, shown in Fig. 5c and S14†). The as-reduced NiCu–ZDC was oxidized, showing a weight-increase at ∼400 °C. However, after DRM in either sour or fresh feeds, huge weight losses were recorded at ∼500 °C, correlating with the oxidation of deposited carbon. Particularly for that without H2S pre-treatment, the weight loss reached 55%. In the catalysts after the EDRM process (J = 1.5 A cm−2), much less carbon was found. Such suppressed carbon deposition was further alleviated by H2S treatment and/or feeding sour CH4–CO2, pertaining to the possible sulfur passivation effects.56,57 The higher grade of coke tolerance in EDRM was ascribed to the steam generated during the preferential oxidation of H2. Fig. 5d compares the equilibrium carbon content (ECC) as a function of H2 (CO) utilization in SOFCs, calculated using HSC Chemistry 5.1. At 825 °C, the ECC was more than 14%, making DRM (0% fuel utilization) extremely prone to graphitization. In the EDRM process, however, when the fuel utilization reached 50% (note that our fuel utilization in the experiment was ∼45% at 1.5 A cm−2), the tendency of coking was substantially reduced by ∼60%, giving 6.5% ECC. Hence, the EDRM process in the TA-SOFC suppressed coke formation both kinetically (via the NiCu catalyst) and thermodynamically.
4. Conclusions
We conclude that electrochemical dry reforming of sour methane in SOFCs consisting of a triple-layer anode enables the co-production of syngas and electricity. The applied NiCu bimetallic catalyst layer was well-incorporated into the anode of the SOFC and possessed an excellent reforming capability and coke/sulfur resistance. The in situ produced H2 is selectively oxidized, resulting in fairly low CO2 emission. The energy required for DRM was partially compensated for and the coproduced steam greatly suppressed the carbon deposition.Acknowledgements
This research was financially supported by the Climate Change and Emissions Management Corporation, Alberta, Canada. N. Yan acknowledges the support from the Sustainable Chemistry research priority area of University of Amsterdam (http://www.suschem.uva.nl).References
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 CAS.
- M. E. S. Hegarty, A. M. O'Connor and J. R. H. Ross, Catal. Today, 1998, 42, 225–232 CrossRef CAS.
- D. J. Wilhelm, D. R. Simbeck, A. D. Karp and R. L. Dickenson, Fuel Process. Technol., 2001, 71, 139–148 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 2000, 63, 159–164 CrossRef CAS.
- C. Agrafiotis, H. von Storch, M. Roeb and C. Sattler, Renewable Sustainable Energy Rev., 2014, 29, 656–682 CrossRef CAS.
- S. Kado, K. Imagawa, A. Kiryu, F. Yagi, T. Minami, H. Kawai, K. Kawazuishi, K. Tomishige, A. Nakamura and Y. Suehiro, Catal. Today, 2011, 171, 97–103 CrossRef CAS.
- E. D. Sloan, Nature, 2003, 426, 353–363 CrossRef CAS PubMed.
- R. A. Alvarez, S. W. Pacala, J. J. Winebrake, W. L. Chameides and S. P. Hamburg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6435–6440 CrossRef CAS PubMed.
- R. W. Howarth, A. Ingraffea and T. Engelder, Nature, 2011, 477, 271–275 CrossRef CAS PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- M. M. Nair, S. Kaliaguine and F. Kleitz, ACS Catal., 2014, 4, 3837–3846 CrossRef CAS.
- J. W. Han, C. Kim, J. S. Park and H. Lee, ChemSusChem, 2014, 7, 451–456 CrossRef CAS PubMed.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- H. Y. Kim, J.-N. Park, G. Henkelman and J. M. Kim, ChemSusChem, 2012, 5, 1474–1481 CrossRef CAS PubMed.
- Z. Li, Y. Kathiraser, J. Ashok, U. Oemar and S. Kawi, Langmuir, 2014, 30, 14694–14705 CrossRef CAS PubMed.
- X. Fan, Z. Liu, Y. Zhu, G. Tong, J. Zhang, C. Engelbrekt, J. Ulstrup, K. Zhu and K. Zhou, J. Catal., 2015, 330, 106–119 CrossRef CAS.
- S. Das, S. Thakur, A. Bag, M. S. Gupta, P. Mondal and A. Bordoloi, J. Catal., 2015, 330, 46–60 CrossRef CAS.
- M. Tan, X. Wang, X. Wang, X. Zou, W. Ding and X. Lu, J. Catal., 2015, 329, 151–166 CrossRef CAS.
- H. Cui, S. Q. Turn and M. A. Reese, Catal. Today, 2009, 139, 274–279 CrossRef CAS.
- T. K. Ghosh and E. L. Tollefson, Can. J. Chem. Eng., 1986, 64, 960–968 CrossRef CAS.
- S. Song, M. Han, J. Zhang and H. Fan, J. Power Sources, 2013, 233, 62–68 CrossRef CAS.
- L. M. Toscani, M. G. Zimicz, J. R. Casanova and S. A. Larrondo, Int. J. Hydrogen Energy, 2014, 39, 8758–8766 CrossRef.
- A. F. Lucrédio, J. M. Assaf and E. M. Assaf, Fuel Process. Technol., 2012, 102, 124–131 CrossRef.
- R. K. Kaila, A. Gutiérrez and A. O. I. Krause, Appl. Catal., B, 2008, 84, 324–331 CrossRef CAS.
- S. T. Misture, K. M. McDevitt, K. C. Glass, D. D. Edwards, J. Y. Howe, K. D. Rector, H. He and S. C. Vogel, Catal. Sci. Technol., 2015, 5, 4565–4574 CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- E. D. Wachsman, C. A. Marlowe and K. T. Lee, Energy Environ. Sci., 2012, 5, 5498–5509 Search PubMed.
- S. P. Jiang, Int. J. Hydrogen Energy, 2012, 37, 449–470 CrossRef CAS.
- C. Guerra, A. Lanzini, P. Leone, M. Santarelli and N. P. Brandon, J. Power Sources, 2014, 245, 154–163 CrossRef CAS.
- Y. Shiratori, et al. , Int. J. Hydrogen Energy, 2013, 38, 10542–10551 CrossRef CAS.
- K. Girona, J. Laurencin, J. Fouletier and F. Lefebvre-Joud, J. Power Sources, 2012, 210, 381–391 CrossRef CAS.
- W. Wang, C. Su, Y. Wu, R. Ran and Z. Shao, J. Power Sources, 2010, 195, 402–411 CrossRef CAS.
- W. Wang, C. Su, R. Ran and Z. Shao, J. Power Sources, 2011, 196, 3855–3862 CrossRef CAS.
- W. Wang, R. Ran and Z. Shao, Int. J. Hydrogen Energy, 2011, 36, 755–764 CrossRef CAS.
- T. Takeguchi, R. Kikuchi, T. Yano, K. Eguchi and K. Murata, Catal. Today, 2003, 84, 217–222 CrossRef CAS.
- Y. Nabae, I. Yamanaka, M. Hatano and K. Otsuka, J. Electrochem. Soc., 2006, 153, A140–A145 CrossRef CAS.
- B. Hua, M. Li, J. Pu, B. Chi and J. Li, J. Mater. Chem. A, 2014, 2, 12576 CAS.
- M. Li, B. Hua, J. L. Luo, S. P. Jiang, J. Pu, B. Chi and J. Li, J. Mater. Chem. A, 2015, 3, 21609 CAS.
- J. J. Baschuk and X. Li, Int. J. Energy Res., 2001, 25, 695–713 CrossRef CAS.
- N. Yan, J.-L. Luo and K. T. Chuang, J. Power Sources, 2014, 250, 212–219 CrossRef CAS.
- C. H. Rochester and R. J. Terrell, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 609–621 RSC.
- D. J. Moon and J. W. Ryu, Catal. Today, 2003, 87, 255–264 CrossRef CAS.
- A. Lanzini and P. Leone, Int. J. Hydrogen Energy, 2010, 35, 2463–2476 CrossRef CAS.
- S. E. Evans, J. Z. Staniforth, R. J. Darton and R. M. Ormerod, Green Chem., 2014, 16, 4587–4594 RSC.
- C. M. Grgicak, M. M. Pakulska, J. S. O'Brien and J. B. Giorgi, J. Power Sources, 2008, 183, 26–33 CrossRef CAS.
- C. M. Grgicak, R. G. Green and J. B. Giorgi, J. Power Sources, 2008, 179, 317–328 CrossRef CAS.
- A. Garcia, N. Yan, A. Vincent, A. Singh, J. M. Hill, K. T. Chuang and J. L. Luo, J. Mater. Chem. A, 2015, 3, 23973–23980 CAS.
- B. Hua, N. Yan, M. Li, Y. Zhang, Y. Sun, J. Li, T. Etsell, P. Sarkar, K. Chuang and J. Luo, Energy Environ. Sci., 2016, 9, 207–215 CAS.
- H. Shi, C. Su, G. Yang, R. Ran, Y. Hao, M. Tade and Z. Shao, AIChE J., 2014, 60, 1036–1044 CrossRef CAS.
- Z. Shao, C. Zhang, W. Wang, C. Su, W. Zhou, Z. Zhu, H. Park and C. Kwak, Angew. Chem., Int. Ed., 2011, 50, 1792–1797 CrossRef CAS PubMed.
- Y. Matsuzaki and I. Yasuda, J. Electrochem. Soc., 2000, 147, 1630–1635 CrossRef CAS.
- A. M. Sukeshini, B. Habibzadeh, B. P. Becker, C. A. Stoltz, B. W. Eichhorn and G. S. Jackson, J. Electrochem. Soc., 2006, 153, A705–A715 CrossRef CAS.
- T. Rosenqvist, J. Iron Steel Inst., 1954, 176, 37–57 CAS.
- T. Osaki, T. Horiuchi, K. Suzuki and T. Mori, Catal. Lett., 1995, 35, 39–43 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1984, 85, 31–43 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta02809h |
| This journal is © The Royal Society of Chemistry 2016 |