
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial doping of carbon nanotubes at the polarisable organic/water interface: a liquid/liquid pseudo-capacitor
P. S.
Toth
*a,
A. N. J.
Rodgers
a,
A. K.
Rabiu
a,
D.
Ibañez
b,
J. X.
Yang
a,
A.
Colina
b and
R. A. W.
Dryfe
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: peter.toth@manchester.ac.uk; robert.dryfe@manchester.ac.uk; Fax: +44 (0) 161 275 4598; Tel: +44 (0) 161 306 4522
bDepartment of Chemistry, Universidad de Burgos, Pza. Misael Bañuelos s/n, Burgos, E-09001, Spain
First published on 11th April 2016
Abstract
The electrochemical reactivity of single-walled carbon nanotube (SWCNT) films, assembled at a polarisable organic/water interface, has been probed using model redox species. Electrons generated by the oxidation of organic 1,1′-dimethylferrocene (DMFc) to DMFc+ can be transferred through the assembled SWCNT layer and reduce aqueous ferricyanide (Fe(CN)63−) to ferrocyanide (Fe(CN)64−), with a doping interaction observed. Several electrochemical techniques, including cyclic voltammetry and electrochemical impedance spectroscopy (EIS), were employed to confirm that the model redox couples dope/charge the SWCNTs. In situ Raman spectro-electrochemistry was also applied to verify the charge transfer processes occurring at the assembled SWCNT films and confirm that the doping effect of the carbon nanotubes is initiated by electrochemical reactions. This doping interaction indicated that the adsorbed SWCNT films can act as a pseudo-capacitor, showing a high area-normalised capacitance. The deeper understanding of the electrochemical properties of SWCNTs, gained from this study, will help determine the performance of this material for practical applications.
Introduction
Over the last few decades,1 carbon nanotubes (CNTs) have received considerable attention from the scientific community due to their varying and fascinating range of properties. These materials are composed of sp2-hybridised carbon sheets (graphene) wrapped into a tube structure, with tube diameters on the order of a few nm's.2,3 A CNT formed from a single wrapped graphene sheet is termed a single-walled CNT (SWCNT), while those formed of multiple layered graphene sheets are termed multi-walled CNTs (MWCNTs).4 Graphene sheets can wrap in various orientations to form CNTs, leading to differing helicities, which influence the electronic density of states within the CNTs and, in turn, lead to either a metallic or semiconducting electronic band structure.2,4 The properties of CNTs are also affected by the open, or closed, nature of the tube ends. In addition to their variety, the mechanical and electronic properties of CNTs are individually impressive, with high tensile strength and Young's modulus, a surface area up to 1500 m2 g−1, thermal conductivity of 6000 W m−1 K−1 and, in metallic CNTs, ballistic electron transfer.3,5 To fully utilise these exceptional and changeable properties, the fundamental study of CNTs is of great importance.Many applications, for which CNTs may be suitable, require assembly of the nanotubes into larger coherent structures. Liquid/liquid systems, composed of immiscible aqueous and organic phases of differing density, are ideally suited for this purpose. The high energy interface between the two liquids favours the attachment and assembly of particles, previously dispersed in one of the two bulk liquid phases, as a means of reducing the overall energy of the system.6–10 Recently, these systems have been applied to the formation of CNT films in situ at the liquid/liquid interface, which can subsequently be extracted and used in a variety of applications.11–17 The versatile chemistry of liquid/liquid systems has also been cleverly utilized by the group of Zarbin to form composite structures such as CNT/polyaniline films.18,19
Dissolution of supporting electrolyte in either liquid phase allows liquid/liquid systems to be electrified. This particular form of liquid/liquid interface is known as the interface between two immiscible electrolyte solutions (ITIES). Application of a Galvani potential difference across the ITIES can then be used to drive reactions between the two phases. Such systems are analogous to the more commonly encountered solid electrode/liquid systems, with current generated by the passage of charge across the ITIES. However, both electrons and ions can contribute to the current, with electron transfer, ion transfer and facilitated ion transfer being the primary processes studied at the ITIES.20–23
Building on the work on liquid/liquid assembled CNTs, some recent studies have utilized ITIES systems to study the electrochemical properties of CNTs and other similar carbon materials. One area of great interest to the ITIES community over the last decade is the study of heterogeneous reduction reactions, primarily the hydrogen evolution and oxygen reduction reactions (HER and ORR, respectively), using lipophilic reducing agents.24–26 Various interfacially adsorbed materials have been used as catalysts for these reactions and the group of Girault found that using MWCNTs27 and reduced graphene oxide (rGO)28 as supports for the catalysts Mo2C and MoS2, respectively, improved the catalytic effect of these materials, compared to their unsupported forms, toward the HER. These effects were attributed to an improved electron transfer from the lipophilic reducing agent to the interfacial catalyst, mediated by the carbon supports. Interfacial graphene29 and graphene oxide (GO)30 have also been found to display inherent catalytic properties toward the ORR at the ITIES. An additional promising, and recently found, candidate for the study of interfacial charge transfer processes is the water/room temperature ionic liquid interface.31,32 Such interfaces can have an impact in areas such as proton-coupled electron transfer, fuel cells, and hydrogen storage, where ionic liquids are used as aprotic solvents.33
In our laboratory, the reduction of aqueous metal salts by a lipophilic electron donor, with the heterogeneous electron transfer proceeding via SWCNTs/graphene adsorbed at the ITIES, has been employed to functionalise these materials with Pt, Pd and Au metal nanoparticles (NPs).34–36 In combination with an ex situ metal deposition step, prior to assembly at the ITIES, this methodology was used to asymmetrically decorate both sides of a chemical vapour deposition (CVD) grown graphene sheet with Au and Pd NPs.37 An interesting consequence of inserting CVD graphene to the ITIES is the proposed suppression of capillary waves, which exist at the bare ITIES and cause roughening of the interface. This phenomenon was suggested by an observed decrease in the interfacial capacitance in the presence of graphene, as measured by electrochemical impedance spectroscopy (EIS).36 Additionally, the growth of a CNT/polypyrrole composite film was controlled electrochemically at the ITIES.38
Raman spectroscopy is one of the most widely used techniques for characterisation of CNTs, with the spectra providing a plethora of information on the electronic structure and diameter of the tubes and their defect density.39 Several bands are present in the Raman spectra of CNTs, with the key bands being the radial breathing mode (RBM), the tangential displacement mode (G), the high-frequency two-phonon mode (2D or G′) band and the defect-induced D band. Electrochemistry and Raman spectroscopy have been combined in the spectro-electrochemical study of the state of CNTs and their composites, supported on solid substrates, under applied potentials.40–44 Very recently, Ibañez et al.45 reported the use of Raman spectro-electrochemistry to study charge transfer processes at the ITIES, leading to the possibility of studying the state of CNTs and graphene during mediation of heterogeneous electron transfer at the ITIES; as the interfacial ET initiated charge transfer will result in a change in the Fermi-level of the assembled carbon nanostructures, thus doping them. All of these phenomena are of interest in electro-catalytic reactions, proton-coupled electron transfers and energy storage using the nanostructured organic/water interface.
In the current work we present a study of the interfacial charging interaction between model redox species and SWCNTs assembled at the ITIES, using several electrochemical techniques and in situ Raman spectro-electrochemistry. Additionally, the interfacial SWCNT density, and thus the SWCNT film thickness, was varied.
Experimental
Materials
Lithium chloride (LiCl, 99.99%); potassium hexacyanoferrate(III) (K3Fe(CN)6, 99%); potassium hexacyanoferrate(II) trihydrate (K4Fe(CN)6, ≥98.5%); 1,1′-dimethylferrocene (DMFc, 97%); 1,2-dichlorobenzene (DCB, ≥99.8%), potassium tetrakis(4-chlorophenyl) borate (KTPBCl, ≥98.0%) and bis(triphenylphosphoranylidene) ammonium chloride (BTPPACl, 97%) were purchased from Sigma-Aldrich, UK. Potassium tetrakis(pentafluorophenyl) borate (KTPFB, ≥98.0%) was obtained from Alfa Aesar, US. All chemicals were used as received. The organic phase electrolytes BTPPATPBCl and BTPPATPFB were prepared as described elsewhere46,47 from BTPPACl, KTPBCl and KTPFB. Elicarb® SWCNTs were purchased from Thomas Swan & Co. Ltd., UK, and CVD and arc discharge (AD) SWCNTs were purchased from Sigma-Aldrich, UK. All metal electrode materials were obtained from Advent Research Materials. Aqueous solutions were prepared using deionized water (18.2 MΩ cm, Milli-Q Direct 8, Merck Millipore). Glassware was cleaned in Piranha solution, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture (by volume) of 30% hydrogen peroxide (H2O2, Fisher Scientific) and concentrated sulphuric acid (H2SO4, Fisher Scientific) – CAUTION required when handling – boiled in ultra-pure water, and dried. The composition of the electrochemical cells is displayed in Scheme 1.
:4 mixture (by volume) of 30% hydrogen peroxide (H2O2, Fisher Scientific) and concentrated sulphuric acid (H2SO4, Fisher Scientific) – CAUTION required when handling – boiled in ultra-pure water, and dried. The composition of the electrochemical cells is displayed in Scheme 1.
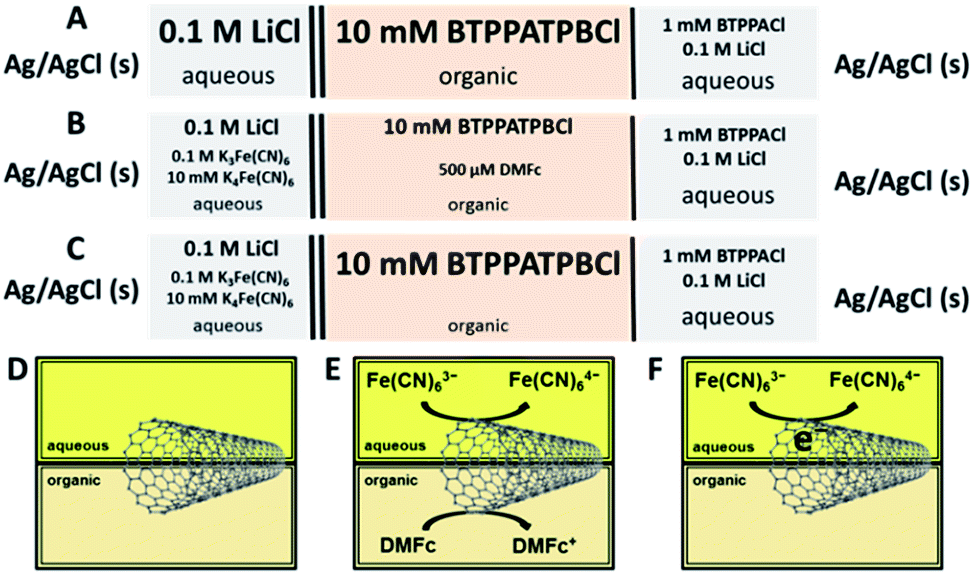 | ||
| Scheme 1 ITIES cell compositions for the supporting blank (A), both aqueous and organic redox couples (B) and only the aqueous redox couple (C) configurations and respective schematics (D–F) when the CNT layers are assembled at the ITIES (DCB/water). | ||
Methods
Electrochemical experiments were performed using a four-electrode configuration with a PGSTAT20 (Eco Chemie Autolab) and a PGSTAT302 (Metrohm-Autolab) Autolab potentiostats. iR compensation was applied for the cyclic voltametric measurements.Ag/AgCl reference electrodes (REs) were directly immersed in the chloride-containing aqueous phase, while an aqueous solution of 0.1 M LiCl and 1 mM BTPPACl was brought into contact with the organic solution to form a second liquid/liquid junction for the organic reference electrode. The three-arm cell used for the liquid/liquid electrochemical measurements, had an interfacial area of 0.74 cm2 and a total solution volume of 2.5 mL. The Galvani potential scale (ΔwoΦ) on all cyclic voltammograms (CVs) was normalized using the formal transfer potential of TMA+, which corresponds to ΔwoΦ° = 0.277 V.48
A three-electrode configuration was employed as a control to determine the diffusion coefficient for DMFc oxidation, using a platinum microdisc electrode (25 micron in diameter; CH Instruments Inc.) as the working electrode and a standard Pt mesh electrode and a Ag wire as the counter and reference electrodes, respectively.
SWCNT powder (2 mg mL−1) was dispersed in DCB using ultrasonication in a bath sonicator (Elmasonic P70H) operating at 37 kHz and 40% power for 12 h. A recirculating cooling system (Julabo F250) was used to maintain a stable temperature of 20 °C throughout sonication. UV-Vis spectroscopy (DH-2000-BAL, Ocean Optics and USB2000 interface, Micropack GmbH) was used to measure the concentration of the SWCNT dispersions.
The assembly of SWCNTs at the organic/water interface in a three-arm cell is described elsewhere.34,35 Briefly, an aliquot of SWCNT dispersion, in a DCB solution containing the organic electrolyte, was placed in contact with an equal volume of aqueous phase, and self-assembly of the SWCNT at the interface was then achieved using a short (less than 5 min) sonication. In situ Raman spectro-electrochemical measurements were carried out using the method reported by Ibañez et al.45 To summarise, Raman spectra were collected using a Confocal Raman Voyage (BWTEK) using a 20× objective with a laser wavelength of 532 nm (operating power of 15 mW). An XYZ piezoelectric positioner (Newport 271), controlled by a Newport motion controller (Newport, ESP 301), was employed to focus the laser beam with micrometric resolution. The organic phase was gellified using high molecular weight polyvinylchloride (PVC), following the methodology reported previously.49
Results and discussion
Electron transfer at interface assembled SWCNT films
The effect of adsorbed SWCNT films on biphasic electron transfer, with one redox reaction in the aqueous phase (aq) and the opposite redox reaction in the organic phase (o), at the ITIES is probed here. Well-known model redox systems were used to study the effect of assembled SWCNT films on such redox processes; explicitly, the reduction of ferricyanide (Fe(CN)63−) to ferrocyanide (Fe(CN)64−) and the oxidation of DMFc to DMFc+ in the aqueous and organic phases,50 respectively, as stated in reaction (1).| DMFc(o) + Fe(CN)63−(w) ⇌ DMFc+(o) + Fe(CN)64−(w) | (1) |
SWCNTs were dispersed in the organic phase at a range of concentrations and, after assembly at the ITIES, produced interfacial SWCNT films with densities of 1.38, 2.77, 5.55, 11.1, and 22.2 μg cm−2, as calculated using the method reported by Rodgers et al.29 The SWCNT film completely covers the interface at the lowest concentration used here (1.38 μg cm−2), so increasing the density of SWCNTs will likely increase the thickness of the interfacial SWCNT film.
Voltammetric measurements were carried out with 0.1 M LiCl and 10 mM BTPPATPBCl supporting electrolytes employed in the aqueous and organic phases, respectively (Fig. 1). A blank CV, in the absence of SWCNTs at the ITIES (Scheme 1A and D), shows a potential window limited by transfer of the aqueous supporting electrolyte ions, Li+ and Cl− (Fig. 1a).
 | ||
| Fig. 1 CVs of interfacial oxidation of organic DMFc by aqueous ferricyanide in the absence (trace b) and presence of varying densities of interfacial SWCNTs (c–g). A CV with only the supporting electrolytes present and in the absence of interfacial SWCNTs (trace a) is shown for comparison. Scan rate = 50 mV s−1. The coverages/densities of SWCNTs at the DCB/water interface were 1.38 μg cm−2 (trace c), 2.77 μg cm−2 (trace d), 5.55 μg cm−2 (trace e), 11.1 μg cm−2 (trace f), and 22.2 μg cm−2 (trace g). | ||
Interfacial electron transfer (ET) occurred when both redox couples were present (Fig. 1b), represented by the faradaic peaks between 0.0 V and +0.1 V, which correspond to heterogeneous DMFc oxidation and re-reduction of DMFc+ (Scheme 1B). A diffusion coefficient for the oxidation (Dox) of DMFc to DMFc+ at the DCB/water interface, was calculated using the Randles–Sevčik equation (eqn (2)):51,52
 | (2) |
| i = 4zFDcr | (3) |
Fig. 1c–g shows reversible CVs obtained in the presence of different coverages/densities of SWCNTs at the interface (b: ET blank = 0 μg cm−2, c: 1.38 μg cm−2, d: 2.77 μg cm−2, e: 5.55 μg cm−2, f: 11.1 μg cm−2, g: 22.2 μg cm−2). The current increases with increasing SWCNT surface coverage/thickness, which may be the result of a doping effect on the SWCNTs, or a result of a change in surface area at the interface.34 Additionally, a small shift can be seen in the peak position, which could also come from doping of the SWCNTs, due to the redox reaction.
In order to probe the charging effect of the interfacial ET process on the SWCNT films, the voltammetric analysis was repeated in the absence of the organic DMFc redox species (Scheme 1C and F). Fig. 2A(b–f) shows cyclic voltammetric characterisation of the cells containing Fe(CN)63−/Fe(CN)64−, in the presence of SWCNT films of differing density/thickness. The current increases as a function of the SWCNT concentration in the presence of the aqueous phase redox couple, suggesting that the potential difference applied across the ITIES causes the reduction of Fe(CN)63− to Fe(CN)64−via the interfacial SWCNTs, thus resulting in electrochemical doping of the SWCNTs.55 An alternative explanation is that the current increase is due to a growth in capacitive current, resulting from the increase in the SWCNT film thickness.
 | ||
| Fig. 2 CVs of the interfacial reduction of Fe(CN)63− to Fe(CN)64− (A, Scheme 1C) and only the supporting electrolytes of 0.1 M LiCl and 10 mM BTPPATPBCl in the organic phases, respectively present in each phase (B, Scheme 1A). Varying densities of interfacial SWCNTs (b–f) were employed. Curve a shows the respective systems in the absence of interfacial SWCNTs. The SWCNT densities at the DCB/water interface were 1.38 μg cm−2 (trace b), 2.77 μg cm−2 (trace c), 5.55 μg cm−2 (trace d), 11.1 μg cm−2 (trace e), and 22.2 μg cm−2 (trace f). Scan rate = 50 mV s−1. | ||
In order to further probe the mechanism of the interfacial charging, several cyclic voltammetric and electrochemical impedance spectroscopy (EIS) measurements were performed in the presence of interfacial assembled SWCNT films of varying density/thickness. The electro-activity of the SWCNT films, with only the supporting electrolytes in both phases (Scheme 1A and D), is shown in Fig. 2B. The similarity in capacitive current, at each different SWCNT film thickness, and the absence of any faradaic peaks (Fig. 2B(b–f)), demonstrates that capacitive charging of the interfacial SWCNTs is not responsible for the current increase seen in Fig. 2A.
EIS measurements were performed in the three different DCB/water configurations: only the supporting electrolytes (Scheme 1A and D), redox species in both phases (Scheme 1B and E), and only the ferri/ferrocyanide redox couple (Scheme 1C and F); and at varying potential differences, corresponding to the completely reduced (−0.1 V), reduced (+0.05 V), oxidised (+0.15 V), and completely oxidised (+0.35 V) forms of DMFc. The representative complex impedance Nyquist plots at +0.15 V and +0.05 V, corresponding to heterogeneous DMFc oxidation and re-reduction of DMFc+, respectively (Scheme 1B), proceeding via a 5.55 μg cm−2 SWCNT film at the DCB/water interface are shown in Fig. 3. The impedance data were analysed and fitted using the equivalent electrical circuit56,57 which is depicted by Fig. 3, where Cint is the capacitance of the interface, RCT is the charge transfer resistance, ZW is the Warburg impedance, Cst is the stray capacitance and Rs is the solution resistance.
 | ||
| Fig. 3 Nyquist plots determined from EIS measurements of the 5.55 μg cm−2 SWCNT film at the DCB/water interface, with both aqueous and organic redox couples present (Scheme 1B). Measurements were performed at +0.15 V and +0.05 V, corresponding to oxidation of DMFc and re-reduction of DMFc+, respectively. The equivalent circuit used to determine interfacial capacitance values is shown. | ||
Calculated Cint values are plotted vs. the interfacial SWCNT coverage (Fig. 4). In the case of the cell containing only supporting electrolytes (Fig. 4A, Scheme 1A and D), no significant change in Cint is seen as a function of increasing SWCNT concentration. A small ascendant trend is initially seen upon increasing the SWCNT density/thickness to 5.55 μg cm−2 in the completely reduced state (−0.1 V, black squares in Fig. 4A), though Cint subsequently decreases upon further increasing the SWCNT density/thickness.
 | ||
| Fig. 4 Interfacial capacitance (Cint) values as a function of the SWCNT density/thickness at the DCB/water interface (1.38 μg cm−2, 2.77 μg cm−2, 5.55 μg cm−2, 11.1 μg cm−2, and 22.2 μg cm−2). The different symbols correspond to the different Galvani potentials, at which the capacitance measurements were performed. The capacitance was measured for different ITIES configurations: only supporting electrolytes (A), both aqueous and organic redox couples (B) and only the aqueous redox couple (C). | ||
However, when redox couples are present in both liquid phases (Fig. 4B, Scheme 1B and E) or only the aqueous ferri/ferrocyanide redox couple is present (Fig. 4C, Scheme 1C and F), an increase of Cint is observed with increasing SWCNT film density/thickness, considering that a thicker SWCNT film would yield a higher effective areal capacitance (analogous to a porous material). For the highest SWCNT coverages (11.1 μg cm−2 and 22.2 μg cm−2) there are exceptions to the trend of increasing Cint in the reduced states: −0.10 V in Fig. 4B (both redox couples) and −0.10 V and +0.05 V in Fig. 4C (only the aqueous redox couple). The general trend, however, indicates an interaction between the electrochemical reduction of ferri/ferrocyanide and the SWCNT films, resulting in their charging.
In situ Raman spectro-electrochemical study at interface-assembled SWCNT films
In a conventional 3-electrode configuration, where SWCNTs are deposited on a solid electrode, the SWCNT Raman bands intensity dependence on electrochemical charging follows a bell-shape curve. Attenuation of the Raman intensity can be explained by filling/depleting of the Van Hove singularities.40In situ Raman spectro-electrochemistry can be applied to the investigation of interfacial ET reactions between DMFc (oxidation to DMFc+) in DCB and Fe(CN)63− (reduction to Fe(CN)64−) in the aqueous phase, by monitoring the evolution of the Raman signals during potentiodynamic measurements in real-time, with a resolution of a few seconds.45 For this reason, interfacial ET in the presence of a SWCNT film was also investigated using in situ Raman spectroscopy. To the best of our knowledge, the use of in situ Raman spectro-electrochemistry to probe ITIES assembled SWCNTs has not been reported previously.The spectro-electrochemical cell composition was: Ag/AgCl(s)|0.1 M K3(Fe(CN)6) + 10 mM K4(Fe(CN)6) + 0.1 M LiCl(aq)|5 mM BTPPATPFB + 10 mM DMFc + 2 wt% PVC(DCB)|1 mM BTPPACl + 10 mM LiCl(aq)|Ag/AgCl(s). Fig. 5 shows the corresponding time-resolved Raman spectra at different potentials during the forward scan. As can be observed, the Raman intensity of all the characteristic SWCNT bands: radial breathing mode (RBM) at 155 cm−1 and 175 cm−1, disordered-induced feature (D band) at 1345 cm−1, tangential displacement mode (G− and G+ bands) at 1570 cm−1 and 1592 cm−1 and D-band overtone (2D band) at 2675 cm−1 decreases as the Galvani potential increases. The changing intensity of the Raman bands is expected to display reversible behaviour. A monotonic blue shifting of the G+ band would indicate that SWCNT film was p-doped.43,44 As can be seen in Fig. 5, the G+ band was not shifted during the electrochemical process. Therefore, it can be concluded that the SWCNT film assembled at the DCB/water interface is n-doped during the interfacial ET reaction. Fig. 6 shows a reversible CV of DMFc oxidation to DMFc+ and subsequent re-reduction, with the intensity of the tangential displacement mode, the G+ band (1592 cm−1), at each Galvani potential, overlayered.
 | ||
| Fig. 5 In situ Raman spectra of SWCNTs assembled at the water/gellified DCB interface, collected during electrochemical cycling in a −0.25 to +0.40 V Galvani potential range (from bottom to top: −0.25 V (trace a), −0.15 V (trace b), −0.05 V (trace c), +0.05 V (trace d), +0.15 V (trace e), +0.25 V (trace f), and +0.40 V (trace g)). 0.1 M K3(Fe(CN)6), 10 mM K4(Fe(CN)6) and 0.1 M LiCl electrolytes and 5 mM BTPPATPFB and 10 mM DMFc electrolytes were present in the aqueous and DCB phases, respectively. The Raman intensity was normalized to the most intense peak. Evolution of the RBM bands at 155 cm−1 and 175 cm−1 (A), D band at 1345 cm−1 (B), G− and G+ bands at 1570 cm−1 and 1592 cm−1 (C) and 2D band at 2675 cm−1 (D). | ||
 | ||
| Fig. 6 Evolution of the G+ band (tangential mode, 1592 cm−1) intensity in the Raman spectrum of SWCNTs at the water/gellified DCB interface during potential cycling (black line). CV peaks correspond to interfacial ET between Fe(CN)63−/Fe(CN)64− and DMFc/DMFc+ redox couples in the aqueous and DCB phases, respectively. The Raman intensity was normalized to the most intense peak. The CV was recorded at 5 mV s−1 starting from ΔwoΦ = −0.25 V (blue line). | ||
During the oxidation of DMFc, from −0.25 V to +0.4 V, Fe(CN)63− reduction occurs concurrently and the intensity of the Raman bands is attenuated. When the DMFc+ is reduced back to DMFc, the G+ band intensity also increases again. This charge transfer occurs over a potential range where the SWCNTs are neither functionalized nor destroyed, and therefore, the Raman intensity of the bands is reversible. The change in Raman intensity is more reversible during the n-doping of the films in aqueous solution at solid/liquid interfaces, anodic polarization in aqueous solution can produce defects by oxidation of the SWCNT, or even film damage when it is illuminated by a laser light.44 It is noteworthy, that generation of Prussian blue (iron(II,III) hexacyanoferrate(II,III)) is observed when the Fe(CN)63− concentration is equal, or in a slight excess, over Fe(CN)64−. In the present case the Prussian blue formation is avoided as the Fe(CN)63−:Fe(CN)64− ratio is 10:1. The in situ Raman data indicates that a net n-doping of the SWCNT film occurs in the presence of both redox couples (aqueous and organic), whereas p-doping is suggested by the EIS data reported in the case of the aqueous redox couple alone, see Fig. 4. This implies that a spontaneous positive charging of the SWCNT film is induced by contact with the aqueous phase redox couple, but a net negative charging of the film occurs when the stronger, organic phase electron donor is present. This mechanism is consistent with the equilibration of Fermi levels recently discussed for adsorbed Au nanoparticles at the ITIES.58
Conclusions
Aqueous and organic model redox species were added to both solution phases to characterise electrochemically active SWCNT films, of varying thicknesses, at the DCB/water interface. Charge transfer through the carbon nanotube films of differing thickness was studied with varying cell compositions and a doping/charging interaction was found between the redox couples and the interfacial SWCNTs using cyclic voltammetry analysis.This doping/charging was indicated by the charge transfer interaction between the electrochemically controlled ferri/ferrocyanide redox reaction and the SWCNT films, causing its doping. A novel in situ Raman spectro-electrochemical technique was exploited to investigate interfacial ET at carbon nanomaterial-decorated ITIES. The in situ Raman results also support the hypothesis of electrochemical doping of the interfacial SWCNT films due to the charge transfer. This doping indicates that the adsorbed SWCNT films can act as a pseudo-capacitor, showing a high area-normalised capacitance, in a novel liquid interface configuration. Moreover, a deeper understanding of how interface-assembled nanomaterials can enhance heterogeneous electron transfer at the ITIES will aid in developing suitable materials to act as interfacial catalysts for biphasic reactions between immiscible aqueous and organic phases.
Acknowledgements
The authors thank the U.K. EPSRC (grants EP/K007033/1, EP/K039547/1, EP/K016954/1) and Ministerio de Economía y Competitividad de España (CTQ2014-55583-R, CTQ2014-61914-EXP) for financial support. A. K. R. also thanks the Educational Trust Fund (Nigeria) for a PhD scholarship. Data files can be obtained from http://www.mub.eps.manchester.ac.uk/robert-dryfe-electrochemistry/. We wish to thank the three reviewers for their comments on the earlier version of this manuscript, which we believe improved the presentation of data significantly.Notes and references
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- P. M. Ajayan and O. Z. Zhou, Top. Appl. Phys., 2001, 80, 391–425 CrossRef CAS.
- V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165–184 RSC.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- E. Pop, D. Mann, Q. Wang, K. E. Goodson and H. J. Dai, Nano Lett., 2006, 6, 96–100 CrossRef CAS PubMed.
- D. Y. Wang, H. W. Duan and H. Möhwald, Soft Matter, 2005, 1, 412–416 RSC.
- L. F. Hu, M. Chen, X. S. Fang and L. M. Wu, Chem. Soc. Rev., 2012, 41, 1350–1362 RSC.
- S. U. Pickering, J. Chem. Soc., Trans., 1907, 91, 2001–2021 RSC.
- B. P. Binks, Phys. Chem. Chem. Phys., 2007, 9, 6298–6299 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- P. Chaturbedy, H. S. S. R. Matte, R. Voggu, A. Govindaraj and C. N. R. Rao, J. Colloid Interface Sci., 2011, 360, 249–255 CrossRef CAS PubMed.
- P. Asuri, S. S. Karajanagi, J. S. Dordick and R. S. Kane, J. Am. Chem. Soc., 2006, 128, 1046–1047 CrossRef CAS PubMed.
- J. Matsui, M. Iko, N. Inokuma, H. Orikasa, M. Mitsuishi, T. Kyotani and T. Miyashita, Chem. Lett., 2006, 35, 42–43 CrossRef CAS.
- Y. J. Zhang, Y. F. Shen, D. Kuehner, S. X. Wu, Z. M. Su, S. Ye and L. Niu, Chem. Commun., 2008, 4273–4275 RSC.
- J. Matsui, K. Yamamoto and T. Miyashita, Carbon, 2009, 47, 1444–1450 CrossRef CAS.
- R. Kudo, J. Matsui, T. Shibata and T. Miyashita, Mol. Cryst. Liq. Cryst., 2010, 519, 9–13 CrossRef CAS.
- C. E. Cava, R. V. Salvatierra, D. C. B. Alves, A. S. Ferlauto, A. J. G. Zarbin and L. S. Roman, Carbon, 2012, 50, 1953–1958 CrossRef CAS.
- R. V. Salvatierra, M. M. Oliveira and A. J. G. Zarbin, Chem. Mater., 2010, 22, 5222–5234 CrossRef CAS.
- V. H. R. de Souza, M. M. Oliveira and A. J. G. Zarbin, J. Power Sources, 2014, 260, 34–42 CrossRef.
- Z. Samec, Pure Appl. Chem., 2004, 76, 2147–2180 CrossRef CAS.
- P. Vanýsek, Electrochemistry on Liquid/Liquid Interfaces, Springer, Berlin, Heidelberg, 1985 Search PubMed.
- P. Vanýsek and L. B. Ramirez, J. Chil. Chem. Soc., 2008, 53, 1455–1463 Search PubMed.
- F. Reymond, D. Fermín, H. J. Lee and H. H. Girault, Electrochim. Acta, 2000, 45, 2647–2662 CrossRef CAS.
- M. A. Mendez, R. Partovi-Nia, I. Hatay, B. Su, P. Y. Ge, A. Olaya, N. Younan, M. Hojeij and H. H. Girault, Phys. Chem. Chem. Phys., 2010, 12, 15163–15171 RSC.
- B. Su, H. H. Girault and Z. Samec, in Catalysis in Electrochemistry, John Wiley & Sons, Inc., 2011, pp. 427–451, DOI:10.1002/9780470929421.ch12.
- A. N. J. Rodgers, S. G. Booth and R. A. W. Dryfe, Electrochem. Commun., 2014, 47, 17–20 CrossRef CAS.
- X. J. Bian, M. D. Scanlon, S. N. Wang, L. Liao, Y. Tang, B. H. Liu and H. H. Girault, Chem. Sci., 2013, 4, 3432–3441 RSC.
- P. Y. Ge, M. D. Scanlon, P. Peljo, X. J. Bian, H. Vubrel, A. O'Neill, J. N. Coleman, M. Cantoni, X. L. Hu, K. Kontturi, B. H. Liu and H. H. Girault, Chem. Commun., 2012, 48, 6484–6486 RSC.
- A. N. J. Rodgers and R. A. W. Dryfe, ChemElectroChem, 2016, 3, 472–479 CrossRef CAS.
- S. Rastgar, H. Deng, F. Cortes-Salazar, M. D. Scanlon, M. Pribil, V. Amstutz, A. A. Karyakin, S. Shahrokhian and H. H. Girault, ChemElectroChem, 2014, 1, 59–63 CrossRef CAS.
- B. M. Quinn, Z. F. Ding, R. Moulton and A. J. Bard, Langmuir, 2002, 18, 1734–1742 CrossRef CAS.
- J. Langmaier and Z. Samec, Anal. Chem., 2009, 81, 6382–6389 CrossRef CAS PubMed.
- E. A. de Eulate, D. S. Silvester and D. W. M. Arrigan, Angew. Chem., Int. Ed., 2015, 54, 14903–14906 CrossRef PubMed.
- P. S. Toth, A. N. J. Rodgers, A. K. Rabiu and R. A. W. Dryfe, Electrochem. Commun., 2015, 50, 6–10 CrossRef CAS.
- P. S. Toth, S. J. Haigh, A. K. Rabiu, A. N. J. Rodgers, A. M. Rakowski and R. A. W. Dryfe, J. Mater. Res., 2015, 30, 2679–2687 CrossRef CAS.
- P. S. Toth, Q. M. Ramasse, M. Velický and R. A. W. Dryfe, Chem. Sci., 2015, 6, 1316–1323 RSC.
- P. S. Toth, M. Velický, Q. M. Ramasse, D. M. Kepaptsoglou and R. A. W. Dryfe, Adv. Funct. Mater., 2015, 25, 2899–2909 CrossRef CAS.
- P. S. Toth, A. K. Rabiu and R. A. W. Dryfe, Electrochem. Commun., 2015, 60, 153–157 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- L. Kavan, P. Rapta and L. Dunsch, Chem. Phys. Lett., 2000, 328, 363–368 CrossRef CAS.
- M. Kalbac, L. Kavan, M. Zukalova and L. Dunsch, Carbon, 2007, 45, 1463–1470 CrossRef CAS.
- A. Colina, V. Ruiz, A. Heras, E. Ochoteco, E. Kauppinen and J. Lopez-Palacios, Electrochim. Acta, 2011, 56, 1294–1299 CrossRef CAS.
- L. Kavan and L. Dunsch, ChemPhysChem, 2011, 12, 47–55 CrossRef CAS PubMed.
- D. Ibañez, E. C. Romero, A. Heras and A. Colina, Electrochim. Acta, 2014, 129, 171–176 CrossRef.
- D. Ibañez, D. Plana, A. Heras, D. J. Fermín and A. Colina, Electrochem. Commun., 2015, 54, 14–17 CrossRef.
- Z. F. Ding, D. J. Fermín, P. F. Brevet and H. H. Girault, J. Electroanal. Chem., 1998, 458, 139–148 CrossRef CAS.
- B. Su, J. P. Abid, D. J. Fermín, H. H. Girault, H. Hoffmannova, P. Krtil and Z. Samec, J. Am. Chem. Soc., 2004, 126, 915–919 CrossRef CAS PubMed.
- B. Hundhammer, C. Muller, T. Solomon, H. Alemu and H. Hassen, J. Electroanal. Chem., 1991, 319, 125–135 CrossRef CAS.
- H. J. Lee, D. J. Fermín, R. M. Corn and H. H. Girault, Electrochem. Commun., 1999, 1, 190–193 CrossRef CAS.
- B. Quinn, R. Lahtinen, L. Murtomäki and K. Kontturi, Electrochim. Acta, 1998, 44, 47–57 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc, 2001 Search PubMed.
- A. A. Stewart, J. A. Campbell, H. H. Girault and M. Edddowes, Ber. Bunsen-Ges., 1990, 94, 83–87 CrossRef CAS.
- A. L. Barker and P. R. Unwin, J. Phys. Chem. B, 2001, 105, 12019–12031 CrossRef CAS.
- J. E. Baur and R. M. Wightman, J. Electroanal. Chem., 1991, 305, 73–81 CrossRef CAS.
- M. D. Scanlon, P. Peljo, M. A. Mendez, E. Smirnov and H. H. Girault, Chem. Sci., 2015, 6, 2705–2720 RSC.
- M. C. Wiles, D. J. Schiffrin, T. J. VanderNoot and A. F. Silva, J. Electroanal. Chem. Interfacial Electrochem., 1990, 278, 151–159 CrossRef CAS.
- V. Mareček, A. Lhotský and S. Račinský, Electrochim. Acta, 1995, 40, 2905–2908 CrossRef.
- E. Smirnov, P. Peljo, M. D. Scanlon and H. H. Girault, ACS Nano, 2015, 9, 6565–6575 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |