
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mixed alloyed chemical composition of chloro-(chloro)n-boron subnaphthalocyanines dictates their physical properties and performance in organic photovoltaic devices†
Jeremy D.
Dang
a,
David S.
Josey
a,
Alan J.
Lough
b,
Yiying
Li
c,
Alaa
Sifate
a,
Zheng-Hong
Lu
c and
Timothy P.
Bender
*abc
aDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College St., Toronto, Ontario, Canada M5S 3E5. E-mail: tim.bender@utoronto.ca
bDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, Ontario M5S 3H6, Canada
cDepartment of Materials Science and Engineering, University of Toronto, 184 College St., Toronto, Ontario, Canada M5S 3E4
First published on 16th May 2016
Abstract
Chloro-boron subnaphthalocyanine (Cl-BsubNc) has recently attracted significant interest as a light-harvesting and charge transporting material in organic photovoltaic devices (OPVs) by enabling an 8.4% efficient planar heterojunction OPV cell. We present herein a variety of experimental data that supports the conclusion that Cl-BsubNc, whether synthesized via literature methods or our in-house methods or purchased commercially, is actually a mixed alloyed composition of Cl-BsubNcs with random amounts of chlorination at the bay position(s) of the BsubNc macrocyclic structure which we hereafter will refer to as Cl–ClnBsubNc(s). We outline our efforts to develop alternative chemical processes, whereby we did obtain samples with lower and higher amounts of bay position chlorination. However, we were unable to obtain a pure, non-bay chlorinated sample of Cl-BsubNc. The positions and frequencies of the peripheral chlorine atoms were determined via single crystal X-ray crystallography of two different mixed alloyed compositions of Cl–ClnBsubNc samples and MS and XPS analysis of all Cl–ClnBsubNc samples. The photo- and electro-physical properties were found to differ amongst the Cl–ClnBsubNc samples with varying amounts of chlorination. These differences also translated into varying performance within planar heterojunction OPVs, whereby a mixture of Cl–ClnBsubNcs with lower amounts of chlorination produced less efficient OPVs (albeit with a higher open circuit voltage) compared to a mixture with higher amounts of chlorination. Additionally, an in-house made sample of Cl–ClnBsubNc, with the highest level of bay position chlorination, yielded the best performing OPVs through an improved fill factor. A commercial sample of Cl–ClnBsubNc also yielded OPVs with efficiencies equivalent to a Cl–ClnBsubNc sample prepared in our laboratory. This mixture of Cl–ClnBsubNcs is therefore likely to be present in the reported 8.4% efficient OPV device. Our results therefore offer a cautionary note that the Cl-BsubNc samples used within the existing literature are likely not a pure chemical composition but are rather mixtures of Cl–ClnBsubNcs with bay position chlorination. Our findings clarify the previous literature results on the chemistry of Cl-BsubNc, firm up the photo- and electro-physical properties of these materials, and offer additional insight into their application as functional materials in efficient OPVs.
Introduction
Boron subnaphthalocyanines (BsubNcs, Fig. 1) are but minor constituents within a broader class of well-known and highly studied compounds known as phthalocyanines (Pcs).1–3 BsubNcs are more structurally similar to another minor constituent of the Pc family, boron subphthalocyanines (BsubPcs, Fig. 1). BsubPcs are comprised of three nitrogen-bridged isoindoline subunits that chelate a single boron atom within their internal cavity.4 BsubPcs are known to be aromatic, non-planar, and cone-shaped molecules.5–8 Despite being first synthesized and characterized in the early 1970s, the majority of the synthesis and application work on BsubPcs has been carried out within the last 10 to 15 years.9–12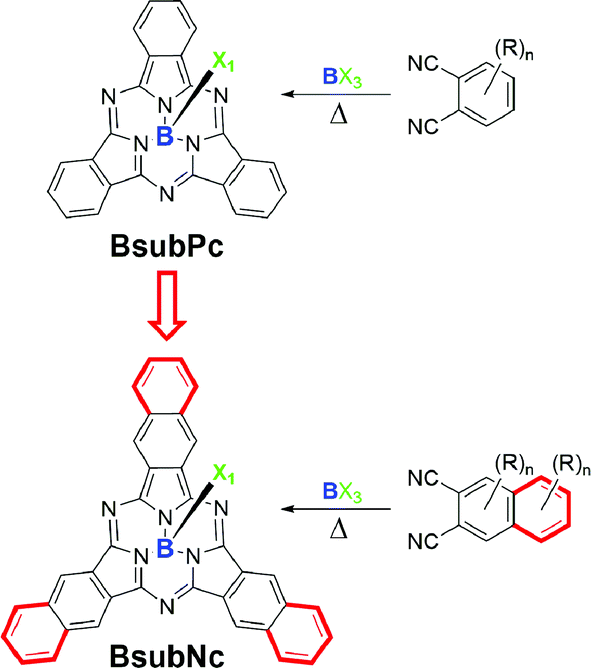 | ||
| Fig. 1 General chemical structures of boron subphthalocyanine (BsubPc) and boron subnaphthalocyanine (BsubNc) where X = Cl, Br, Ph, or other. R group designations omitted in places for clarity. | ||
Halo-BsubPcs are relatively easy to prepare by the reaction of phthalonitrile (an ortho-dinitrile) with a boron trihalide templating agent such as BCl3 (ref. 10 and 13) or BBr3 (ref. 14 and 15) in an aromatic solvent resulting in the formation of Cl-BsubPc and Br-BsubPc, respectively, in high yield and high purity (Fig. 1). The halide of no other metal from the periodic table is known to template the formation of BsubPcs, whereas all other elements template the formation of Pcs.
Like BsubPcs, BsubNcs have been reportedly prepared by heating 2,3-dicyanonaphthalene (also an ortho-dinitrile) with a boron templating agent (e.g. PhBCl2, BBr3, BCl3) in an aromatic solvent (Fig. 1). A relatively limited amount of work has been reported in the literature on the synthesis of BsubNcs compared to BsubPcs. In summary, the first report on the synthesis of a BsubNc was by Hanack and Rauschnabel in 1995 (Table S1,† entry 1), where they treated 2,3-dicyanonaphthalene and an alkylated 2,3-dicyanonaphthalene derivative with PhBCl2 in boiling naphthalene to produce the respective Ph-BsubNcs, albeit in only analytical amounts.16,17 In 1999, Kobayashi et al. reported the use of BBr3 as the boron templating agent to synthesize Br-BsubNc in a 34.6% yield (Table S1,† entry 2),9 although Torres et al.18 had questioned the purity of this sample. In 2000, both Kennedy13 and Torres18 reported the synthesis of Cl-BsubNc in 53% and 35%, respectively, using BCl3 as the boron template and 2,3-dicyanonaphthalene as the starting material (Table S1,† entry 3 & 4). In 2007 Giribabu et al. reportedly employed microwave irradiation to afford a tert-butylated Cl-BsubNc in 82% yield (Table S1,† entry 5).19 More recently, Takao et al. prepared two peripherally fluorinated Cl-BsubNc derivatives, Cl-F6BsubNc (26% yield) and Cl-F12BsubNc (44% yield), from their respective fluorinated 2,3-dicyanonaphthalene via multistep synthesis (Table S1,† entry 6). Takao et al. also treated the two products with AgBF4 to substitute the axial chloride with a fluoride.20 In addition to the synthesis of ‘standard’ BsubNcs from 2,3-dicyanonaphthalene, Kobayashi et al. also prepared chiral analogs of BsubNcs via the reaction of 1,2-dicyanonaphthalene with BCl3 to form chiral Cl-1,2-BsubNc and subsequent phenoxy derivatives.21
A review of the above mentioned studies (see Table S1†) reveals that detailed characterization of the BsubNcs is generally limited. 1H NMR data have been numerically reported (entry 1, 3, and 4) but spectra were not published to graphically illustrate the purity of the sample(s). High resolution mass spectrometry (HRMS) data have not been reported for any of the BsubNcs; low resolution mass spectrometry (LRMS) data were however reported (entry 3 and 4). Elemental analysis (EA) results have been reported for BsubNcs from entries 2 and 4 , however, the EA data were only supplied by the former entry and the analysis was not within an acceptable range from the calculated values of the respective proposed molecular formula (C36H18N6BBr) for publication (i.e. ±0.4%). 1H and 19F NMR data are reported (both numerically and graphically) along with HRMS data by Takao et al. (entry 6).20
BsubPcs have recently been applied as functional materials in organic electronic devices most notably in organic photovoltaic devices (OPVs).22–31 Although the recent efforts of our group29,30 and others25–28,31 have shown that a variety of BsubPcs can have application in OPVs, the majority of the applications have focused on the use of the prototypical derivative Cl-BsubPc.32 Through their use, power conversion efficiencies as high as 4.69% have been achieved.31
At the same time, Cl-BsubNc has also been applied in OPVs.27,31,33–39 As a result of the more-extended π-conjugation system compared to Cl-BsubPc, Cl-BsubNc has a λmax of absorption that is significantly red-shifted to the mid 600 nm region in solution13 and at 686 nm in the solid-state,33 making Cl-BsubNc able to capture regions of red light for OPV applications. The first examples of Cl-BsubNc in OPVs showed that its substitutions for Cl-BsubPc as an electron donating and charge generating material paired with fullerene (C60) resulted in small improvements in photocurrent generation.33–35,37 This substitution reduced the spectral overlap between the donor and acceptor, but lost the photocurrent contributions in the 500–600 nm region from the Cl-BsubPc. Gommans et al. were the first to pair a BsubPc with Cl-BsubNc, reclaiming the BsubPc region of the spectrum but losing the blue region contributions from C60.27
Recently, Cheyns and Cnops et al. have demonstrated that the inclusion of Cl-BsubNc into a planar heterojunction (PHJ) OPV enables power conversion efficiencies (PCEs) as high as 6.4% (ref. 39) and 8.4%.31 These measured efficiencies are amongst the highest achieved for PHJ OPVs. Cheyns, Cnops et al. show evidence that the high PCEs are attributable to the complementary absorption profiles of BsubPc and BsubNc, broadening the overall absorption range of the OPV. To achieve the 8.4% efficiency, an energy-relay cascade was used to transfer excitons to a single dissociating interface, avoiding the drop in VOC typically seen in other 3-layer OPVs where multiple dissociating interfaces are incorporated.31
Now considering the chemical nature of the Cl-BsubNc used in the above mentioned OPVs, in three cases, Cl-BsubNc was obtained from a commercial source with an advertised “75% dye content” (i.e. 75% purity).27,33,34 In two of these three studies,27,34 the Cl-BsubNc was used “as is” while in the third,33 Cl-BsubNc was purified via thermal gradient sublimation prior to use. This commercial supplier no longer sells Cl-BsubNc. In four other studies, Cl-BsubNc was obtained from a second (and current) commercial supplier who does not advertise their purity.36–38,40 In two other papers, the source of the Cl-BsubNc was not reported but is assumed to be synthesized in the respective laboratories.31,39
Concurrent with the communication from Verreet et al.39 and Cnops et al.31 our group was investigating the process chemistry surrounding Cl-BsubNc. What we found was that the literature procedure for producing Cl-BsubNc in fact produces a product mixture with a considerable amount of chlorination of the BsubNc chromophore. We also determined that the Cl-BsubNc offered by a commercial supplier has equivalent amounts of chlorination. We have identified that chlorination is present exclusively at the bay position of the BsubNc chromophore. This is in marked contrast to our past experience with the synthesis of Cl-BsubPc13 using a similar synthetic protocol whereby no peripheral chlorination is present or has ever been observed. This is of general concern given that peripheral chlorination of Pcs is known to affect the resulting OPV characteristics.41,42 Very recently, Kahn et al. published a paper outlining the use of XPS to determine the presence of peripheral chlorination within a sample of Cl-BsubNc also from a commercial supplier.43
Herein, we outline our multi-year synthetic and device integration study, how we arrived at these conclusions and how our conclusions further support the very recent observations of Kahn et al.43 Included herein is extensive documentation of our conclusions and our efforts in modifying the synthetic and work-up procedures to obtain a highly pure (or purer) version of Cl-BsubNc with little chlorination and alternately a highly chlorinated sample(s) of Cl-BsubNc to provide points of contrast and comparison. We also present the subsequent detailed physical characterization of chlorinated Cl-BsubNcs obtained using our new processes including their incorporation into PHJ OPVs and their direct comparison with the “literature procedure” Cl-BsubNc and “commercially available” Cl-BsubNc samples. Points of comparison also include the changes in the fundamental photo- and electro-physical properties as a function of the amounts of bay position chlorination.
Results and discussion
Cl-BsubNc synthesis
Our initial investigation began with the synthesis of Cl-BsubNc via the literature method of Torres et al.,18 where 2,3-dicyanonaphthalene was reacted with BCl3 in a mixture of chlorobenzene and toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 130 °C (Table S2† – Method 1.1). This synthetic procedure is directly analogous to the one used reproducibly for the synthesis of Cl-BsubPc.13 In our hands, this procedure resulted in the formation of five Cl-BsubNc compounds as determined by HPLC-UV/vis analysis of the reaction mixture with Rt ∼ 3.6, 4.4, 5.4, 6.9, and 7.1 min, each with an absorption profile characteristic of a BsubNc chromophore (Fig. S1†). The reported purification method, Soxhlet extraction using hexane followed by chromatography on silica gel using toluene, did not produce a chromatographically pure sample of Cl-BsubNc, rather it still gave a mixture of five BsubNc products with some other non-BsubNc impurities remaining. Two additional reactions based on the modification of this method were carried out; an increase in the molar equivalencies of BCl3 from 1.0 to 2.5 (Table S2† – Method 1.2) and substitution of toluene for para-xylene (Table S2† – Method 1.3). Each led to the formation of the same five BsubNc compounds.
:1 v/v) at 130 °C (Table S2† – Method 1.1). This synthetic procedure is directly analogous to the one used reproducibly for the synthesis of Cl-BsubPc.13 In our hands, this procedure resulted in the formation of five Cl-BsubNc compounds as determined by HPLC-UV/vis analysis of the reaction mixture with Rt ∼ 3.6, 4.4, 5.4, 6.9, and 7.1 min, each with an absorption profile characteristic of a BsubNc chromophore (Fig. S1†). The reported purification method, Soxhlet extraction using hexane followed by chromatography on silica gel using toluene, did not produce a chromatographically pure sample of Cl-BsubNc, rather it still gave a mixture of five BsubNc products with some other non-BsubNc impurities remaining. Two additional reactions based on the modification of this method were carried out; an increase in the molar equivalencies of BCl3 from 1.0 to 2.5 (Table S2† – Method 1.2) and substitution of toluene for para-xylene (Table S2† – Method 1.3). Each led to the formation of the same five BsubNc compounds.
To determine if the formation of the five BsubNc compounds was exclusive under these conditions, we moved to the method described by Kennedy13 whereby 2,3-dicyanonaphthalene was reacted with BCl3 in dried 1,2-dichlorobenzene at 180 °C (Table S3† – Method 2.1). This reaction also produced the same five Cl-BsubNc compounds (Fig. S2†) albeit with a higher overall conversion. Their reported purification method – chromatography on Al2O3(IV) using toluene – also did not produce a chromatographically pure sample of Cl-BsubNc.
Based on the fact that Cl-BsubNc is vacuum sublimable,27,31,33,36–39 we turned to the use of train sublimation23 in an attempt to purify these samples of Cl-BsubNc. Train subliming crude Cl-BsubNc prepared via an adaptation of the Kennedy method (Table S3† – Method 2.3) was successful in removing most of the non-Cl-BsubNc impurities from the crude mixture after two successive train sublimation runs. While this technique was not successful in separating each of the five Cl-BsubNc compounds, the sample became suitable for analysis by low resolution mass spectroscopy (LRMS) and X-ray crystallography.
LRMS results from the sublimed Cl-BsubNc mixture showed five clusters of peaks of interest (Fig. 2). The first cluster is consistent with the +BsubNc fragment of Cl-BsubNc (m/z 545.2). The presence of this peak indicates that simple fragmentation of the axial chloride from Cl-BsubNc occurs using our MS method. The second cluster is consistent with the mass of Cl-BsubNc (m/z 580.1) however the ratio of the isotopic peaks does not. The presence of the remaining higher m/z peaks is explainable with that observation. The third, fourth, and fifth clusters at progressively higher m/z ratios are consistent with mono-chlorinated (m/z 614.1), di-chlorinated (m/z 648.1), and tri-chlorinated (m/z 682.0) Cl-BsubNc derivatives, which we will from here on refer to as Cl–Cl1BsubPc, Cl–Cl2BsubNc, and Cl–Cl3BsubNc, respectively. Assuming that each chlorinated BsubNc also fragments, the presence of the respective fragment one cluster lower explains the inconsistency in the isotopic ratios mentioned above. The ratio of the isotopic peaks for Cl–Cl3BsubNc is consistent with the structure indicating that the fragment of a higher chlorinated species is not present. Due to the relative impurity of the sample (i.e. mixture of BsubNc compounds), a HRMS analysis could not be performed. It should be noted that the number of Cl–ClnBsubNc products via LRMS (i.e. 4) is one lower than that by HPLC (i.e. 5). This could likely be explained by a set of structural isomers having slightly different Rts (6.9 and 7.1 min). Another possible explanation is that a MS peak for a fifth Cl–ClnBsubNc was not observed due to its intensity being below the detection limit of our LRMS instrument. Regardless of the analysis, the sample is a mixture consisting of differing amounts of peripheral chlorinates.
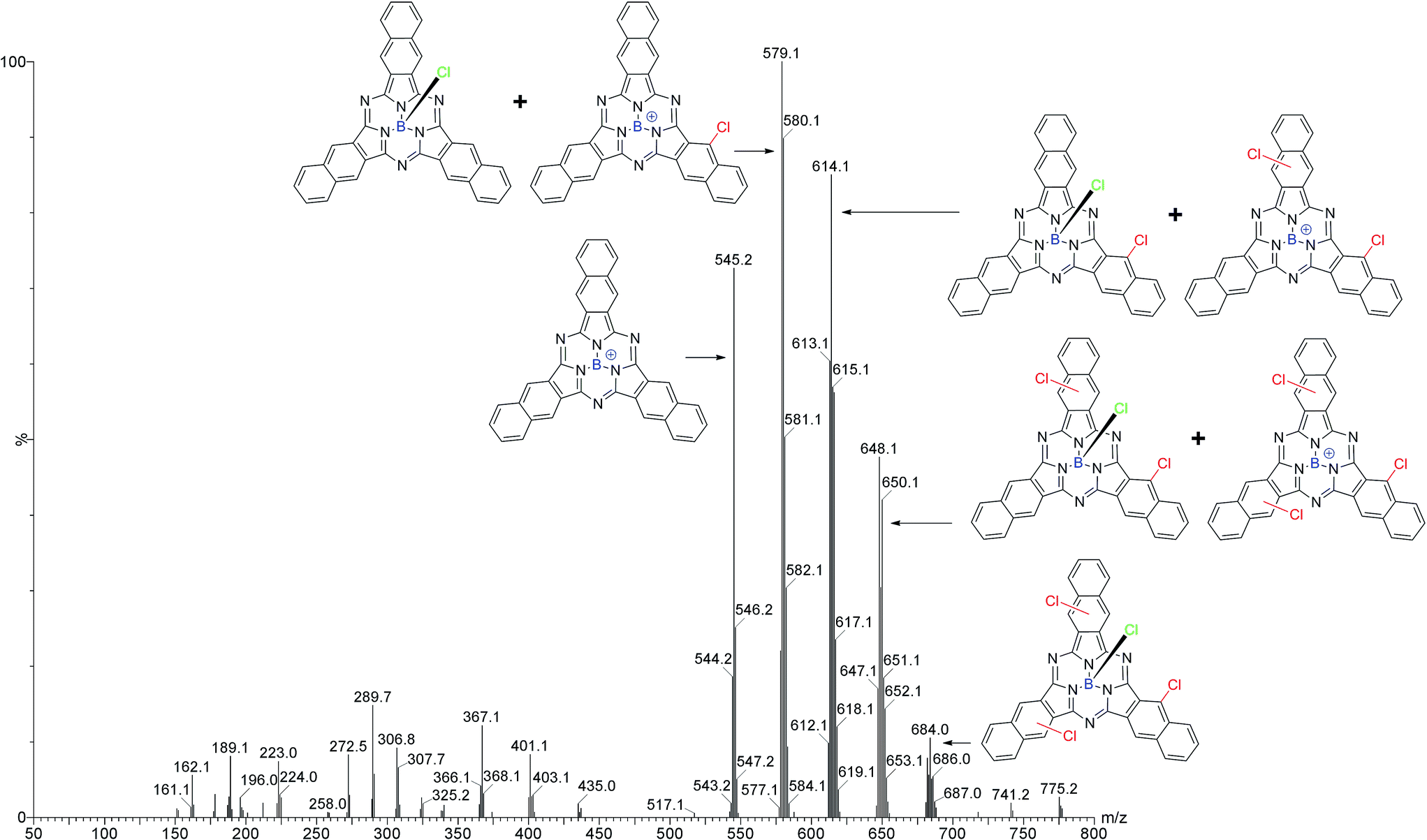 | ||
| Fig. 2 Mass spectrum of a sublimed sample of Cl–ClnBsubNc made via an adaptation of the Kennedy method indicating the presence of Cl–Cl1BsubPc, Cl–Cl2BsubNc, and Cl–Cl3BsubNc. | ||
The train sublimation process also produced crystals suitable for X-ray diffraction. While this does mark the first time a diffractable crystal of Cl-BsubNc has been analyzed and reported (Fig. 3a, Table S4–S9, and Fig. S3†), what was of more interest was the fact that the crystal was also shown to have contained mixtures of Cl-BsubNc and chlorinated Cl-BsubNcs. We will therefore from here on refer to this crystal as the literature–Cl–ClnBsubNc (Cl–ClnBsubNc produced from the literature procedure). The X-ray diffraction analysis indicated that the crystal was a mixture of compounds with differing levels of peripheral chlorination, whereby the chlorine atoms were only found to occupy the bay positions (C4, C4A, C9, C9A, C16, and C16A) of the BsubNc molecular fragment. The structure was also found to be symmetric and hence, there are three different sites for potential chlorine occupancies (C4, C9, and C16 and their symmetric sites). The probability of a chlorine atom at C4, C9, and C16 was found to be 23.6%, 16.9%, and 16.1%, respectively, resulting in a total occupancy of 1.13 peripheral chlorines per molecule and an average molecular formula of C36H16.87BCl2.13N6.
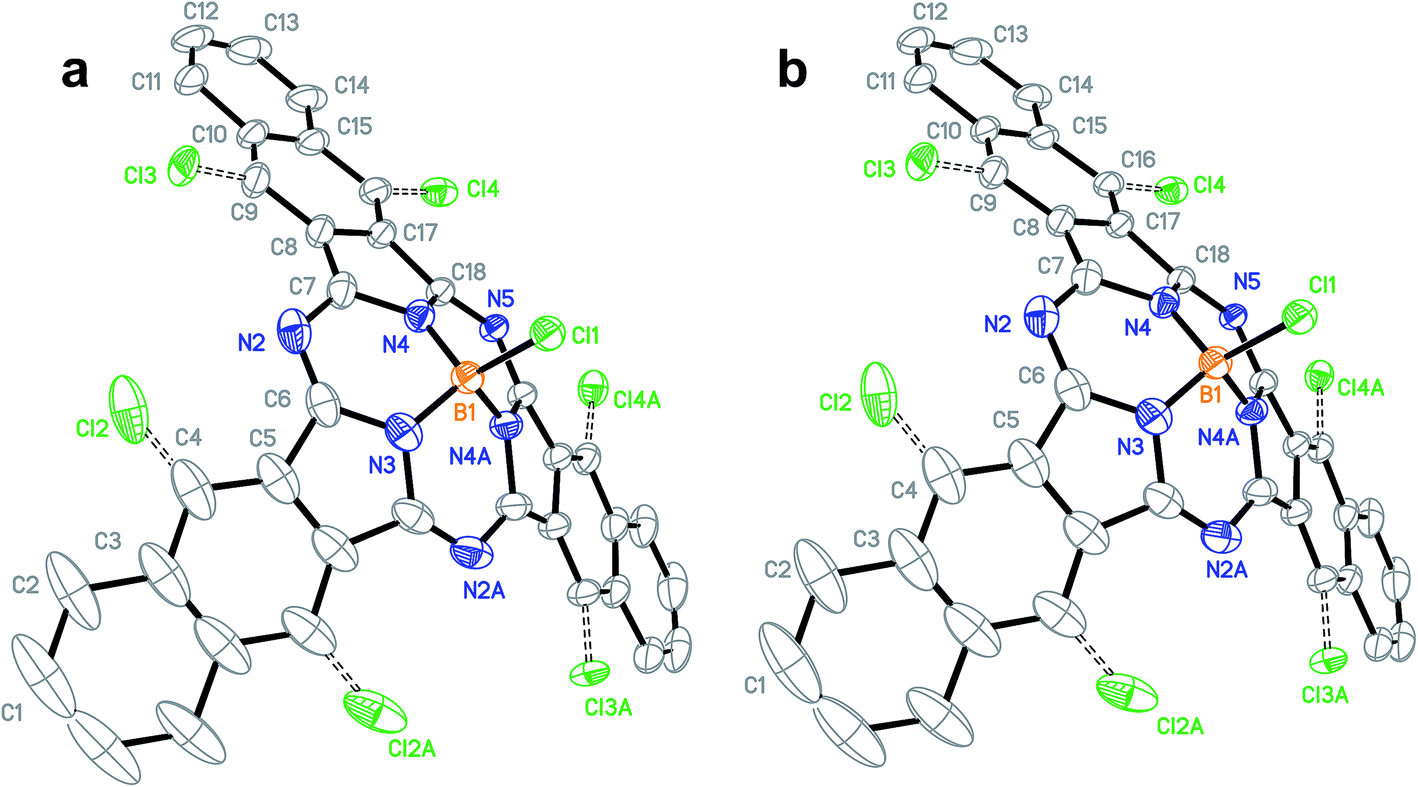 | ||
| Fig. 3 Ellipsoid plot (50% probability) showing the structure and atom numbering scheme of Cl–ClnBsubNc crystals obtained from (a) the literature method (CCDC deposition number: 1452383†) and (b) the nitrobenzene method (CCDC deposition number: 1452384†). Hydrogen atoms have been omitted for clarity. Colors: boron – orange; nitrogen – blue; carbon – black; chlorine – green. | ||
Thereafter we set out to develop synthetic conditions that might limit the amount of bay position chlorination. It has been reported that peripheral chlorination (or halogenation) can be reduced by using an electron-rich aromatic co-solvent to scavenge any chlorinating species.44,45 For this role, we chose 1-methylnaphthalene (1-MNAP) as a co-solvent45 and 2,3-dicyanonaphthalene was treated with BCl3 in a 1,2,4-trichlorobenzene:1-MNAP mixture (4:1 v/v) at 130 °C (Table S10† – Method 3.6). The introduction of 1-MNAP into the process completely suppressed the formation of three of the five Cl-BsubNc peaks leading to just two Cl-BsubNcs being present in the HPLC analysis (Fig. S4a†). Moreover, the integration of the peak at 3.6 min is much greater than the peak at 4.4 min which is opposite to what is observed above. Attempts to isolate a pure sample of Cl-BsubNc were still however unsuccessful. We tried train sublimation to purify the mixture. Like the previous example (Method 2.3), the technique was not successful in separating the two Cl-BsubNc compounds. Moreover, the sublimation yield was found to be extremely low (highest obtained yield of 3.7%) making it very challenging and laborious to obtain adequate amounts of sublimed material. However, a LRMS analysis (Fig. S4b†) on the sublimed mixture was consistent with the target Cl-BsubNc (m/z 580.1) and a trace of Cl–Cl1BsubNc (m/z 614.1). This result combined with the greater HPLC peak intensity at 3.6 min versus 4.4 min allowed for the identification of the Cl-BsubNcs at 3.6 and 4.4 min to be Cl-BsubNc (non-chlorinated) and Cl–Cl1BsubNc (mono-chlorinated), respectively.
The low yield of this process is not entirely attributable to the sublimation step. While the conversion by HPLC was measured to be ∼58% this does not accurately reflect the actual conversion. A considerable amount of black, insoluble mass is formed during the reaction, making it necessary to do filtration and wash with hot toluene to separate the black mass from the extracted Cl-BsubNc products.
We therefore next moved to reduce the amount of black mass produced during the process. Our hypothesis was that the Lewis acidic BCl3 was promoting the polymerization of 2,3-dicyanonaphthalene and producing the black mass. We scoped the reaction conditions whereby the molar concentration of 2,3-dicyanonaphthalene, the molar equivalencies of the boron template, and the solvent system were changed (Table S11† – Methods 4.1–4.15). Unfortunately, none of the attempts were successful in producing a non-chlorinated Cl-BsubNc; either a mixture of Cl–ClnBsubNcs or no Cl-BsubNc was formed and in all cases a considerable amount of black mass was still produced. We also added ethylene glycol to the reaction mixture in an attempt to lower the reactivity/Lewis acidity of BCl3 through a chloride-oxygen ligand displacement reaction(s) (Table S12† – Methods 5.1–5.10). In short, none of the reactions were successful in producing exclusively the desired, non-peripherally chlorinated Cl-BsubNc and nor did it they reduce the amount of black mass produced.
Considering that the addition of 1-MNAP as a chlorinating species scavenger was the most promising by producing just two Cl–ClnBsubNcs, we turned to the exploration of other potentially more effective chlorinating species scavengers. Potential scavengers were selected to be liquids with a boiling point greater than 180 °C (the maximum temperature that the reaction was heated to) and since the exact mechanism of the chlorination was not known, scavengers were selected to specifically target Cl+ (cationic) and Cl˙ (radical) species. Four scavengers were selected: dipentene (Cl+), (−)-β-pinene (Cl+), 1-octadecene (Cl+), and p-cymene (Cl+ and Cl˙) (Table S13† – Methods 6.1–6.4). The addition of 1-octadecence and dipentene resulted in complete stalling of the reaction; no Cl-BsubNc was produced. The reactions containing either (−)-β-pinene or p-cymene resulted in the formation of the same two Cl-BsubNc compounds (Rt = 3.6 and 4.4 min, Cl-BsubNc and Cl–Cl1BsubNc) that were observed in the 1-MNAP experiment. The apparent conversion by HPLC was poor for the (−)-β-pinene experiment (17%) and higher for the p-cymene experiment (77%), however there was still a considerable amount of black mass produced. A comparison of the HPLC analysis for the 1-MNAP (Fig. S4a†) and p-cymene process (Fig. S5a†) shows that the intensity of the peak at 4.4 min (Cl–Cl1BsubNc) relative to the peak at 3.6 min (Cl-BsubNc) is lower for the p-cymene reaction. It therefore appears that p-cymene is slightly more effective at scavenging chlorine species than 1-MNAP. For this reason, p-cymene was investigated furthermore.
Sublimation of the crude Cl-BsubNc material prepared via the p-cymene process was still very low yielding (highest yield obtained was 6.1%), but not as low as the 1-MNAP case (highest yield obtained was 3.7%). Furthermore, the technique was again not successful in separating the two BsubNc compounds (as indicated by HPLC analysis). LRMS analysis (Fig. S5b†) on the sublimed mixture was consistent with Cl-BsubNc (m/z 580.1). A MS peak for Cl–Cl1BsubNc (m/z 614.1) species was not observed but this was likely due to its intensity being below the detection limit of our LRMS instrument.
We then set out to synthesize a comparative Cl–ClnBsubNc sample with a maximized level of peripheral chlorination. For this purpose, we chose an aromatic solvent with limited or no chlorine scavenging properties. Three different electron-poor solvents – nitrobenzene (NB), 1,2,4-trichlorobenzene, and 2,4-dinitro-1-fluorobenzene – were chosen (Table S14† – Methods 7.1–7.4). The experiment carried out in NB was the most interesting as it produced a significantly different HPLC chromatogram than any of the aforementioned processes (Fig. S6a†). Five BsubNc compounds were detected (Rt = 4.4, 5.4, 6.7, 6.9, and 8.7 min) like the literature process, but the peak at 3.6 min is absent and a new peak at 8.7 min is formed. Moreover, the peaks at the higher Rts are more intensive than those at the lower Rts, indicating that this mixture is composed of a higher composition of chlorinated species. Like all cases mentioned above, train sublimation was not successful in separating any of the BsubNc compounds. Nevertheless, sublimation of the crude product was found to be higher-yielding (average: 10%) than the p-cymene or 1-MNAP process. LRMS analysis on the sublimed mixture was consistent with the presence of Cl–Cl1BsubNc (m/z 614.1), Cl–Cl2BsubNc (m/z 648.1), Cl–Cl3BsubNc (m/z 682.0), Cl–Cl4BsubNc (m/z 716.0), and Cl–Cl5BsubNc (m/z 749.9) (Fig. S6b†). It should be emphasized that the non-chlorinated Cl-BsubNc was not formed in the NB process as evident in the absence of its HPLC peak at 3.6 minutes (Fig. S6a†) and the m/z 580.1 peak (Fig. S6b†).
The sublimation process also produced single crystals suitable for X-ray diffraction (Fig. 3b, Table S15–S20, and Fig. S7†). Like the previous analysis outlined above, the X-ray analysis of the crystals determined that they were a mixture of BsubNcs with differing levels of peripheral chlorination, where the chlorine atoms were present exclusively at the bay position. Unlike the previous sample for literature–Cl-BsubNc, the level of peripheral chlorination is much higher for the nitrobenzene–Cl-BsubNc (2.96 vs. 1.13 peripheral chlorines per molecule). This results in an average molecular formula of C36H15.04BCl3.96N6 and is consistent with the LRMS results presented above. Beyond this analysis, both were orthorhombic crystals of the Pnma space group. Unit cell dimensions were found to only be different by a marginal amount (0.28 Å, 0.09 Å, and 0.38 Å for a, b, and c respectively, Table S21†). The crystals from the nitrobenzene process were of 0.07 mg cm−3 higher calculated density owing to the higher chlorine content. Additionally, having two crystal structures provides a point of comparison for the respective solid-state arrangements as well. It was determined that each crystal had the same solid-state arrangement (Fig. S8 & S9†). Again, it is important to note that these are diffractable crystals consisting of a mixture of compounds.
To summarize the synthetic process development, we were able to produce four samples of Cl–(Clx)BsubNc with varying degrees of peripheral chlorination; a sample of Cl-BsubNcs made via the Kennedy's method is a mixture of three compounds: Cl-BsubNc, Cl–Cl1BsubNc, and Cl–Cl2BsubNc; a sample using the 1-MNAP procedure is a similar mixture of two compounds: Cl-BsubNc and Cl–Cl1BsubNc; a sample using the p-cymene method which is also a mixture of two compounds: Cl-BsubNc and Cl–Cl1BsubNc; and a sample using the nitrobenzene (NB) procedure is a mixture of five compounds: Cl–Cl1BsubNc, Cl–Cl2BsubNc, Cl–Cl3BsubNc, Cl–Cl4BsubNc, and Cl–Cl5BsubNc. We will term each of these samples “literature–Cl–ClnBsubNc”, “1-MNAP–Cl–ClnBsubNc”, “p-cymene–Cl–ClnBsubNc”, and “nitrobenzene–Cl–ClnBsubNc, respectively. In the 1-MNAP- and p-cymene–Cl–ClnBsubNc samples, the relative amount of Cl-BsubNc and Cl–Cl1BsubNc are slightly different. Finally, we were only able to produce adequate amounts of train sublimed samples of “literature–Cl–ClnBsubNc”, “p-cymene–Cl–ClnBsubNc”, and “nitrobenzene–Cl–ClnBsubNc” for further investigation. “1-MNAP–Cl–ClnBsubNc” could not be obtained due to its very poor sublimation yield (highest obtained yield = 3.7%). Numerous attempts were made to optimize the yield of 1-MNAP–Cl–ClnBsubNc, but none of them could produce a yield greater than 3.7%. We surmise that the low sublimation yields are attributed to a narrow temperature window for the desired sublimation process to occur and the undesired decomposition process. The latter is based on the fact that the residual material in the boat following a sublimation process is black, insoluble char. Finally and more generally, we believe that based on our results there is little prospect for the development of a synthetic or purification process that will yield pure Cl-BsubNc.
Analysis of commercial Cl-BsubNc samples
We also acquired a sample of commercial Cl-BsubNc (termed “commercial–Cl–ClnBsubNc”) and analyzed it by HPLC and LRMS. HPLC (Fig. S10a†) revealed the presence of the same five BsubNc compounds produced from the literature–Cl–ClnBsubNc process. Also, the LRMS analysis results of commercial–Cl–ClnBsubNc matched that of the literature–Cl–ClnBsubNc (Fig. S10b†). Combined, these analyses demonstrate that the commercial source of Cl–ClnBsubNc was not a pure sample, rather a mixture of peripherally chlorinated BsubNc products.X-ray photoelectron spectroscopy (XPS) analysis
Core level XPS (Cl 2p) analysis was also performed on thermally evaporated, vacuum-deposited thin films of the Cl–ClnBsubNcs (Fig. 4, Table S22, and Fig. S11†). For all Cl–ClnBsubNcs, two distinct chemical states are found for chlorine, chlorine bonded to boron (Cl–B, binding energy = 199.5–199.6 eV) and chlorine bonded to carbon (Cl–C, binding energy = 200.5–200.8 eV). These results are consistent with those of Kahn et al. who found two distinct chlorine peaks at 199.3 eV and 200.5 eV.43 XPS analysis for boron and nitrogen (Fig. S12–14†) is also consistent with the chemical structure of BsubNc.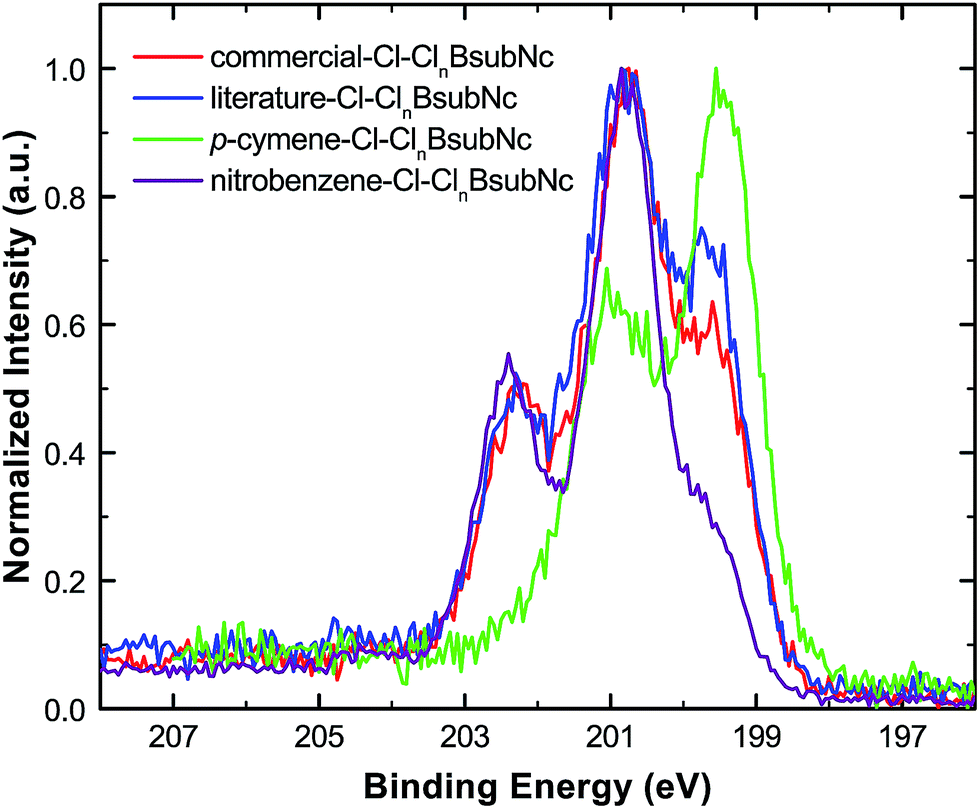 | ||
| Fig. 4 Overlay of the literature–Cl–ClnBsubNc (blue), commercial–Cl–ClnBsubNc (red), p-cymene–Cl–ClnBsubNc (green), and nitrobenzene–Cl–ClnBsubNc (purple) normalized Cl 2p core-level XPS spectra. | ||
Bay position chlorination was further quantified by examining the deconvoluted XPS Cl peak areas (Table S23†) and their relative ratios (Cl–C/Cl–B). The ratios are found to be 1.21, 1.61, 0.18, and 4.17 for literature-, commercial-, p-cymene-, and nitrobenzene–Cl–ClnBsubNc, respectively, while Kahn et al.43 found a ratio of ∼1.5 for commercial Cl-BsubNc. Alternatively, the number of bay position chlorine atoms can also be calculated by considering the Cltotal/Ntotal ratio by multiplying by 6 (i.e. the total number of nitrogens per molecule) and subtracting 1 (i.e. the number of chlorine bonded to boron). This approach produces similar numbers of 1.40, 1.76, 0.32, and 4.40 for literature-, commercial–, p-cymene-, and nitrobenzene–Cl–ClnBsubNc, respectively (Table S23†). Overall, these measurements are consistent with the differing degree of bay position chlorination among the samples.
1H NMR analysis
1H NMR analysis was carried out for the four synthetic Cl–ClnBsubNc samples, each doubly sublimed along with the commercial sample (Fig. S15†). For literature-, 1-MNAP- and p-cymene–Cl–ClnBsubNc, the 1H NMR spectra are very similar in terms of chemical shift and multiplicity. Integration of the bay position 1H resonances found at ∼9.45 ppm indicates an increased presence of the bay position protons for the 1-MNAP–Cl–ClnBsubNc and p-cymene–Cl–ClnBsubNc, an observation consistent with the preceding data. A noticeable observation is the presence of additional peaks (i.e. impurities) in the spectrum of commercial–Cl–ClnBsubNc. The 1H NMR spectrum of nitrobenzene–Cl–ClnBsubNc, on the other hand, is very different compared to the other three spectra for the synthetic samples. There are additional peaks in the aromatic region consistent with a large number of chlorinated BsubNc isomers with asymmetry. Assignment of the resonances and quantification is problematic. Kahn et al. have briefly reported similar results on the purity of a commercial source of Cl-BsubNc via1H NMR. They had concluded the same and that the commercial Cl-BsubNc sample is a mixture of chlorinated species and outlined that full detailed NMR investigation is currently underway within their group.43Photophysical analyses of Cl–ClnBsubNcs
Given there is a discrepancy in the literature regarding the photophysical properties of BsubNcs,9,18 UV-vis absorption spectra of literature–Cl–ClnBsubNc, commercial–Cl–ClnBsubNc, p-cymene–Cl–ClnBsubNc, and nitrobenzene–Cl–ClnBsubNc were acquired in toluene solutions at room temperature (Table 1 and Fig. S16a†). Due to the challenges associated with subliming 1-MNAP–Cl–ClnBsubNc, this compound was excluded from this photophysical and subsequent study. The solution absorption profiles of the literature–Cl–ClnBsubNc and commercial–Cl–ClnBsubNc are found to be nearly identical, both with a λmax value of 656 nm. In contrast to this, p-cymene–Cl–ClnBsubNc has a blue-shifted λmax value of 651 nm while nitrobenzene–Cl–ClnBsubNc has a red-shifted λmax value of 664 nm. The shift in the λmax,abs values is not surprising to us as we have observed a similar effect for the analogous Cl-BsubPc, whereby peripheral chlorination red-shifts the λmax,abs: Cl-BsubPc, Cl–Cl6BsubPc, and Cl–Cl12BsubPc at 565, 574, and 593 nm, respectively (Table S24†). Torres et al. have reported a higher λmax,abs value of 661 nm for their Cl-BsubNc.18 The extinction coefficients (ε) for the four Cl–ClnBsubNcs were found to be about the same and also consistent with the value reported by Torres et al.18| Compound | λ max,abs |λmax,absb (nm) | Extinction coefficient (ε, M−1 cm−1) | λ max,PL , |λmax,PLb,c (nm) | Stokes shifta|Stokes shiftb (nm) | Φ PL , (%) |
|---|---|---|---|---|---|
| a In toluene solution. b Solid state (film). c λ exc = 630 nm. d Relative to a oxazine 170 standard using an excitation wavelength of 630 nm. e Data taken from Torres et al.18 | |||||
| Literature–Cl–ClnBsubNc | 656|688 | 78400 |
666|746 | 10|58 | 27 |
| Commercial–Cl–ClnBsubNc | 656|691 | 78400 |
666|738 | 10|47 | 24 |
| p-Cymene–Cl–ClnBsubNc | 651|685 | 79200 |
662|746 | 11|61 | 29 |
| Nitrobenzene–Cl–ClnBsubNc | 664|698 | 77800 |
674|750 | 10|52 | 21 |
| Torres' Cl-BsubNc | 661e | 79400e |
677e | 16 | 22e |
The photoluminescence (PL) spectra of the four Cl–ClnBsubNc samples were also acquired in toluene solutions at room temperature (Table 1 and Fig. S16b†). Like the results from the absorption study, the PL spectra are nearly identical for both literature–Cl–ClnBsubNc and commercial–Cl–ClnBsubNc (λmax,PL = 666 nm) while a blue-shift in the spectrum of p-cymene- (λmax = 662 nm) and a red-shift in the spectrum of nitrobenzene–Cl–ClnBsubNc (λmax = 674 nm) is observed. Despite the differences in the λmax of absorption and emission spectra, all the Stokes shifts are found to be similar (10–11 nm). Torres et al. have reported a slightly more red-shifted λmax,PL of 677 nm for their Cl-BsubNc, resulting in a marginally larger measured Stokes shift of 16 nm.36 Fluorescence quantum yields (ΦPL) were also measured and they were found to be between 24 and 29% relative to a standard of oxazine 170 (Table 1 and eqn (S1)†). These values are in line with that reported by Torres et al. (ΦPL = 0.22).18
Solid state (film) UV-vis absorption and PL spectra were also acquired and compared to the solution state (Table 1 and Fig. S17†). As expected, the spectra are broader but are still very similar in shape and form. A moderate bathochromic shift of 32–35 nm in the absorption spectra and an even greater bathochromic shift of 72–84 nm in the PL spectra are observed relative to their respective solution state spectra. Large Stokes shifts of 47–61 nm are seen in the solid state and are indicative of intermolecular organization and aggregation.
Electrochemical analyses of Cl–ClnBsubNcs
The electrochemical properties of the four Cl–ClnBsubNc samples were then analysed via cyclic voltammetry (CV) in degassed DCM solution containing 0.1 M tetrabutylammonium perchlorate (TBAPC) (Table 2 and Fig. S18a–20a†). All potentials (E) were corrected to the half-wave reduction potential (E1/2,red) of decamethylferrocene (−0.012 V vs. Ag/AgCl46). Literature-, commercial-, and p-cymene–Cl–ClnBsubNc have a nearly identical reversible first (E1ox) oxidation and an irreversible second (E2ox) oxidation. On the reduction side, a single pseudo-reversible reductive process is also observed. Unlike what is observed in the oxidation regime, the reduction potentials are found to be different depending on the nature of the BsubNc mixture; literature- (−0.87 V) and commercial–Cl–ClnBsubNc (−0.89 V) shared a similar reduction potential (E1red) while p-cymene Cl–ClnBsubNc has a much higher potential (−0.99 V). A cyclic voltammogram of nitrobenzene–Cl–ClnBsubNc could not be obtained due to the poor signal-to-noise ratio likely caused by its observed poor solubility in DCM. While chlorination did not impact the oxidative potential and behavior of Cl–ClnBsubNcs, chlorination did lower the reduction potential.| Compound | E 1ox|E2oxb (V) | E 1red (V) | E 1ox|E2oxc (V) | E 1red|E2red|E3redc (V) |
|---|---|---|---|---|
| a E = redox potential from CV. b In degassed DCM solution relative to Ag/AgCl. c In degassed DMF solution relative to Ag/AgCl. d Peak potential. e Half-wave potential. f Minor peak. g Data taken from Nonell et al.50 | ||||
| Literature–Cl–ClnBsubNc | +0.83e|+1.28d | −0.87d | +0.84d | −0.92d|−1.06d,f|−1.36d,f |
| Commercial–Cl–ClnBsubNc | +0.84e|+1.29d | −0.89d | +0.85d | −0.91d|−1.07d,f|−1.37d,f |
| p-Cymene–Cl–ClnBsubNc | +0.82e|+1.28d | −0.99d | +0.75d|+1.11d | −0.94d|−1.06d,f|−1.38d,f |
| Nitrobenzene–Cl–ClnBsubNc | — | — | +0.86d|+1.11d | −0.81d|−1.28d,f |
| Nonell et al. | — | — | +0.68g | −0.85g |
Due to the poor solubility of nitrobenzene–Cl–ClnBsubNc in DCM solution, CV analyses of the four samples were thereafter redone in degassed N,N-dimethylformamide (DMF) solution (Table 2 and Fig. S21a–S24a†). Within DMF, all redox events were found to be irreversible or pseudo-reversible at best. Literature- and commercial–Cl–ClnBsubNc have nearly the same voltammograms with similar E1ox, E1red, E2red, and E3red values while noticeable differences are observed for p-cymene- and nitrobenzene–Cl–ClnBsubNc. For p-cymene–Cl–ClnBsubNc, it has a lower E1ox (+0.75 V) and also has a E2ox (+1.11 V). In the reduction regime, there are three reductive events (−0.94, −1.06, −1.38 V). For nitrobenzene–Cl–ClnBsubNc, there are two oxidative events where E1oxid (+0.86 V) and E2oxid (+1.11 V) are in line with the values seen for literature-/commercial–Cl–ClnBsubNc and p-cymene–Cl–ClnBsubNc, respectively. On the reduction side, two peaks (−0.81, −1.28 V) are noted with E1red being lower than that of literature-, commercial-, or p-cymene–Cl–ClnBsubNc. Overall, these results do not totally match the CV results obtained in DCM suggesting that the electrochemical properties of Cl–ClnBsubNc is solvent-dependent as expected.47–49 Additionally, these results are not consistent with the electrochemical properties of Cl-BsubNc previously reported by Nonell et al. measured in degassed DMF with 0.1 M tetrabutylammonium hexafluorophosphate (TBAHF)50 using Cl-BsubNc prepared according to the procedure published of Torres et al.18 This sample likely has the same level of peripheral chlorination as our literature- and commercial–Cl–ClnBsubNc samples. A single irreversible oxidation at +0.68 V and a pseudo-reversible reduction at −0.85 V were reported, both lower than the E1ox and E1red of literature- or commercial–Cl–ClnBsubNc measured herein.
Differential pulse voltammetry (DPV, 0.1 M TBAPC) was also performed (Table 3, Table S25, and Fig. S18b–24b†). DPV redox potentials (E′) were found to be generally lower than CV redox potentials (E) in either DCM or DMF. In DCM, additional peaks were detected via DPV in the reduction region for p-cymene- and commercial–Cl–ClnBsubNc (Fig. S19b and S20b,† respectively). In DMF, additional peaks were detected via DPV in the oxidation region for literature- and commercial–Cl–ClnBsubNc and in the reduction region for nitrobenzene–Cl–ClnBsubNc (Fig. S21b, S24b, and S23b,† respectively). Despite these differences, the findings within the DPV dataset are in line with those within the CV dataset of the same solvent system.
| Compound | E 1′ox|E2′oxb (V) | E 1′red|E2′red|E3′redb (V) | IE (HOMO)e (eV) |
|---|---|---|---|
| a E′ = redox potential from DPV. b In degassed DMF solution relative to Ag/AgCl. c Peak potential. d Minor peak. e Measured by UPS. f Data taken from Kahn et al.43 | |||
| Literature–Cl–ClnBsubNc | +0.75c|+1.00c,d | −0.87c|−1.02c|−1.33c | 5.32 |
| Commercial–Cl–ClnBsubNc | +0.76c|+1.02c,d | −0.86c|−1.01c|−1.29c | 5.31 |
| p-Cymene–Cl–ClnBsubNc | +0.69c|+1.01c | −0.93c|−1.08c|−1.35c | 5.29 |
| Nitrobenzene–Cl–ClnBsubNc | +0.78c|+1.02c | −0.79c|−0.98c,d|−1.26c,d | 5.42 |
| Kahn et al. | — | — | 5.35f |
Considering that the four samples under study are mixtures (i.e. not a single, pure compound) it should be noted that this would or could affect the electrochemical signals' dimensions (broadness, height, resolution, etc.) and thus, the apparent peak potential values. For this reason, three other processing methods were performed on the DPV dataset to determine if the results acquired from the apex peak method are plausible (Table S26†) and in short, these further studies supported the overall trends. Further details about these processing methods can be found in the ESI.†
Ultraviolet photoelectron spectroscopy (UPS) analysis
Given the application of interest for Cl–ClnBsubNcs is within the thin solid film of an organic photovoltaic device (OPV), we then examined each of the four mixtures using ultraviolet photoelectron spectroscopy (UPS). UPS was performed on thermally evaporated, vacuum-deposited thin films to determine the HOMO energy level (i.e. ionization energy) of each sample and to further demonstrate their electronic property differences (Fig. 5, Table 3, and Fig. S25†). The HOMO level is found to be the shallowest for p-cymene–Cl–ClnBsubNc (5.29 eV), about the same for literature- (5.32 eV) and commercial–Cl–ClnBsubNc (5.31 eV), and the deepest for nitrobenzene–Cl–ClnBsubNc (5.42 eV). Kahn et al. have also reported a very similar value of 5.35 eV for a commercial sample of Cl-BsubNc.43 A trend where the HOMO level deepens with increasing chlorination is evident here. A similar effect was reported for BsubPcs, where the UPS-determined HOMO level shifted away from the vacuum line with increasing fluorination.23 Also, the peak at a binding energy of ∼4.5 eV is more pronounced in the nitrobenzene–Cl–ClnBsubNc sample, an observation that is likely attributed to the higher chlorinated species. Further UPS details can be found in the ESI.†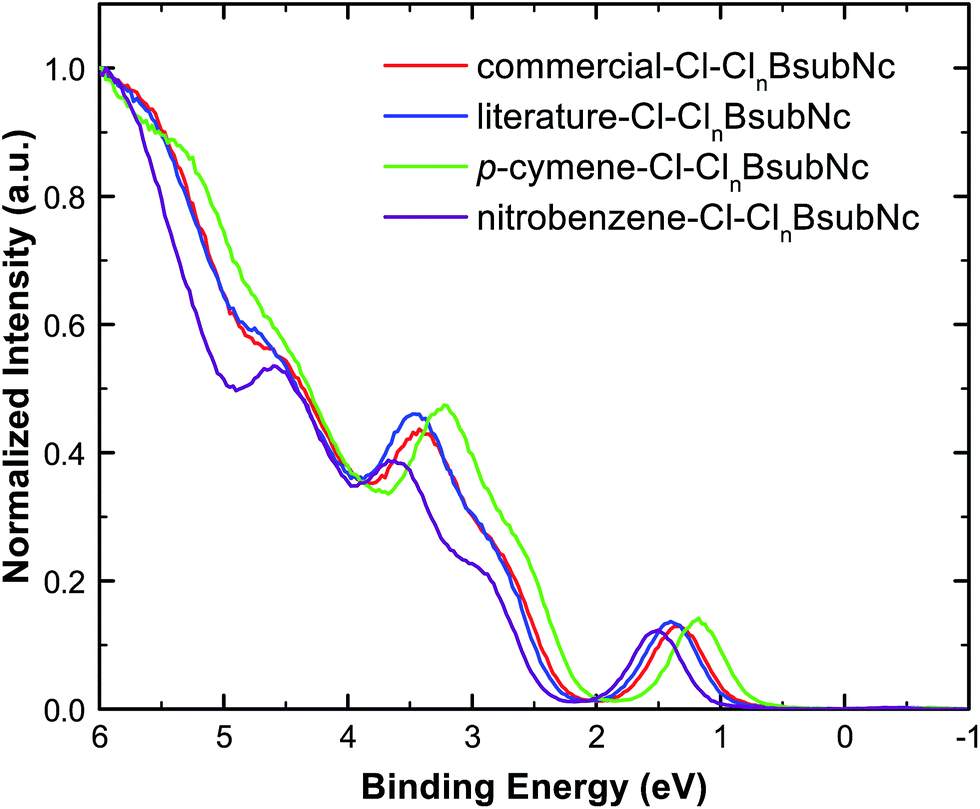 | ||
| Fig. 5 Overlay of the literature–Cl–ClnBsubNc (blue), commercial–Cl–ClnBsubNc (red), p-cymene–Cl–ClnBsubNc (green), and nitrobenzene–Cl–ClnBsubNc (purple) normalized UPS valence band spectra. | ||
OPV device integration of Cl–ClnBsubNcs
Although we could not purify any of our synthetic samples to an absolutely pure single Cl-BsubNc compound, we did examine the performance of each within a PHJ OPV to determine if there are any performance differences attributable to the differing amounts of peripheral chlorination. Given that diffractable crystals were obtained via sublimation and XPS results obtained from sublimed films were consistent, we were not concerned with any alteration of the chemical composition in the fabrication of PHJ OPVs. We also incorporated the “commercial–Cl–ClnBsubNc” into OPVs as an additional point of comparison.Four sets of OPV devices were therefore fabricated, pairing each sample of Cl–ClnBsubNc as the electron acceptor with sexithiophene (α-6T) as an electron donor, with the following device configuration: indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/sexithiophene (α-6T, 55 nm)/Cl–ClnBsubNc (25 nm)/bathocuprione (BCP, 10 nm)/silver (Ag, 80 nm). These devices mimic our previous work using Cl-BsubPc paired with α-6T51 and are consistent with and comparable to the PHJ OPVs fabricated by Cnops et al.31 Current density–voltage (J–V) characteristics (Fig. 6a) were measured under 100 mW cm−2 of simulated solar illumination (AM1.5). The measured external quantum efficiency (EQE) spectra are shown in Fig. 6b. Data are derived from the measurement of at least 6 PHJ OPV devices and therefore the standard deviation for each device metric is also presented (Table 4).
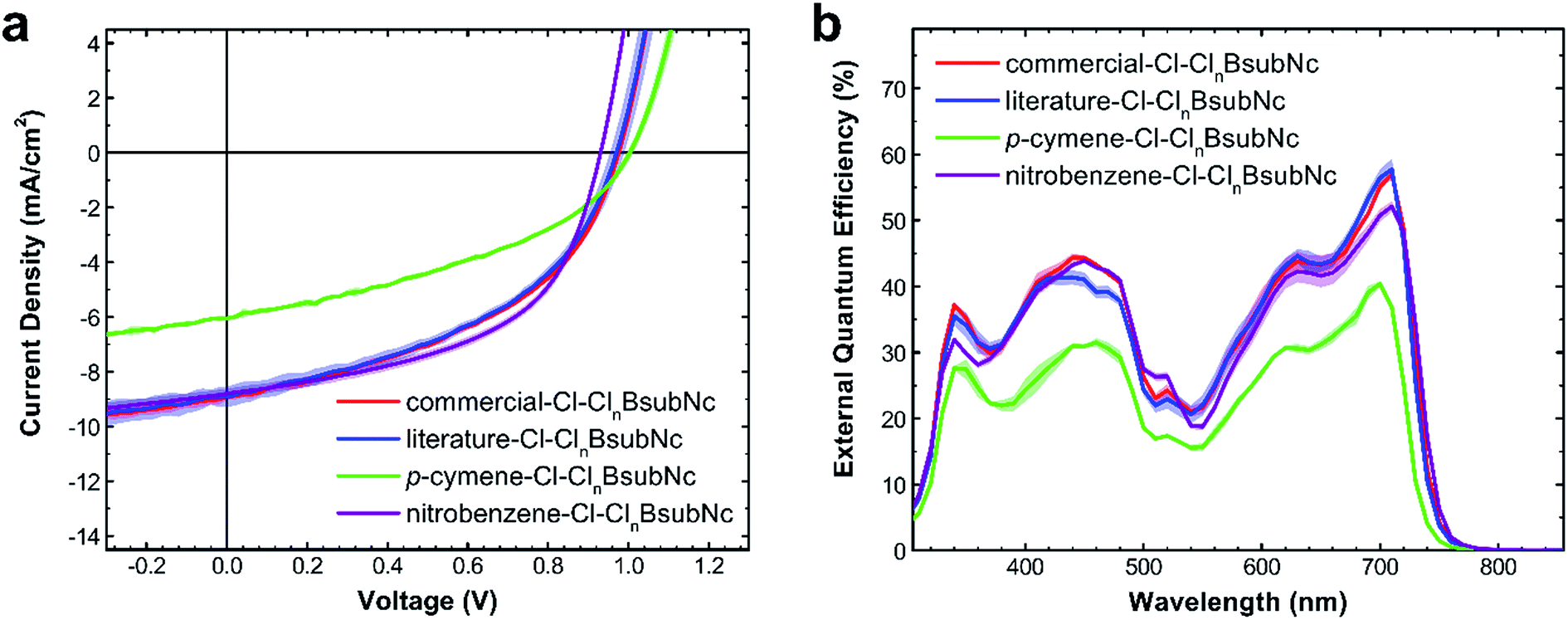 | ||
| Fig. 6 (a) J/V characteristics and (b) external quantum efficiency (EQE) spectra of p-cymene–Cl–ClnBsubNc, literature–Cl–ClnBsubNc, and commercial–Cl–ClnBsubNc. PHJ OPVs of the following configuration: ITO/PEDOT:PSS/α-6T (55 nm)/Cl–ClnBsubNc (25 nm)/BCP (10 nm)/Ag (80 nm) where the Cl–ClnBsubNc layer is made of nitrobenzene–Cl–ClnBsubNc p-cymene–Cl–ClnBsubNc, literature–Cl–ClnBsubNc, or commercial–Cl–ClnBsubNc. | ||
| Acceptor | V OC (V) | J SC (mA cm−2) | FFc | PCEd (%) |
|---|---|---|---|---|
| a Open-circuit voltage. b Short-circuit current. c Fill factor. d Power conversion efficiency. | ||||
| p-Cymene–Cl–ClnBsubPc | 1.004 (0.009) | 6.05 (0.07) | 0.40 (0.01) | 2.44 (0.13) |
| Literature–Cl–ClnBsubPc | 0.972 (0.018) | 8.92 (0.37) | 0.45 (0.01) | 3.90 (0.12) |
| Commercial–Cl–ClnBsubPc | 0.976 (0.005) | 8.91 (0.13) | 0.46 (0.01) | 3.96 (0.07) |
| Nitrobenzene–Cl–ClnBsubPc | 0.930 (0.002) | 8.80 (0.19) | 0.53 (0.01) | 4.32 (0.11) |
The OPVs prepared with literature–Cl–ClnBsubNc and commercial–Cl–ClnBsubNc samples perform nearly identically. The performance of these devices fabricated in our laboratory is not quite as high as for those made by Cnops et al.,31 but the losses can be attributed to a less extensive optimization of the layer thicknesses undertaken in our fabrication system. A much lower performance is achieved for devices made with the p-cymene–Cl–ClnBsubNc sample, with less bay position chlorination, where both the short-circuit current (JSC) and the fill factor (FF) fall significantly short of the OPVs derived from Cl–ClnBsubNcs with higher levels of chlorination. Conversely a small yet statistically relevant increase in VOC was observed, as was suggested by Khan et al.43 Now considering nitrobenzene–Cl–ClnBsubNc, while we saw an equally small reduction in the VOC of the PHJ OPV and a small reduction in the JSC, a notable increase in the fill factor therefore resulted in the highest PCE. The observed small changes in VOC are in line and proportional with the measured HOMO by UPS.
These results suggest that inclusion of the bay position chlorinated Cl–ClnBsubNc derivatives in a mixture with Cl-BsubNc improves the overall performance of the layer in OPVs. This is conceptually similar to the work of Fleetham et al. who showed an enhancement in OPV performance using a mixture of peripherally chlorinated zinc phthalocyanine (ZnPc) and pure ZnPc.41,42
Traditionally it has been believed that impurities in organic semiconductors will always act as traps, hindering device performance.52,53 Street et al. have recently challenged that notion, demonstrating that different materials, specifically fullerene derivatives, can be blended together to achieve an alloying effect of their electronic properties, so long as their size allows them to easily intermix without disrupting the local order of one another.54,55 The crystal structure of literature–Cl–ClnBsubNc shows co-crystallization of the different bay position chlorinated Cl-BsubNc compounds, indicating that the local order is not disrupted by the presence of bay position chlorination. Therefore, an alloying effect similar to what Street et al.54,55 observed for fullerenes in bulk heterojunction OPVs could be occurring. This could explain why the performance of a Cl–ClnBsubNc mixture in OPVs is not crippled by impurity trap states rather it is improved. While a comparison against absolutely pure Cl-BsubNc was not possible, and may not be possible, the strong performance in OPVs utilizing “Cl-BsubNc” reported in literature may in fact be caused by the alloying together of slightly different electronic states.
Conclusions
In this paper, we outline the proof that synthetic and commercial samples of Cl-BsubNc are actually a mixed alloyed chemical composition with random and significant amounts of chlorination in the bay position of the BsubNc chromophore. We also outline our efforts to prepare a single, pure Cl-BsubNc compound instead of a mixture of Cl–ClnBsubNc compounds with differing levels of bay position chlorination. After realizing that this was unfeasible, we shifted our focus into the synthesis of Cl–ClnBsubNc mixtures with less and more chlorination to ultimately determine any differences in the basic properties and performance within OPV devices. Single crystals of literature–Cl–ClnBsubNc and nitrobenzene–Cl–ClnBsubNc were obtained, analyzed and were shown to have chlorines at the bay positions of the BsubNc molecular fragment with differing amounts (1.13 vs. 2.96 average chlorines per molecule, respectively). Among the three Cl–ClnBsubNcs and a commercial sample, the photo- and electro-physical change in properties were found to be translated to differing performances in planar heterojunction organic photovoltaic devices (PHJ OPVs) when applied as an electron accepting material paired with α-sexithiophene (α-6T, as an electron donor material). PHJ OPVs made from Cl–ClnBsubNc with lower amounts of chlorine (p-cymene–Cl–ClnBsubNc) was found to perform poorly when compared against PHJ OPVs made from Cl–ClnBsubNc with higher/highest amounts of chlorination (nitrobenzene–Cl–ClnBsubNc). Ultimately nitrobenzene–Cl–ClnBsubPc was found to perform moderately better than Cl–ClnBsubNc made via the literature procedure (literature–Cl–ClnBsubNc) or a commercially-available Cl–ClnBsubNc sample (commercial–Cl–ClnBsubNc). However, we feel that these rather small differences do not call into question the 8.4% efficient PHJ OPV results of Cnops et al.,31 rather they offer a cautionary note as to the actual chemical composition of the “Cl-BsubNc” used previously in their study, other studies presented in the literature, and any moving forward. Our current work is focused on obtaining Cl-BsubNc with the bay positions chemically blocked so as to avoid random bay position chlorination.Notes and references
- L. Edwards and M. Gouterman, J. Mol. Spectrosc., 1970, 33, 292–310 CrossRef CAS
.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS
- P. Peumans and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 126–128 CrossRef CAS
- A. Meller and A. Ossko, Monatsh. Chem., 1972, 103, 150–155 CrossRef CAS
- H. Kietaibl, Monatsh. Chem., 1974, 105, 405–418 CrossRef CAS
- A. S. Paton, G. E. Morse, A. J. Lough and T. P. Bender, CrystEngComm, 2011, 13, 914–919 RSC
- J. D. Virdo, Y. H. Kawar, A. J. Lough and T. P. Bender, CrystEngComm, 2013, 15, 3187–3199 RSC
- G. E. Morse, I. Gong, Y. Kawar, A. J. Lough and T. P. Bender, Cryst. Growth Des., 2014, 14, 2138–2147 CAS
- N. Kobayashi, T. Ishizaki, K. Ishii and H. Konami, J. Am. Chem. Soc., 1999, 121, 9096–9110 CrossRef CAS
- C. G. Claessens, D. Gonzalez-Rodriguez, B. del Rey, T. Torres, G. Mark, H.-P. Schuchmann, C. von Sonntag, J. G. MacDonald and R. S. Nohr, Eur. J. Org. Chem., 2003, 2547–2551, DOI:10.1002/ejoc.200300169
- D. Gonzalez-Rodriguez, T. Torres, D. M. Guldi, J. Rivera, M. A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 6301–6313 CrossRef CAS PubMed
- D. Gonzalez-Rodriguez, T. Torres, M. M. Olmstead, J. Rivera, M. Angeles Herranz, L. Echegoyen, C. Atienza Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 10680–10681 CrossRef CAS PubMed
- C. D. Zyskowski and V. O. Kennedy, J. Porphyrins Phthalocyanines, 2000, 4, 707–712 CrossRef CAS
- R. Potz, M. Goldner, H. Huckstadt, U. Cornelissen, A. Tutass and H. Homborg, Z. Anorg. Allg. Chem., 2000, 626, 588–596 CrossRef CAS
- J. D. Dang, J. D. Virdo, B. H. Lessard, E. Bultz, A. S. Paton and T. P. Bender, Macromolecules, 2012, 45, 7791–7798 CrossRef CAS
- J. Rauschnabel and M. Hanack, Tetrahedron Lett., 1995, 36, 1629–1632 CrossRef CAS
- M. Geyer, F. Plenzig, J. Rauschnabel, M. Hanack, B. Del Rey, A. Sastre and T. Torres, Synthesis, 1996, 1139–1151, DOI:10.1055/s-1996-4349
- S. Nonell, N. Rubio, B. del Rey and T. Torres, Perkin 2, 2000, 1091–1094 RSC
- L. Giribabu, C. V. Kumar, A. Surendar, V. G. Reddy, M. Chandrasekharam and P. Y. Reddy, Synth. Commun., 2007, 37, 4141–4147 CrossRef CAS
- Y. Takao, T. Masuoka, K. Yamamoto, T. Mizutani, F. Matsumoto, K. Moriwaki, K. Hida, T. Iwai, T. Ito, T. Mizuno and T. Ohno, Tetrahedron Lett., 2014, 55, 4564–4567 CrossRef CAS
- S. Shimizu, A. Miura, S. Khene, T. Nyokong and N. Kobayashi, J. Am. Chem. Soc., 2011, 133, 17322–17328 CrossRef CAS PubMed
- D. D. Diaz, H. J. Bolink, L. Cappelli, C. G. Claessens, E. Coronado and T. Torres, Tetrahedron Lett., 2007, 48, 4657–4660 CrossRef CAS
- G. E. Morse, M. G. Helander, J. F. Maka, Z.-H. Lu and T. P. Bender, ACS Appl. Mater. Interfaces, 2010, 2, 1934–1944 CAS
- M. G. Helander, G. E. Morse, J. Qiu, J. S. Castrucci, T. P. Bender and Z.-H. Lu, ACS Appl. Mater. Interfaces, 2010, 2, 3147–3152 CAS
- K. L. Mutolo, E. I. Mayo, B. P. Rand, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2006, 128, 8108–8109 CrossRef CAS PubMed
- H. Gommans, D. Cheyns, T. Aernouts, C. Girotto, J. Poortmans and P. Heremans, Adv. Funct. Mater., 2007, 17, 2653–2658 CrossRef CAS
- H. Gommans, T. Aernouts, B. Verreet, P. Heremans, A. Medina, C. G. Claessens and T. Torres, Adv. Funct. Mater., 2009, 19, 3435–3439 CrossRef CAS
- P. Sullivan, A. Duraud, I. Hancox, N. Beaumont, G. Mirri, J. H. R. Tucker, R. A. Hatton, M. Shipman and T. S. Jones, Adv. Energy Mater., 2011, 1, 352–355 CrossRef CAS
- G. E. Morse, J. L. Gantz, K. X. Steirer, N. R. Armstrong and T. P. Bender, ACS Appl. Mater. Interfaces, 2014, 6, 1515–1524 CAS
- N. Beaumont, J. S. Castrucci, P. Sullivan, G. E. Morse, A. S. Paton, Z.-H. Lu, T. P. Bender and T. S. Jones, J. Phys. Chem. C, 2014, 118, 14813–14823 CAS
- K. Cnops, M. A. Empl, P. Heremans, B. P. Rand, D. Cheyns and B. Verreet, Nat. Commun., 2014, 5, 3406 Search PubMed
- G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055–5068 CAS
- B. Verreet, S. Schols, D. Cheyns, B. P. Rand, H. Gommans, T. Aernouts, P. Heremans and J. Genoe, J. Mater. Chem., 2009, 19, 5295–5297 RSC
- B. Ma, C. H. Woo, Y. Miyamoto and J. M. J. Frechet, Chem. Mater., 2009, 21, 1413–1417 CrossRef CAS
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740–1747 CrossRef CAS PubMed
- D. Cheyns, B. P. Rand and P. Heremans, Appl. Phys. Lett., 2010, 97, 033301 CrossRef
- C. Kulshreshtha, G. W. Kim, R. Lampande, D. H. Huh, M. Chae and J. H. Kwon, J. Mater. Chem. A, 2013, 1, 4077–4082 CAS
- G. Chen, H. Sasabe, T. Sano, X.-F. Wang, Z. Hong, J. Kido and Y. Yang, Nanotechnology, 2013, 24, 484007 CrossRef PubMed
- B. Verreet, K. Cnops, D. Cheyns, P. Heremans, A. Stesmans, G. Zango, C. G. Claessens, T. Torres and B. P. Rand, Adv. Energy Mater., 2014, 4, 1301413 Search PubMed
- S. M. Menke and R. J. Holmes, ACS Appl. Mater. Interfaces, 2015, 7, 2912–2918 CAS
- T. B. Fleetham, N. Bakkan, J. P. Mudrick, J. D. Myers, V. D. Cassidy, J. Cui, J. Xue and J. Li, J. Mater. Sci., 2013, 48, 7104–7114 CrossRef CAS
- T. B. Fleetham, J. P. Mudrick, W. Cao, K. Klimes, J. Xue and J. Li, ACS Appl. Mater. Interfaces, 2014, 6, 7254–7259 CAS
- J. Endres, I. Pelczer, B. P. Rand and A. Kahn, Chem. Mater., 2016, 28, 794–801 CrossRef CAS
- J. R. Stork, R. J. Potucek, W. S. Durfee and B. C. Noll, Tetrahedron Lett., 1999, 40, 8055–8058 CrossRef CAS
- G. Martin, G. Rojo, F. Agullo-Lopez, V. R. Ferro, J. M. Garcia de la Vega, M. V. Martinez-Diaz, T. Torres, I. Ledoux and J. Zyss, J. Phys. Chem. B, 2002, 106, 13139–13145 CrossRef CAS
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS
- K. M. Kadish and M. M. Morrison, J. Am. Chem. Soc., 1976, 98, 3326–3328 CrossRef CAS PubMed
- N. G. Tsierkezos, J. Solution Chem., 2007, 36, 289–302 CrossRef CAS
- D. Ajloo, B. Yoonesi and A. Soleymanpour, Int. J. Electrochem. Sci., 2010, 5, 459–477 CAS
- N. Rubio, A. Jimenez-Banzo, T. Torres and S. Nonell, J. Photochem. Photobiol., A, 2007, 185, 214–219 CrossRef CAS
- D. S. Josey, J. S. Castrucci, J. D. Dang, B. H. Lessard and T. P. Bender, ChemPhysChem, 2015, 16, 1245–1250 CrossRef CAS PubMed
- R. F. Salzman, J. Xue, B. P. Rand, A. Alexander, M. E. Thompson and S. R. Forrest, Org. Electron., 2005, 6, 242–246 CrossRef CAS
- D. M. Pai, J. F. Yanus and M. Stolka, J. Phys. Chem., 1984, 88, 4714–4717 CrossRef CAS
- R. A. Street, D. Davies, P. P. Khlyabich, B. Burkhart and B. C. Thompson, J. Am. Chem. Soc., 2013, 135, 986–989 CrossRef CAS PubMed
- R. A. Street, P. P. Khlyabich, A. E. Rudenko and B. C. Thompson, J. Phys. Chem. C, 2014, 118, 26569–26576 CAS
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, syntheses, high-performance liquid chromatography (HPLC) chromatograms, mass spectra, UV-vis absorption spectra, fluorescence spectra, X-ray photoelectron spectroscopy (XPS), ultraviolet photoelectron spectroscopy (UPS), nuclear magnetic resonance (NMR) spectra, voltammetry, and X-ray crystallography. CCDC 1452383 and 1452384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ta02457b |
| This journal is © The Royal Society of Chemistry 2016 |