
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of hierarchical porous Co3O4 nanoboxes as efficient cathode catalysts for Li–O2 batteries†
Jian
Zhang‡
ab,
Zhiyang
Lyu‡
b,
Feng
Zhang‡
bc,
Liangjun
Wang
d,
Peng
Xiao
b,
Kaidi
Yuan
d,
Min
Lai
a and
Wei
Chen
*bde
aSchool of Physics and Optoelectronic Engineering, Nanjing University of Information Science & Technology, Nanjing 210044, Jiangsu, China
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: phycw@nus.edu.sg
cSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210046, China
dDepartment of Physics, National University of Singapore, 2 Science Drive 3, 117542 Singapore
eNational University of Singapore (Suzhou) Research Institute, 377 Lin Quan Street, Suzhou Industrial Park, Jiangsu Prov. 215123, China
First published on 11th April 2016
Abstract
Rechargeable Li–O2 batteries with remarkably high theoretical energy densities have attracted extensive attention. However, to enable Li–O2 batteries for practical applications, numerous challenges need to be overcome, e.g. high overpotential, low rate capability, and poor cycling stability. The key factor to tackle these issues is to develop highly-efficient cathode catalysts. Moreover, cathode catalysts with a porous structure and large surface area are favorable in Li–O2 batteries. In this paper, hierarchical porous Co3O4 nanoboxes with well-defined interior voids, functional shells and a large surface area have been facilely synthesized via an ion exchange reaction between Prussian blue analogue nanocubic precursors and OH− at a low temperature (60 °C). The obtained products possess hierarchical pore sizes and an extremely large surface area (272.5 m2 g−1), which provide more catalytically active sites to promote the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) as a Li–O2 battery cathode, as well as facilitating the diffusion of oxygen and the electrolyte. The hierarchical porous Co3O4 nanobox cathode shows enhanced discharge capacity, reduced overpotential, improved rate performance and cycle stability, in comparison with the EC-300J carbon cathode. The superb performance of the hierarchical porous Co3O4 nanoboxes, together with the facile fabrication approach, presents an alternative method to develop advanced cathode catalysts for Li–O2 batteries.
1. Introduction
Excessive use of fossil fuels has caused serious environmental problems, such as air pollution and global warming, etc.1 Electric vehicles hold the potential for reducing the use of fossil fuels, cutting carbon dioxide emissions, and improving air quality. Power supply is one of the key parts for realizing the wide application of electric vehicles. Therefore, many efforts have been devoted to the development of high capacity energy storage systems.2–6 Rechargeable Li–O2 batteries with remarkably high theoretical energy densities have attracted extensive attention.7,8 The theoretical energy density of Li–O2 batteries is about 3500 W h kg−1, which is about 4 to 6 times higher than that of conventional Li-ion batteries.9 However, Li–O2 batteries still suffer from many challenges for practical applications, such as electrolyte instability, poor cycle stability and high overpotential.10–12 One effective way to overcome these problems is to develop highly-efficient cathode catalysts. Many catalysts, such as noble metals and metal oxides,13–16 non-noble metal oxides,17–19 and carbon-based materials20–22 have been developed as cathode catalysts for Li–O2 batteries in order to improve battery performance.Hollow-structured catalysts with well-defined interior voids, functional shells and large surface areas are favorable in the application of Li–O2 batteries, regardless of what type of catalysts are developed. They can facilitate the effective transportation of oxygen and the electrolyte and provide sufficient catalytically active sites to promote the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), thereby leading to improved battery performance.23,24 Even mixed with carbon additives to prepare the cathodes, a well-defined hollow structure can act as a framework to build fine porous conducting matrixes, which ensures easy infiltration of the electrolyte and the diffusion of oxygen. The presence of the hollow structure can also minimize undesirable clogging of the cathode; thereby, dramatically improving the cycle stability of the Li–O2 batteries. Prussian blue analogues (PBAs) with controlled morphology have been recognized as promising precursors to synthesize porous and hollow metal oxides,25–28 such as Fe2O3, FexCo3−xO4, CuO/Cu2O. Among them, Co–Mn–O nanocubes and hierarchical porous δ-MnO2 nanoboxes have been demonstrated as efficient cathode catalysts for Li–O2 batteries.29,30
Co3O4 has been demonstrated as a potential cathode catalyst for rechargeable Li–O2 batteries due to its high catalytic ability, controllable size and morphology, and various approaches have been developed to synthesize Co3O4-based catalysts.31–34 However, most of the existing processes for the synthesis of Co3O4-based catalysts are based on hydrothermal and calcination methods which result in final products with small surface areas and non-porous structures. There has been limited success in the facile synthesis of hollow Co3O4 materials which can act as good cathode catalysts for Li–O2 batteries.
In this work, we developed a facile method to synthesize porous Co3O4 nanoboxes via an ion exchange reaction between PBA nanocubic precursors and OH− at a low temperature (60 °C). The as-synthesized Co3O4 nanoboxes possessed a hierarchical porous structure and large surface area up to 272.5 m2 g−1. When employed as the cathode catalyst for Li–O2 batteries, the porous Co3O4 nanoboxes showed high catalytic activity towards both ORR and OER with a reduced overpotential up to 300 mV, and presented enhanced capacity and improved rate performance. The synergistic effects of large specific surface area, porous structure, and high electrocatalytic activity of the porous Co3O4 nanobox electrode ensured a Li–O2 battery with excellent cycle stability (168 cycles at a limited capacity of 500 mA h g−1 and 124 cycles at 1000 mA h g−1).
2. Experimental
2.1 Synthesis procedures
2.2 Instruments for characterization
The morphology of the as-prepared samples was examined by scanning electron microscopy (SEM) on a JEOL JSM 6700F and transmission electron microscopy (TEM) on a JEOL 2010 microscope. The composition of the samples was analyzed by energy-dispersive X-ray spectroscopy (EDX) attached to the TEM instrument. The X-ray diffraction (XRD) patterns of the samples were collected from a PANalytical Empyren DY 708 diffractometer with Cu radiation (Cu Kα = 0.15406 nm). The Brunauer–Emmett–Teller (BET) surface area was measured by nitrogen sorption at 77 K on a surface area & pore size analyzer (NOVA 2200e). X-ray photoelectron spectroscopy (XPS) analyses were obtained on an XR 50 HP X-ray source and Phoibos HSA3500 analyzer.2.3 Li–O2 cell assembly and battery test
Generally, the catalyst slurry was prepared by mixing 40% catalyst with 50% Ketjenblack EC-300J (EC-300J) carbon (Cabot Carbon Ltd) and 10% polytetrafluoroethylene (PTFE), or 90% EC-300J carbon with 10% PTFE. The air cathodes were fabricated by coating the catalyst slurry on a carbon paper current collector homogenously. The mass loading of the slurry on the cathode was about 0.8–1.2 mg cm−2, and the discharge capacity was calculated based on the total weight of the catalyst and carbon additives. All the Li–O2 batteries were assembled by using coin cells in a glove box under an argon atmosphere. Glass fiber membranes were used as the separator and 1 M lithium trifluoromethanesulfonate/tetraethylene glycol dimethyl ether (LiCF3SO3/TEGDME) was used as the electrolyte. Galvanostatic discharge–charge tests of the Li–O2 battery were carried out on a LAND multi-channel battery testing system.3. Results and discussions
3.1 Synthesis, morphological, structural characterization
The fabrication of the hollow Co3O4 nanoboxes was carried out through a two-step route. Firstly, nanosized PBA precursors were synthesized through a self-assembly solution method.35,36 The morphology of the as-synthesized precursors was examined by SEM and TEM and is shown in Fig. 1a and b. These precursors presented a well-defined cubic shape with an average size of 150–250 nm and smooth facets. The XRD pattern (Fig. 1c) evidenced that the precursors were KCo[Fe(CN)6]·3H2O instead of Co3[Fe(CN)6]2·nH2O, consistent with the previous report.35 The existence of the K element was also confirmed by EDX and XPS measurements, as shown in Fig. S1a and S2a.† In the next step, the as-prepared precursors were mixed with NaOH solution to conduct the ion exchange reaction. In the NaOH solution, the ion exchange reaction happened as described below:| KCo[Fe(CN)6](s) + 2OH−(aq) → Co(OH)2(s) + Fe(CN)63− + K+. |
 | ||
| Fig. 1 SEM images (a, d and g), TEM images (b, e and h) and XRD patterns (c, f and i) of the obtained nanocube-like PBA precursors (a–c), yolk-shelled nanoboxes (d–f) and hierarchical porous Co3O4 nanoboxes (g–i). | ||
It was found that when 20 mL of 0.025 M NaOH solution was used, a yolk-shelled structural compound was obtained as shown in Fig. 1d and e. The XRD pattern (Fig. 1f) indicated that the compound was a mix of ingredients comprising Co3O4 and KCo[Fe(CN)6]·3H2O. On increasing the amount of 0.025 M NaOH solution to 40 mL, perfect hollow nanoboxes were obtained, as shown in Fig. 1g and h. The as-prepared nanoboxes analyzed by XRD were indexed to Co3O4 (PDF #74-2120) instead of Co(OH)2 (Fig. 1i). This was possibly due to the fact that Co(OH)2 can be easily oxidized by the dissolved oxygen during the reaction process.37,38 Therefore, it can be proposed that the ion exchange reaction firstly took place at the interface between the solid PBA nanocubic precursors and NaOH solution when the concentration of NaOH was 0.025 M, and a thin layer of Co3O4 was formed. With the reaction proceeded, the solid KCo[Fe(CN)6]·3H2O continued dissolve and diffuse outward to react with OH− to form Co(OH)2. Subsequently, it was oxidized by oxygen to form Co3O4.39 If the amount of OH− was adequate, the ion exchange reaction proceeded until all the PBAs were consumed and well-defined nanoboxes were finally formed (Fig. 1g and h). Otherwise, an incomplete ion exchange reaction took place and a yolk-shelled structural compound was obtained (Fig. 1d and e). The ion exchange reaction was also evidenced by EDX and XPS analysis. As shown in Fig. S1 and S2,† the characteristic peaks of K and Fe in the EDX spectrum and dominant K 2p, Fe 2p and N 1s peaks in the XPS spectra disappeared after adding 40 mL of 0.025 M NaOH, indicating the removal of K+ and [Fe(CN)6]3−. The XPS measurements (Fig. S3†) further confirmed the formation of Co3O4. The two peaks at 794.7 and 779.5 eV correspond to the Co 2p3/2 and Co 2p1/2 of Co3O4, which were in good agreement with previous reports.40,41
The HR-TEM of the as-prepared hollow Co3O4 nanoboxes is shown in Fig. 2. The surface of the nanoboxes is dominated by the assembly of nanosheets and nanoneedles (Fig. 2b and c). A clear lattice spacing of 0.202 nm was also observed in the high-resolution TEM image (Fig. 2d), in good agreement with the inter-plane spacing of the (400) plane of Co3O4 (PDF #74-2120). The SAED pattern (Fig. 2a inset) showed that the Co3O4 was polycrystalline. The specific surface area and porosity of the Co3O4 nanoboxes were determined by N2 adsorption–desorption measurements. A type IV nitrogen adsorption–desorption isotherm with a H3-shaped hysteresis loop is shown in Fig. 2e. The BET specific surface area of the porous Co3O4 nanoboxes was up to 272.5 m2 g−1. To our knowledge, this is one of the largest surface areas reported among Co3O4 materials.34,37,42 The pore size distribution calculated from the Barrett–Joyner–Halenda (BJH) method is shown in the inset of Fig. 2e. A wide range of pore sizes from 20 to 140 nm was observed. The sharp peak centred at about 60 nm was related to the hollow structure of the nanoboxes, and the mesopores existing in the porous Co3O4 nanoboxes resulted from the accumulated pores of inter-nanoboxes and inter-nanosheets on the surface of the nanoboxes. The pore volume of the porous Co3O4 nanoboxes was about 0.63 cm3 g−1.
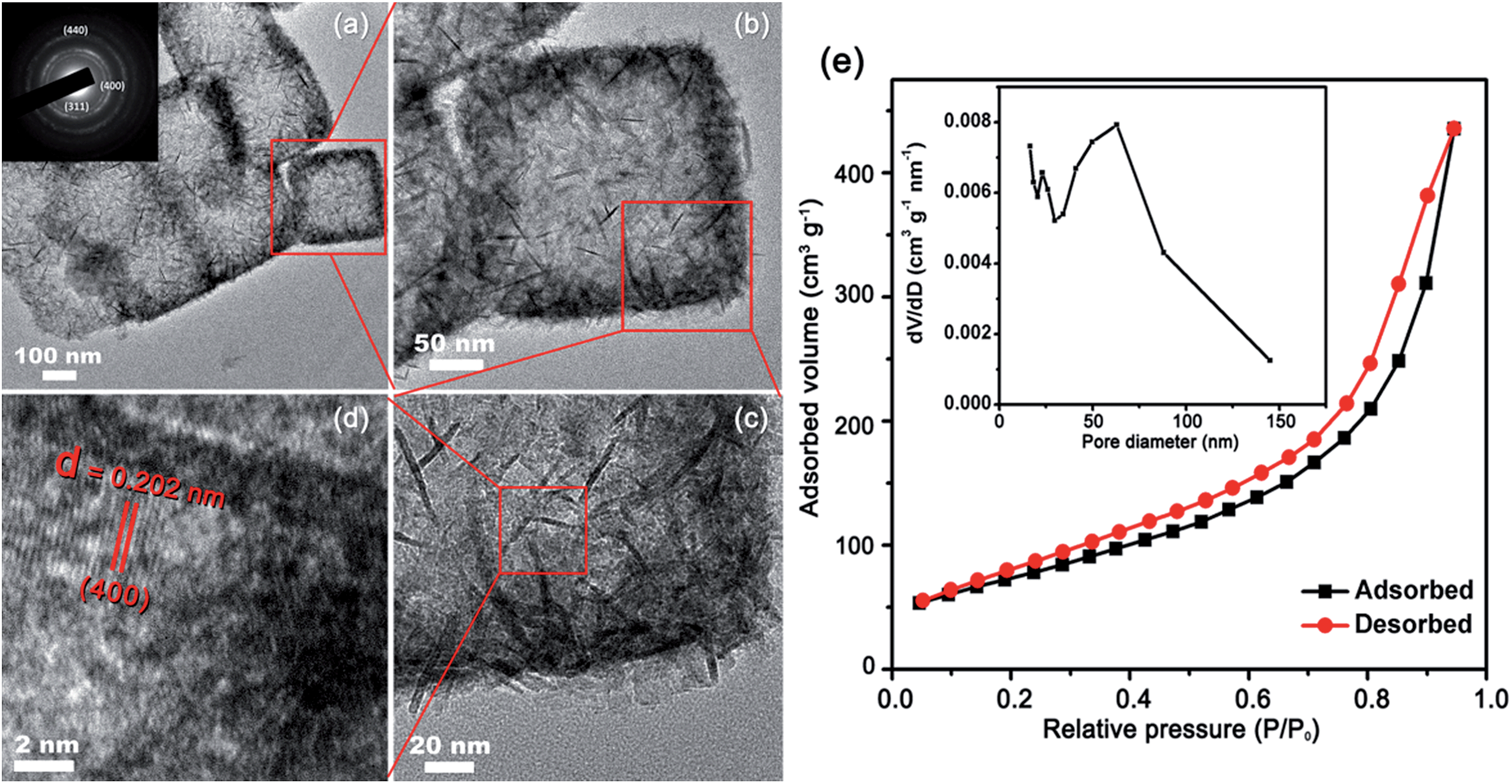 | ||
| Fig. 2 HR-TEM images (a–d) of hierarchical porous Co3O4 nanoboxes, inset in (a) is SAED pattern; (e) nitrogen adsorption–desorption isotherms and pore size distribution (inset) of the hierarchical porous Co3O4 nanoboxes. | ||
We also investigated the correlation of the morphology of Co3O4 with the reaction conditions such as reaction time, reaction temperature and the NaOH concentration, as shown in Fig. 3. It can be seen in Fig. 3a that similar Co3O4 nanoboxes were obtained when the reaction time was extended to 120 min, which indicated that no further reaction took place after the completion of the ion exchange reaction. Fig. 3b and c show the products obtained at room temperature and 100 °C, respectively. When mixing the PBA nanocubic precursors with NaOH at room temperature, no obvious change in the PBA nanocubic precursors was observed (Fig. 3b), indicating that no ion exchange reaction occurred at room temperature. However, when the temperature was increased to 100 °C, Co3O4 nanoboxes formed via the assembly of small nanoparticles were observed, as shown in Fig. 3c. These results suggested that temperature was one of the dominant factors for the ion exchange reaction. At room temperature, the thermal energy was not sufficient to drive the ion exchange reaction; while a high temperature (100 °C) can accelerate the reaction and lead to the formation of localized nanoparticles. The concentration of NaOH also had a crucial effect on the ion exchange reaction. Both the PBAs and Co(OH)2 were insoluble in water. The proceeding of the ion exchange reaction was attributed to the high concentration of NaOH, which promoted the forward reaction to form the Co(OH)2. As shown in Fig. 3d, if the concentration was relatively low (0.01 M), negligible change in the PBA precursors was observed. When the concentration of NaOH was increased to 0.05 M, a hollow structure was maintained. However, the surface of the nanoboxes was partially damaged (Fig. 3e), which may be due to the rapid ion exchange reaction rate. At the NaOH concentration of 0.1 M, the inward diffusion of OH− and the rate of ion exchange reaction were significantly accelerated, thereby completely destroying the nanobox structure and causing the formation of scattered nanoparticles (Fig. 3f). These results indicated that the key to obtaining the hollow Co3O4 nanoboxes was the precise control of the reaction temperature and the concentration of the alkaline solution.
 | ||
| Fig. 3 TEM images of the final products under various experimental conditions: (a) react for 120 min; (b) at room temperature; (c) at 100 °C; (d–f) with 40 mL 0.01 M, 0.05 M and 0.1 M NaOH, respectively. | ||
3.2 Battery testing
The battery performance of the porous Co3O4 nanoboxes catalyst was evaluated by galvanostatic discharge–charge measurements in coin-cell type Li–O2 batteries. EC-300J carbon was used as both the conductive additive and the reference cathode for comparison. Fig. 4a shows the first discharge–charge profiles of the Li–O2 cells with porous Co3O4 nanoboxes and EC-300J carbon electrodes at the current density of 0.08 mA cm−2. In the first discharge process, the porous Co3O4 nanobox electrode delivered a specific capacity of about 4032 mA h g−1, which was much larger than that of the EC-300J carbon electrode (2535 mA h g−1). The enhanced capacity was attributed to the large surface area of the porous Co3O4 nanobox catalyst, which provided sufficient space to afford the deposition of Li2O2. In addition, the relatively higher catalytic activity of the porous Co3O4 nanobox catalyst also promoted the ORR. More importantly, the high catalytic activity of the porous Co3O4 nanoboxes gave rise to an increased discharge potential and reduced charge potential of the Li–O2 battery. As can be seen in Fig. 4a, the voltage gap of the battery with the porous Co3O4 nanoboxes was about 1.39 V, which was about 300 mV lower than that of the EC-300J electrode. The reduced overpotential meant enhanced round-trip efficiency (the ratio of discharge to charge voltage). As shown in Fig. 4b and S4a,† the rate performance of the porous Co3O4 nanoboxes was further investigated at the current densities of 0.04, 0.16 and 0.24 mA cm−2, respectively. The batteries with porous Co3O4 nanobox electrodes exhibited capacities of 4876 mA h g−1 at 0.04 mA cm−2, 3250 mA h g−1 at 0.16 mA cm−2 and 2075 mA h g−1 at 0.24 mA cm−2. It should be noted that these capacities were much higher than those of the EC-300J carbon electrodes at the same current density (Fig. 4b and S4b†). The porous Co3O4 nanobox electrode also exhibited better capacity retention than the EC-300J carbon electrode under all investigated current densities (Fig. S4c†). These results further indicated that the porous Co3O4 nanobox electrode had better rate performance. The improved rate performance of the porous Co3O4 nanobox electrode was attributed to the porous structure facilitating the diffusion of oxygen and electrolyte during the discharge process. In addition, the high catalytic activity of the catalysts, which can promote the sluggish ORR even at high current densities, was also beneficial to improve the battery performance. To investigate the stability of the porous Co3O4 nanobox electrode, the long-term performance of the porous Co3O4 nanobox electrode was evaluated following a widely used method by limiting the discharge capacity.23,43 When the discharge potential dropped below 2.0 V, we considered the cells to have failed. Fig. 4c and d show the selected cycled voltage profiles of the batteries with porous Co3O4 nanobox electrodes at the current density of 0.16 mA cm−2 with limited capacities of 500 mA h g−1 and 1000 mA h g−1, respectively. As can be seen in Fig. 4c, there was no loss of discharge capacity until the 168th cycle with a limited capacity of 500 mA h g−1. When increasing the curtailing capacity to 1000 mA h g−1, the battery with the porous Co3O4 nanobox electrode could still be cycled for 125 cycles (Fig. 4d). In contrast, as shown in Fig. S5,† the cycle numbers of the EC-300J electrodes decreased to 20 and 11 with the limited capacities of 500 mA h g−1 and 1000 mA h g−1, respectively, which were much smaller than those of the porous Co3O4 nanobox electrode. These results reveal that the porous Co3O4 nanobox electrode had excellent cycle stability. | ||
| Fig. 4 (a) First discharge–charge curves of Li–O2 batteries with the hierarchical porous Co3O4 nanobox and EC-300J carbon electrodes at 0.08 mA cm−2; (b) discharge capacity of Li–O2 battery cells with different electrodes at various current densities; (c) and (d) cyclic performance of the hierarchical porous Co3O4 nanobox electrode at 0.16 mA cm−2 with limited capacities of (c) 500 mA h g−1 and (d) 1000 mA h g−1, respectively. | ||
4. Conclusion
In conclusion, we have developed a facile and controllable method to synthesize porous Co3O4 nanoboxes at a low temperature (60 °C) by using Prussian blue analogues as the precursor. The correlation between the morphology and the experimental conditions was also investigated in detail. The obtained products possessed hierarchical pores and an extremely large surface area (272.5 m2 g−1), which favour oxygen transportation and provide more catalytically active sites to promote ORR and OER. By using the porous Co3O4 nanoboxes as the cathode catalyst, the Li–O2 batteries showed improved battery performance with higher discharge capacity, lower overpotential, enhanced rate performance and superior cycle stability. The facile fabrication method reported here also represented an alternative to develop metal oxide nanomaterials for potential applications in Li–O2 batteries, as well as other applications such as supercapacitors and Li-ion batteries.Acknowledgements
The authors acknowledge the financial support from the Singapore MOE grant R143-000-593-112 and China Scholarship Council.Notes and references
- I. Dincer, Renewable Sustainable Energy Rev., 2000, 4, 157–175 CrossRef.
- H. Zhang, S. Jing, Y. Hu, H. Jiang and C. Li, J. Power Sources, 2016, 307, 214–219 CrossRef CAS.
- Y. Dai, L. Chen, V. Babayan, Q. Cheng, P. Saha, H. Jiang and C. Li, J. Mater. Chem. A, 2015, 3, 21337–21342 CAS.
- H. Jiang, H. Zhang, Y. Fu, S. Guo, Y. Hu, L. Zhang, Y. Liu, H. Liu and C. Li, ACS Nano, 2016, 10, 1648–1654 CrossRef CAS PubMed.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- R. Black, S. H. Oh, J.-H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef CAS PubMed.
- Y. Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J. G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- F. Li, D.-M. Tang, Y. Chen, D. Golberg, H. Kitaura, T. Zhang, A. Yamada and H. Zhou, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed.
- E. Yilmaz, C. Yogi, K. Yamanaka, T. Ohta and H. R. Byon, Nano Lett., 2013, 13(10), 4679–4684 CrossRef CAS PubMed.
- P. Tan, W. Shyy, T. S. Zhao, X. B. Zhu and Z. H. Wei, J. Mater. Chem. A, 2015, 3, 19042–19049 CAS.
- W. Zhou, Y. Cheng, X. Yang, B. Wu, H. Nie, H. Zhang and H. Zhang, J. Mater. Chem. A, 2015, 3, 14556–14561 CAS.
- B. Wu, H. Zhang, W. Zhou, M. Wang, X. Li and H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 23182–23189 CAS.
- J. Zhang, Y. Zhao, X. Zhao, Z. Liu and W. Chen, Sci. Rep., 2014, 4, 6005–6010 CAS.
- M. Hong, H. C. Choi and H. R. Byon, Chem. Mater., 2015, 27, 2234–2241 CrossRef CAS.
- Y. Cao, M.-s. Zheng, S. Cai, X. Lin, C. Yang, W. Hu and Q.-f. Dong, J. Mater. Chem. A, 2014, 2, 18736–18741 CAS.
- X. Lin, L. Zhou, T. Huang and A. Yu, J. Mater. Chem. A, 2013, 1, 1239–1245 CAS.
- S. H. Liu, Z. Y. Wang, C. Yu, Z. B. Zhao, X. M. Fan, Z. Ling and J. S. Qiu, J. Mater. Chem. A, 2013, 1, 12033–12037 CAS.
- J.-J. Xu, Z.-L. Wang, D. Xu, F.-Z. Meng and X.-B. Zhang, Energy Environ. Sci., 2014, 7, 2213–2219 CAS.
- F. Li, D.-M. Tang, T. Zhang, K. Liao, P. He, D. Golberg, A. Yamada and H. Zhou, Adv. Energy Mater., 2015, 5, 1500294 Search PubMed.
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- L. Hu and Q. Chen, Nanoscale, 2014, 6, 1236–1257 RSC.
- L. Hu, P. Zhang, H. Zhong, X. Zheng, N. Yan and Q. Chen, Chem.–Eur. J., 2012, 18, 15049–15056 CrossRef CAS PubMed.
- X.-Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331–5335 CrossRef CAS PubMed.
- J. Zhang, Y. Luan, Z. Lyu, L. Wang, L. Xu, K. Yuan, F. Pan, M. Lai, Z. Liu and W. Chen, Nanoscale, 2015, 7, 14881–14888 RSC.
- J. Zhang, L. Wang, L. Xu, X. Ge, X. Zhao, M. Lai, Z. Liu and W. Chen, Nanoscale, 2015, 7, 720–726 RSC.
- Y. Cui, Z. Wen and Y. Liu, Energy Environ. Sci., 2011, 4, 4727–4734 CAS.
- J. Zeng, C. Francia, J. Amici, S. Bodoardo and N. Penazzi, J. Power Sources, 2014, 272, 1003–1009 CrossRef CAS.
- J. Zhu, X. Ren, J. Liu, W. Zhang and Z. Wen, ACS Catal., 2015, 5, 73–81 CrossRef CAS.
- F. Wang, Z. Wen, C. Shen, K. Rui, X. Wu and C. Chen, J. Mater. Chem. A, 2015, 3, 7600–7606 CAS.
- M. Hu, S. Ishihara, K. Ariga, M. Imura and Y. Yamauchi, Chem.–Eur. J., 2013, 19, 1882–1885 CrossRef CAS PubMed.
- M. Hu, N. L. Torad and Y. Yamauchi, Eur. J. Inorg. Chem., 2012, 2012, 4795–4799 CrossRef CAS.
- Y. Li, B. Tan and Y. Wu, J. Am. Chem. Soc., 2006, 128, 14258–14259 CrossRef CAS PubMed.
- Y. Li and Y. Wu, Chem. Mater., 2010, 22, 5537–5542 CrossRef CAS.
- L. Zhang, H. B. Wu and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- Y. Tan, Q. Gao, C. Yang, K. Yang, W. Tian and L. Zhu, Sci. Rep., 2015, 5, 12382 CrossRef CAS PubMed.
- S. Zhang, J.-j. Shan, Y. Zhu, A. I. Frenkel, A. Patlolla, W. Huang, S. J. Yoon, L. Wang, H. Yoshida, S. Takeda and F. Tao, J. Am. Chem. Soc., 2013, 135, 8283–8293 CrossRef CAS PubMed.
- W.-H. Ryu, T.-H. Yoon, S. H. Song, S. Jeon, Y.-J. Park and I.-D. Kim, Nano Lett., 2013, 13, 4190–4197 CrossRef CAS PubMed.
- Y. Qin, J. Lu, P. Du, Z. Chen, Y. Ren, T. Wu, J. T. Miller, J. Wen, D. J. Miller, Z. Zhang and K. Amine, Energy Environ. Sci., 2013, 6, 519 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta01995a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |