
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali-stable and highly anion conducting poly(phenylene oxide)s carrying quaternary piperidinium cations†
Hai-Son
Dang
and
Patric
Jannasch
*
Department of Chemistry, Polymer and Materials Chemistry, Lund University, P.O. Box 124, Lund 221 00, Sweden. E-mail: patric.jannasch@chem.lu.se; Fax: +46-46-222-4012
First published on 30th June 2016
Abstract
New durable and hydroxide ion conducting anion-exchange membranes (AEMs) are currently required in order to develop alkaline fuel cells into efficient and clean energy conversion devices. In the present work we have attached quaternary piperidinium (QPi) cations to poly(2,6-dimethyl-1,4-phenylene oxide)s (PPOs) via flexible alkyl spacer chains with the aim to prepare AEMs. The bromine atoms of bromoalkylated PPOs were displaced in Menshutkin reactions to attach one or two QPi groups, respectively, via heptyl spacers. The cationic polymers have excellent solubility in, e.g., methanol, dimethylsulfoxide and N-methyl-2-pyrrolidone at room temperature, and form tough and transparent membranes. AEMs with bis-QPi side chains efficiently form ionic clusters and reach very high hydroxide ion conductivities, up to 69 and 186 mS cm−1 at 20 and 80 °C, respectively. The AEMs further have excellent alkaline stability, and 1H NMR analysis showed no degradation of the AEMs after storage in 1 M NaOH at 90 °C during 8 days. Thermogravimetry indicated decomposition only above 225 °C. The AEM properties were further tuned by controlled formation of bis-QPi crosslinks through an efficient reaction between bromoalkylated PPO and 4,4′-trimethylenebis(1-methylpiperidine) during a reactive membrane casting process. In conclusion, alkali-stable and highly conductive AEMs can be prepared by placing cycloaliphatic quaternary ammonium cations on flexible side chains and crosslinks.
Introduction
During the last decade fuel cells have attracted a significant interest as one of the most promising class of technologies for environmentally benign power generation.1 For example, proton-exchange membrane fuel cells (PEMFCs) have been extensively developed over the last years and are now on the verge of broad commercialization for automotive applications.2 However, the absolute dependence on noble metal electrocatalysts and the lack of fuel flexibility are two significant remaining limitations. These shortcomings have resulted in an increasing interest in alkaline membrane fuel cells (AMFCs).3 Alkaline conditions allow faster oxygen reduction kinetics, more efficient water management, a much wider choice of fuels, and the possibility to make use of non-precious metal catalysts based on, e.g., Ni and Ag.4,5 Conversely, the hydrogen oxidation reaction becomes slower at high pH.The research for new improved AEM materials is currently intense and the area has progressed significantly during the last 5 years. However, there is still a lack of durable AEMs that combine high OH− conductivity with sufficient stability under alkaline conditions.4–12 Because of its high basicity and nucleophilicity, OH− tends to attack and degrade archetypal anion-exchange groups, as well as polymer backbones which are activated for basic (nucleophilic) hydrolysis.4,7,8 A wide range of different high-performance aromatic polymers, including polysulfones, polyethers, polyphenylenes, and polystyrenes have been modified with cationic groups and evaluated as AEMs.4,7,8,11 Among these, poly(2,6-dimethyl-1,4-phenylene oxide) (PPO) has excellent chemical, thermal and mechanical properties.13 Moreover, PPO has very good solubility in organic solvents, excellent film-formability upon casting, and is reported to possess a high stability under alkaline conditions.14 A further advantage is that functional groups can be introduced in both aromatic and benzylic positions using a wide range of chemical modification techniques.15 This opens up for many synthetic possibilities to target new high-performance PPO-based AEMs.
By employing standard procedures to introduce the cationic groups, e.g., chloromethylation and subsequent reaction with trimethylamine, benzyl trimethylammonium (BTA) groups are usually formed directly on aromatic polymer backbones. Unfortunately, quaternary ammonium (QA) groups in benzylic positions have in most cases proven to be quite sensitive towards nucleophilic attack by OH−, and also seem to activate the cleavage of adjacent ether links in aromatic polymer backbones.16–18 QA groups with long alkyl chain segments are generally sensitive to Hofmann β-eliminations. Recently however, β-protons have been found to be far less sensitive to nucleophilic attack than previously suggested.4,19 Many studies have now shown that the alkaline stability of AEMs is significantly improved by inserting flexible alkyl spacer chains in-between the polymer backbone and the QA groups, thereby avoiding any benzylic positions.20–35 In addition, the presence of flexible spacers seems to increase the local mobility of the cations which in turn facilitates clustering of the ions in the AEM. This is likely to favor the formation of a well hydrated and percolating OH− conducting phase domain.27,28 Hibbs has for example synthesised polyphenylenes tethered with trimethylammonium ions via hexyl spacers and reported a significant increase in the alkaline stability compared to corresponding polymers with BTA groups.25 We have previously reported on PPOs with pendant trimethylammonium ions attached via heptyl spacers and found efficient ionic clustering, enhanced OH− conductivity, and significantly improved alkaline stability in relation to PPOs with BTA groups.27,28 Lee and co-workers prepared fluorene-based polymers with trimethylammonium ions attached via hexyl spacer chains and reported high OH− conductivity and excellent alkaline stability.34 The high alkaline stability of alkyl trimethylammonium cations has been rationalized by Pivovar et al. who via density functional theory calculations showed that Hofmann elimination is the most harmful degradation route for the alkyl trimethylammonium cations.36 However, because of steric effects, the energy barrier against elimination was found to increase drastically when the number of carbon atoms in the alkyl chain is increased to four.
A large number of different cationic groups, such as various QA, imidazolium, guanidinium and phosphonium groups, have been attached to different polymer backbones for the development of AEMs.4,7,8 Somewhat surprisingly, cycloaliphatic QAs have been given very little attention in the development of improved AEMs. The few exceptions are piperazinium,35 quaternized 1,4-diazabicyclo[2.2.2]octane (DABCO),37–39 and morpholinium.40 However, these QAs all contain a heteroatom close the cationic centre which is likely to decrease the stability.19 Recently, Marino and Kreuer studied the stability of a large number of organic cationic model compounds in NaOH concentrations up to 10 M and temperatures up to 160 °C.19 The degradation was monitored by NMR analysis to follow degradation pathways and to determine the half-times of the compounds. They reported that, with the exception of aromatic cations, most cationic compounds exhibited an unexpected high stability. Cycloaliphatic QAs were identified as especially alkali-stable, most probably because they contain β-protons with the C–C bond rotationally restricted by the ring geometry.19 At 160 °C in 6 M NaOH, the half-times of N,N-dimethylpiperidinium and N,N-dimethylpyrrolidinium were 87 and 37 h, respectively. Thus, the former 6-membered cyclic QA was found to be significantly more stable than the latter 5-membered one. Nucleophilic attack and substitution at one of the methyl groups dominated the degradation of the piperidinium cation, while the pyrrolidinium cation in addition degraded through ring-opening substitution.19 The half-times of these fully aliphatic mono-cyclic QA compounds can be compared with those of benzyl trimethylammonium and N-benzyl-N-methylpiperidinium at mere 4.2 and 7.3 h, respectively, under the same conditions.19 An N-spirocyclic QA compound based on piperidine (6-azonia-spiro[5.5]undecane) displayed an even higher half-time, 110 h. However, the incorporation of N-spirocyclic QA groups into polymer structures and AEMs is rather complicated.41 In contrast, modifications with aliphatic mono-cyclic QA groups are quite straight-forward and less costly.
With the aim to efficiently combine high OH− conductivity and high alkaline stability, we have in the present work tethered quaternary piperidinium (QPi) groups to PPO via flexible heptyl spacers and evaluated their properties as AEMs. Starting from PPOs functionalized with different concentrations of 7-bromoheptyl side chains, either one or two QPi groups were attached via Menshutkin reactions using 1-methylpiperidine and 4,4′-trimethylenebis(1-methylpiperidine) (BMP), respectively, to displace the bromine atoms. In the latter case, the terminal piperidine rings were subsequently quaternized using iodomethane. The trimethylene chain in-between the QPi rings ensures that the cationic charges are sufficiently separated not to limit the ionic dissociation and charge carrier formation by counter ion condensation.42–44 We hypothesized that by placing two QPi groups on each spacer unit, the ionic clustering would increase and the membrane properties improve. In addition, crosslinked AEMs were prepared by reacting bromoalkylated PPO with BMP during a reactive membrane casting process, followed by complete quaternization of the remaining piperidine rings. AEMs with different QPi configurations, ionic concentrations and degrees of crosslinking were prepared by solvent casting and carefully characterized with respect to ion-exchange capacity (IEC), crosslink density, ionic segregation, thermal and alkaline stability, water uptake and anionic conductivity.
Experimental section
Materials
Tetrahydrofuran (THF, HPLC grade, Honeywell) was dried over molecular sieves (4 Å, 8–12 mesh, Acros) before use. PPO (Sigma-Aldrich, Mn = 20 kg mol−1, MwMn−1 = 2.3), n-butyllithium (n-BuLi, 2.5 M, solution in hexanes, Acros), 1,6-dibromohexane (98%, Acros), N-methyl-2-pyrrolidone (NMP, reagent grade, Acros), 1-methylpiperidine (99%, Sigma), BMP (98%, Sigma), iodomethane (99%, Sigma-Aldrich), diethyl ether (99+%, Sigma-Aldrich), methanol (HPLC grade, Honeywell) and 2-propanol (HPLC grade, Honeywell) were used as received. Sodium nitrate (99%, Sigma) and sodium bromide (99.5%, Sigma-Aldrich) were dried under vacuum before use, and silver nitrate (99.995%, Sigma-Aldrich) was dried under vacuum in the dark before use.Polymer synthesis
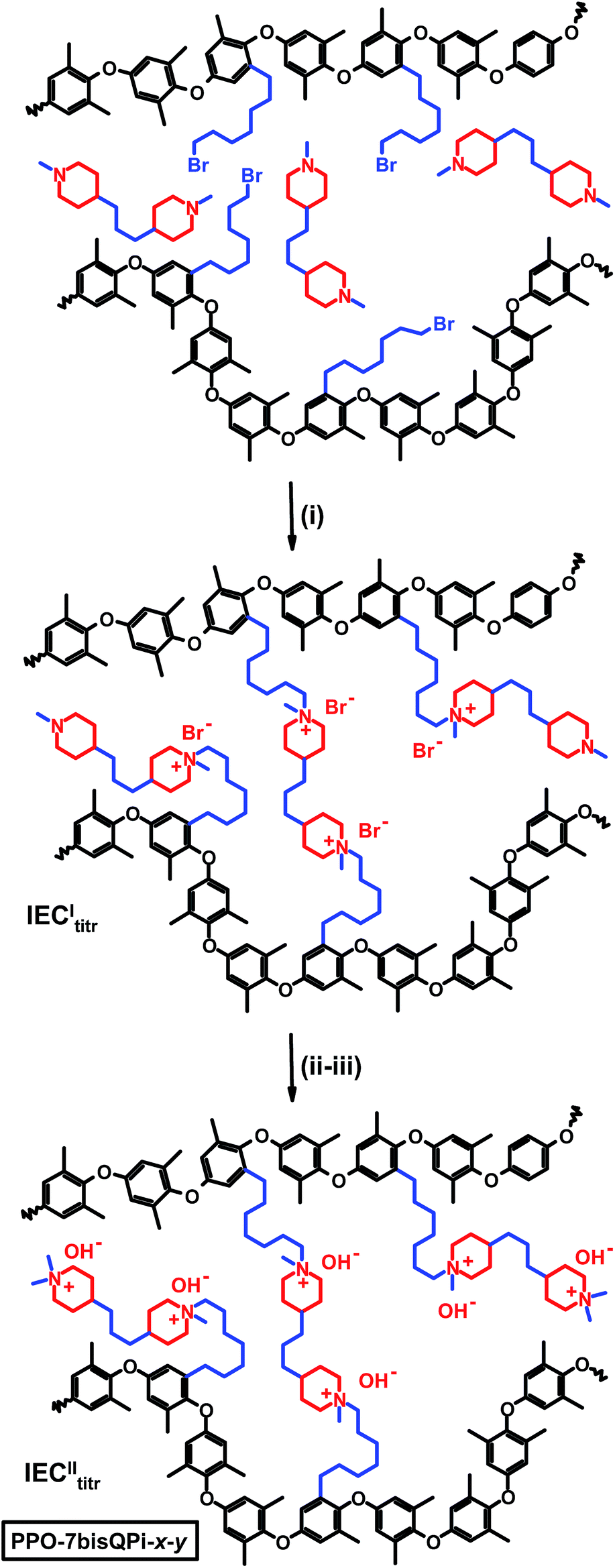
Five PPOs were functionalized with QPi groups via heptyl spacer chains, i.e., one sample tethered with single QPi groups and four samples with dual QPi groups (Scheme 1). The former sample was prepared by reaction of bromoalkylated PPO with 1-methylpiperidine, while the PPOs with bis-QPi side chains were prepared by first reacting the bromoalkylated PPOs with BMP to form the first QPi group, followed by quaternization of the remaining tertiary piperidine ring using iodomethane.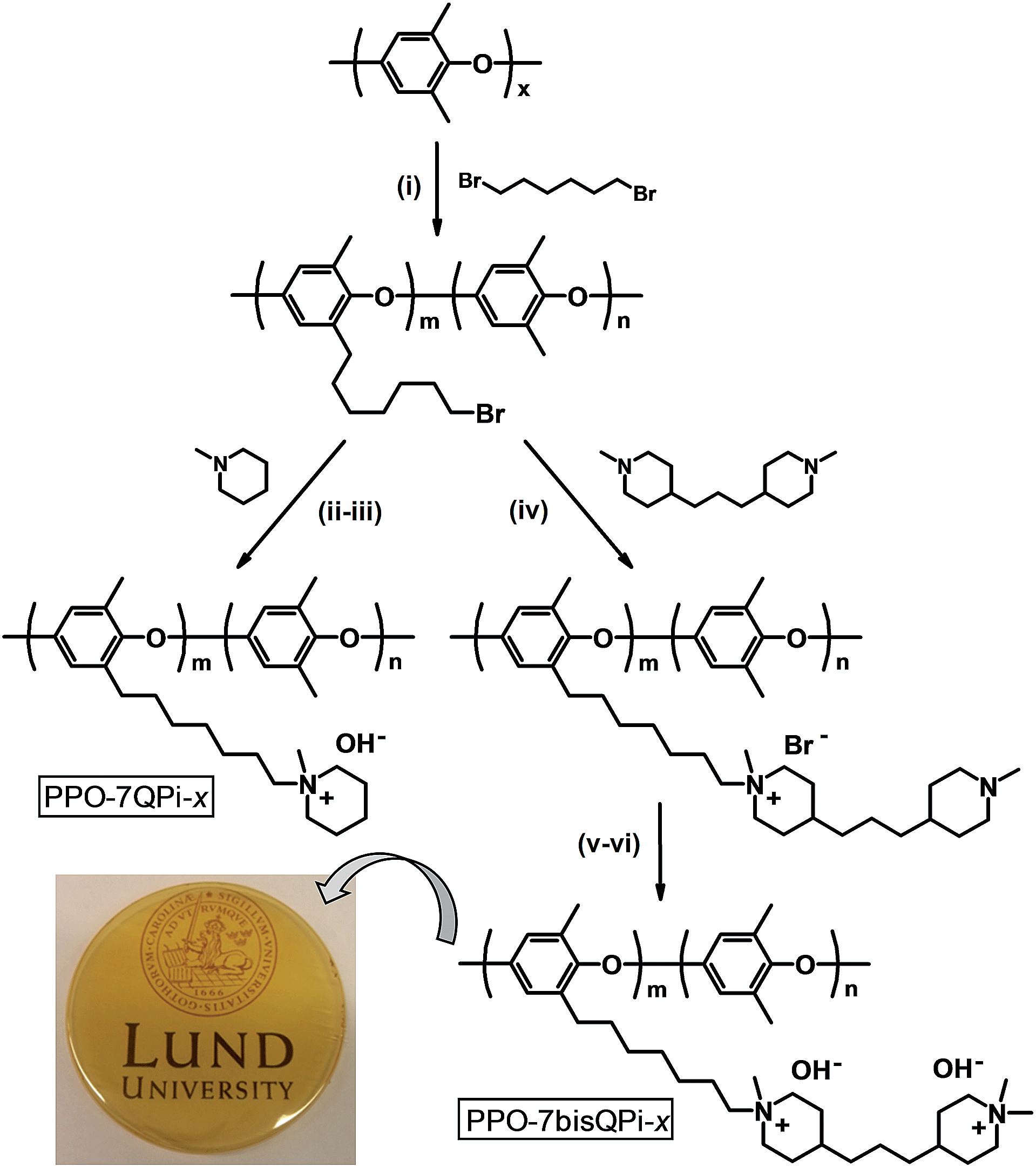 | ||
| Scheme 1 Synthesis of PPO functionalized with mono- and bis-QPi groups via alkyl spacers. Photograph shows membrane PPO-7bisQPi-1.7 cast from NMP at 80 °C [key: (i) n-BuLi, THF, −70 °C, (ii) NMP, 85 °C, (iii) aqueous NaOH, 20 °C, (iv) NMP, 85 °C, (v) MeI, NMP, 40 °C, (vi) aqueous NaOH, 20 °C]. | ||
The bromoalkylated samples were prepared by lithiation of PPO and reaction with an excess of 1,6-dibromohexane, similar to the procedure we reported previously.28 The samples were designated as PPO-7Br-DB where DB is the degree of bromoalkylation, i.e., the percentage of bromoalkylated repeating units of the PPO. The value of DB was kept at 11, 15, 17, 19 and 30 mol%, respectively, to target final ion exchange capacity (IEC) values between 1.4 and 2.0 mequiv. g−1 in the OH− form. Here, the preparation of the sample PPO-7Br-19 will be described as an example. A 4-neck 500 mL round bottomed flask containing an oval magnetic stirring bar and fitted with thermometer, a rubber septum and an argon inlet-outlet was charged with PPO (3 g, 24.97 mmol repeat units) and dry THF (300 mL). The mixture was degassed by 3 vacuum-argon replacements, and stirred at 60 °C until homogeneous. The solution was allowed to cool to room temperature and was then carefully degassed by 7 vacuum-argon replacements. Remaining impurities (including traces of water) were first titrated by a few droplets of n-BuLi via syringe until a slightly yellow color appeared to indicate the formation of lithiated PPO. Next, 10 mL n-BuLi solution (25 mmol) was added drop-wise and the resulting solution was kept for 3 h with stirring. The orange solution was then brought to −70 °C using a dry ice/2-propanol bath. Next, an 200 mol% excess of 1,6-dibromohexane was quickly and all at once added and the color of the solution immediately turned colorless, indicating the fast reaction of the lithiated PPO with the electrophile. The reaction was left overnight at room temperature under stirring. The resulting clear colorless solution was then added drop-wise into methanol to precipitate the product as a white fine powder. The product was filtered off, washed repeatedly (at least 3 times) with fresh methanol and dried at 50 °C until constant weight was obtained after about 48 h.
The samples functionalized with mono- and bis-QPi groups were designated as PPO-7QPi-x and PPO-7bisQPi-x, respectively, where x is the IEC value in the OH− form as determined via Mohr titrations. Samples in the Br− form were given the suffix -Br. The PPO-7QPi-1.7 sample was synthesized from PPO-7Br-30 via a Menshutkin reaction with 1-methylpiperidine. A 300% excess of the cyclic amine was added to an NMP solution containing 5 wt% of the brominated PPO. The reaction solution was kept in a sealed vessel at 85 °C during 7 days under stirring to achieve full conversion. Next, the orange reaction solution was added drop-wise to diethyl ether under vigorous stirring. The precipitate was collected on a glass filter and washed several times with fresh diethyl ether. After drying under vacuum for 2 days at room temperature, a slightly yellow powder of the product in the Br− form was obtained.
In the synthesis of the PPO-7bisQPi-x samples, a volume of 14 mL of an NMP solution containing 5 wt% bromoalkylated PPO was added drop-wise to 2.4 mL (corresponding to ∼1000 mol% excess to avoid crosslinking reactions) of vigorously stirred BMP to perform the first Menshutkin reaction. The homogeneous and slightly yellow solution was kept in a sealed vessel at 85 °C for 7 days under vigorous stirring to achieve full conversion. Next, the solution was added drop-wise to diethyl ether to precipitate the intermediate semi-quaternized product as a white powder. After filtration, the powder was washed thrice with diethyl ether and dried at room temperature under vacuum. In the second Menshutkin reaction, a 200 mol% excess of iodomethane was added to a solution containing 5 wt% of the polymer intermediate in NMP. The solution was covered with aluminum foil and kept at 40 °C during 2 days under stirring. The clear red-orange solution was slowly added drop-wise to diethyl ether to precipitate the product as a slightly yellow powder. The PPO-7bisQPi-x sample, at this stage containing both Br− and I− counter ions, was then filtered, extensively washed with diethyl ether, and dried before immersion in 1 M aqueous NaBr during 48 h under stirring. After thorough washing in de-ionized water, the product in the Br− form was collected by filtration and dried under vacuum at room temperature during at least 2 days before analysis and further use.
Structural characterization
1H-NMR spectra of the polymer samples in CDCl3 or DMSO-d6 were acquired using a Bruker DR X 400 NMR spectrometer at 400.13 MHz. The molecular weight and polydispersity (PDI, MwMn−1) of the neat PPO and all the bromoalkylated samples were determined by size-exclusion chromatography (SEC) using a setup equipped with three Shodex gel columns (KF-805, -804 and -802.5) in series and a refractive index detector. Four polystyrene standards (Mn = 650 kg mol−1 from Water Associates, Mn = 96 and 30 kg mol−1 from Polymer Laboratories, and Mn = 3.18 kg mol−1 from Agilent Technologies) were used for calibration. Dry samples were first dissolved in chloroform and then passed through a Teflon filter (0.45 μm) to remove any insoluble impurities. Next, SEC measurements were performed with chloroform as the mobile phase at a constant elution rate of 1.0 mL min−1.Casting of non-crosslinked AEMs
Non-crosslinked AEMs were prepared by dissolving dry PPO-7QPi-x and PPO-7bisQPi-x samples in the Br− form (0.15 g) in 3 g NMP under stirring at room temperature. Each homogeneous solution was then poured onto a Petri dish with a 5 cm diameter, and the AEM was formed by allowing the solvent to evaporate in a well-ventilated casting oven at 80 °C during 48 h. The resulting film was placed in water, gently peeled off the Petri dish and washed several times before storage in de-ionized water. The membrane thickness was approximately 60 μm. The AEM in the OH− form was obtained after immersion in thoroughly degassed 1 M aqueous NaOH, followed by washing with thoroughly degassed de-ionized water to remove the excess NaOH.Reactive casting of crosslinked AEMs
AEMs crosslinked with bis-QPi units were prepared in a reactive solution casting process. In the first step, crosslinked AEMs were prepared by casting films of PPO-7Br-DB and an excess of BMP from NMP solutions at 85 °C. The molar ratio of [Br]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BMP] was kept at 1:1, 1:2 and 1:4, respectively. Furthermore, two precursor PPO-7Br-DB samples with a DB of 20 and 30%, respectively, were employed. Mohr titrations of these intermediate AEMs after leaching excess BMP by immersion/swelling in THF gave IECItitr. In the second step, the residual tertiary methylpiperidine groups were converted to QPi groups via reaction with an excess of iodomethane with the membranes immersed and swollen in NMP at 40 °C. Mohr titrations of these final AEMs gave IECIItitr, i.e., the concentration of QPi groups formed during both the first and second step. The crosslinked AEMs were designated as PPO-7bisQPi-x-y where x denotes the final ion exchange capacity (IECIItitr) in the OH− form and y denotes the crosslink density, i.e. mol crosslinking bis-QPi units per g dry AEM, calculated as: y = 2IECItitr − IECIItitr/2. Samples in the Br− form were given the suffix -Br.
:[BMP] was kept at 1:1, 1:2 and 1:4, respectively. Furthermore, two precursor PPO-7Br-DB samples with a DB of 20 and 30%, respectively, were employed. Mohr titrations of these intermediate AEMs after leaching excess BMP by immersion/swelling in THF gave IECItitr. In the second step, the residual tertiary methylpiperidine groups were converted to QPi groups via reaction with an excess of iodomethane with the membranes immersed and swollen in NMP at 40 °C. Mohr titrations of these final AEMs gave IECIItitr, i.e., the concentration of QPi groups formed during both the first and second step. The crosslinked AEMs were designated as PPO-7bisQPi-x-y where x denotes the final ion exchange capacity (IECIItitr) in the OH− form and y denotes the crosslink density, i.e. mol crosslinking bis-QPi units per g dry AEM, calculated as: y = 2IECItitr − IECIItitr/2. Samples in the Br− form were given the suffix -Br.
An example using PPO-7Br-20 is described here. In the first step, PPO-7Br-20 solutions with concentration of ∼5 wt% were prepared in three different vials by dissolving 0.45 g of PPO-7Br-20 in 9 g of NMP at room temperature. Next, BMP was added to the solutions in different amounts to obtain molar [Br]:[BMP] ratios of 1:1, 1:2 and 1:4, respectively. The solutions were then kept under stirring at room temperature. This period had to be kept below 1 h to avoid the risk of premature crosslinking. The homogenous solutions were poured onto Petri dishes and films were cast in an oven at 85 °C during 48 h, and then kept at 105 °C during 48 h to ensure complete reaction. The intermediate AEMs were removed from the Petri dish, rinsed with deionized water, and dried with tissue paper to remove excess water. It was immersed in 100 mL THF (exchanged with fresh THF every day) during 4 days under stirring at room temperature. Next, the membrane was dried one day each at 20 and 50 °C, respectively, before further characterization and use. To carry out the second step, the intermediate AEMs were immersed in a glass flask containing 70 mL of NMP and an excess of iodomethane 10.65 g (0.075 mmol) was added. The reaction solutions were gently stirred during 4 days at 40 °C in darkness to ensure complete quaternization of the remaining piperidine groups. Subsequently, the final AEMs were carefully rinsed with deionized water and immersed in 400 mL of 1 M NaBr for 3 days at 40 °C to complete the ion exchange. Finally, the yellow AEMs in Br− form were thoroughly washed with deionized water and stored at room temperature before evaluation.
Thermal characterization
The thermal decomposition of all the polymers under nitrogen atmosphere was studied by thermogravimetrical analysis (TGA) using a TA instruments TGA Q500. Samples were dried in a vacuum oven at 50 °C for at least 48 h. Prior to measurement, all the samples were preheated for 10 min at 150 °C in the furnace to remove traces of water. Next, the data was recorded from 20 to 600 °C at a temperature ramp of 10 °C min−1 under a constant N2 flow of 60 mL min−1. The thermal decomposition temperature (Td,95) was taken at the point where 5% weight loss was noted. The glass transition temperature (Tg) values of the different bromoalkylated PPOs was investigated by differential scanning calorimetry (DSC) using a model Q2000 DSC analyzer from TA instruments. Measurements were done during heating–cooling–heating cycles between 50 and 280 °C where the upper temperature limit was set by Td,95.Determination of ion exchange capacity and water uptake
The theoretical IEC was calculated using the integrals of the 1H NMR signals from the alkylbrominated PPO polymers. Subsequently, the IEC of the AEMs was determined by Mohr titrations. After drying for 2 days at 50 °C, a membrane sample (∼50 mg) in the Br− form was immersed 0.2 M aqueous NaNO3 (25.00 mL) for 48 h to achieve full ion exchange. At least 3 volumes of 5.0 mL were then titrated using potassium chromate (K2CrO4) as indicator, before calculating the average IEC value.The water uptake was measured by first determining the dry weight (WBr) of membrane pieces in Br− form after storage under vacuum at 50 °C during at least 48 h. The dry weight of the membranes in OH− form (WOH) was then precisely calculated using the titrated values of the IEC and WBr. Next, the dried membranes in Br− form were individually immersed in 200 mL of degassed 1 M aqueous NaOH in a sealed desiccator for at least 24 h under nitrogen flow. The membranes were then quickly transferred to a beaker containing degassed de-ionized water and kept for at least 2 h under nitrogen. The procedure was repeated 4–5 times until the rinse water reached neutral pH. Next, the membranes were stored in degassed de-ionized water under nitrogen flow during at least 24 h to reach equilibrium at room temperature. The membranes were then taken out and, after quickly removing the surface water by tissue paper, the wet weight in the OH− form (W′OH) was quickly measured. The water uptake (WU) was then calculated as:
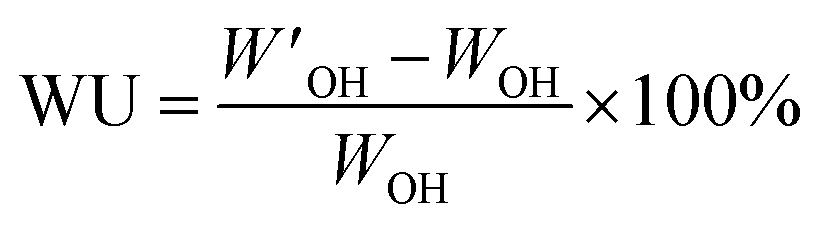 | (1) |
Using a similar procedure, water uptake measurements were carried out at different temperatures (40, 60, and 80 °C) by keeping the membrane samples in a 100 mL round-bottomed flask with inlet/outlet for argon/vacuum. The hydration number (λ), defined as the number of water molecules per functional group, was calculated as:
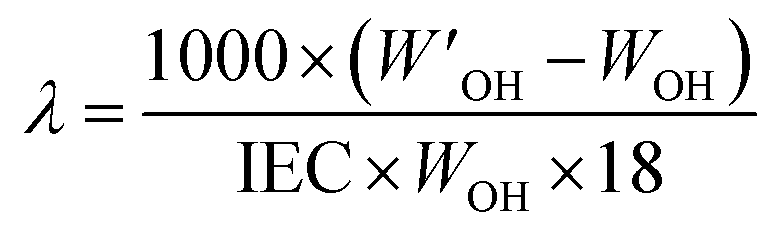 | (2) |
Conductivity measurements
Anion conductivity measurements under fully hydrated conditions were carried out by electrochemical impedance spectroscopy (EIS) employing a Novocontrol high resolution dielectric analyzer 1.01S in the frequency range 10−1 to 10−7 Hz at 50 mV. AEM samples in the Br− form were each immersed in 1 M NaOH during at least 24 hours under a nitrogen atmosphere to ensure complete ion exchange. The membranes were subsequently carefully washed with degassed distilled water and stored in degassed distilled water for at least 2 h under nitrogen atmosphere. The washing and storing steps were repeated 4–5 times until the rinsing water reached a neutral pH. The OH− conductivity of fully hydrated AEMs was finally collected using a two-probe method under fully immersed conditions during the thermal cycle: 20 → 80 → −20 → 80 °C.X-ray scattering
Small angle X-ray scattering (SAXS) measurements were employed to investigate the ionic clustering and morphology of dry AEMs in the Br− form. The SAXS measurements were performed using a SAXSLab ApS instrument (JJ-X-ray, Denmark) equipped with a microfocus sealed X-ray tube and a Pilatus detector. The scattering vector (q) was calculated as: | (3) |
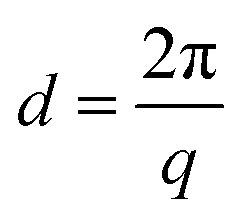 | (4) |
Evaluation of alkaline stability
The chemical stability of the AEMs under alkaline conditions was studied by 1H NMR spectroscopy and measurements of IEC, OH− conductivity and thermal decomposition.For the NMR evaluation, two weighted samples (∼10 mg) of each membrane were immersed in 350 mL of 1 M aq. NaOH. The solution was degassed and left under a nitrogen flow under stirring at 90 °C. To ensure the accuracy of the concentration, the NaOH solution was exchanged every second day. After 4 and 8 days of immersion, respectively, the AEM samples were taken out, washed repeatedly with de-ionized water, and immersed in 100 mL 1 M NaBr under stirring at 60 °C during 2 days. Next, the samples were immersed in de-ionized water for at least 24 h and carefully washed to remove any remaining NaBr. The samples were dried at 50 °C for 24 h and weighted again before the NMR measurements were carried out as described above.
For the conductivity measurements, samples were first kept for 8 days in 1 M NaOH at 90 °C, then transferred to a glass vial containing 100 mL degassed 1 M aq. NaOH, and stored at room temperature overnight under nitrogen. Next, the samples were washed with de-ionized water and stored in 100 mL degassed de-ionized water for at least 3 h. The washing and storing was repeated at least 5 times under nitrogen before the conductivity measurements were carried out under immersed conditions as described above.
IEC measurements were carried out after 8 days of immersion in 1 M NaOH at 90 °C. The samples were then washed carefully with de-ionized water and immersed in 100 mL 1 M aq. NaBr at 60 °C under stirring for 2 days. Next, the samples were immersed in de-ionized water for at least 24 h and carefully washed with de-ionized water to remove any remaining NaBr. The sample was dried at 50 °C for at least 48 h. The measurement procedure was then the same as described above.
For the TGA evaluation, weighted samples of the membranes (∼10 mg) were immersed in 350 mL of 1 M aq. NaOH. The solutions were degassed and left under a nitrogen flow and stirring at 90 °C. To ensure the concentration accuracy, the NaOH solutions were exchanged every second day. After 8 days of immersion the AEM samples were taken out, washed repeatedly with de-ionized water, and immersed in 100 mL 1 M NaBr under stirring at 60 °C for 2 days. Next, the samples were immersed in de-ionized water for at least 24 h and carefully washed to remove any remaining NaBr. The TGA measurements were done as described above after drying the samples at 50 °C for 24 h.
Results and discussion
Polymer synthesis and characterization
A series of five PPO samples were functionalized with QPi groups via heptyl spacer chains, i.e., one carrying mono-QPi groups and four samples with different contents of bis-QPi groups (Scheme 1). In the first step, PPO was bromoalkylated by benzylic lithiation and subsequent reaction with 1,6-dibromohexane, as reported previously.27,28 Although requiring high-purity reagents and necessary safety precautions, organolithium chemistry is nowadays widely used on an industrial scale to produce, e.g., bulk polymers such as synthetic rubbers and styrenic thermoplastic elastomers.45 In the present work, PPO was dissolved in THF and lithiated by n-BuLi to various predetermined degrees at −70 °C. A 200% excess of 1,6-dibromohexane in relation to the benzyllithium was added instantaneously and all at once to immediately quench all the lithiated sites in order to prevent potential crosslinking reactions that may otherwise occur because of the difunctionality of the dihalide. Analysis by SEC indicated an apparent increase in the Mn and PDI value of the polymers after the bromoalkylation (Table 1).Branching through a limited level of inter-polymer coupling most probably caused this, as indicated by the appearance of high-molecular weight fractions in the SEC traces after the bromoalkylation (ESI, Fig. S1†). However, no sign of gel formation was observed in any of the samples. Under the reaction conditions employed the highest attainable DB was ∼30%. Assuming close to complete lithiation (i.e., one Li+ per repeating PPO unit) and that only the benzylic positions are reactive, this limiting value agreed very well with the expected ratio between arylic and benzylic lithiation for PPO (approx. 65:35) found previously under similar conditions.46–48 A DB value of ∼30% corresponded to a maximum theoretical IEC of 1.7 and 2.6 mequiv. g−1 for PPOs tethered with mono- and bis-QPi groups, respectively.
The successful bromoalkylation of PPO was confirmed by 1H NMR spectroscopy, as shown in Fig. 1b. In addition to the signals observed for the neat PPO, a new signal ascribed to the –CH2Br protons appeared at 3.4 ppm. Signals from additional methylene protons of the heptyl side chains were seen between 1.2 and 2.0 ppm. In addition, the signal of the benzylic methylene protons was observed just above 2.4 ppm.
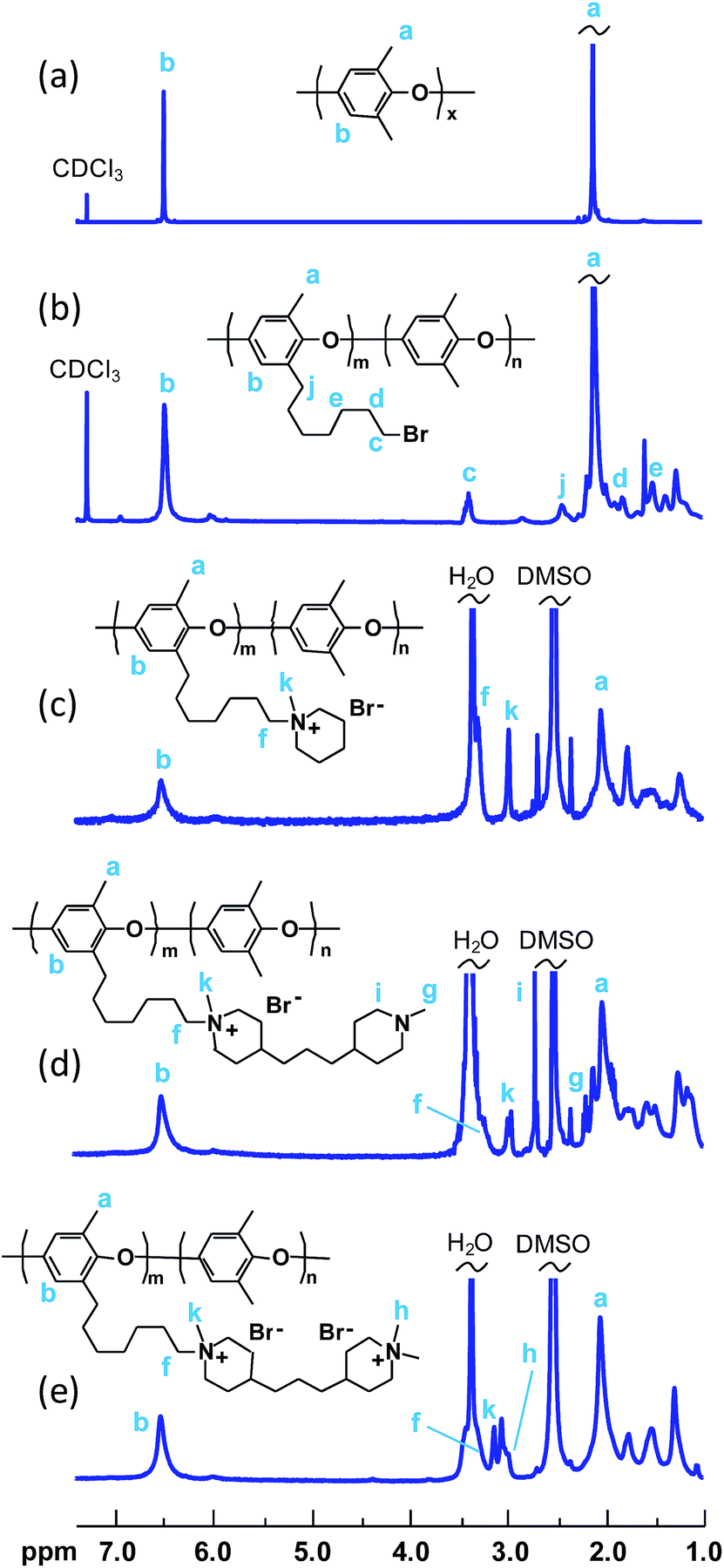 | ||
| Fig. 1 1H NMR spectra of native PPO (a), bromoalkylated PPO (PPO-7Br-19) (b), PPO with mono-QPi side chains (PPO-7QPi-1.7Br) (c), semi-quaternized intermediate (d), and PPO with bis-QPi side chains (PPO-7bisQPi-1.8Br) (e). | ||
The values of DB were calculated from the ratio of the integrated signals of the –CH2Br protons at 3.4 ppm and the aromatic protons at 6.4 ppm. Five different PPO samples with DB values ranging from 11 to 30% were synthesized (Table 1) at yields very close to 100%, and were designated as PPO-7Br-DB. DSC showed that the introduction of the flexible bromoheptyl side chains drastically reduced the Tg because of internal plasticization, from 216 °C for the native PPO to 158 °C for sample PPO-7Br-30 (ESI, Fig. S2†). Concurrently, the value of Td,95 decreased by 74 °C (Table 1).
The bromoalkylated PPOs were all functionalized via Menshutkin reactions involving 1-methylpiperidine and BMP in NMP at 85 °C to form side chains carrying one or two QPi groups, respectively (Scheme 1). In the latter case, the second QPi group on the spacer unit was formed in a subsequent Menshutkin reaction where the remaining tertiary piperidine group was quaternized with iodomethane in NMP at 40 °C. In Fig. 1c, the 1H NMR spectrum of PPO-7QPi-1.7 with single QPi groups on the spacers revealed new signals at 3.0 and 3.3 ppm from the –N+CH3 and –N+CH2– protons, respectively, of the QPi groups (the former unfortunately overlapped by the water signal). In addition, new signals from the cycloaliphatic methylene protons of the QPi units appeared between 3.3 and 3.4 ppm.
Fig. 1d shows the 1H NMR spectrum of the intermediate PPO derivative carrying one QPi and one tertiary piperidine ring per spacer unit after reaction with BMP. As seen, signals from the –N+CH3 and –N+CH2– protons appeared at 3.0 and 3.1 ppm, respectively. Additional signals from the many aliphatic units of the side chain were observed between 1.0 and 2.6 ppm. After the quaternization reaction with iodomethane and complete ion exchange to Br− counter ions, the number of signals from aliphatic groups decreased, as expected. In addition, at least two separate signals appeared between 3.0 and 3.4 ppm from the two chemically different QPi groups on the spacer units (Fig. 1e). Mohr titrations to determine the Br− content of all the QPi functional PPO materials confirmed the complete quaternization. Within the error of the method, the values were in excellent agreement with the theoretical ones calculated from the 1H NMR data of the corresponding bromoalkylated PPO (Table 2). Hence, the samples had IEC values ranging from 1.4 to 2.0 mequiv. g−1 in the OH− form.
| AEM | Precursor polymer | IECNMRa [mequiv. g−1] | IECtitra [mequiv. g−1] | T d,95 [°C] | q max [nm−1] | d [nm] | λ |
|---|---|---|---|---|---|---|---|
| a IEC in the OH− form (values within parenthesis are in the Br− form). b Measured by TGA. c Immersed in the OH− form at 20 °C. | |||||||
| PPO-7QPi-1.7 | PPO-7Br-30 | 1.7 (1.5) | 1.7 (1.5) | 247 | 1.89 | 3.3 | 12 |
| PPO-7bisQPi-1.4 | PPO-7Br-11 | 1.4 (1.3) | 1.4 (1.3) | 252 | 1.28 | 4.9 | 15 |
| PPO-7bisQPi-1.7 | PPO-7Br-15 | 1.7 (1.5) | 1.7 (1.5) | 225 | 1.33 | 4.7 | 18 |
| PPO-7bisQPi-1.8 | PPO-7Br-17 | 1.8 (1.6) | 1.8 (1.6) | 235 | 1.33 | 4.7 | 18 |
| PPO-7bisQPi-1.9 | PPO-7Br-19 | 2.0 (1.8) | 1.9 (1.7) | 238 | 1.35 | 4.7 | 24 |
Membrane preparation and morphology
All the QPi functionalized materials were soluble in DMSO and NMP at room temperature. Samples with IECs above 1.4 mequiv. g−1 were also completely soluble in methanol. The QPi functional PPOs can thus be employed in the fabrication of both AEMs and active catalyst layers for different electrochemical applications. In the present study, yellow-brownish and transparent AEMs were cast from NMP solutions at 80 °C (Scheme 1). The membranes were flexible and creasable, and could be bent 180° multiple times without damage (ESI, Fig. S3†). SAXS was employed to study the ability of the QPi groups to phase separate and form ionic clusters in the PPO matrix of dry AEMs in the Br− form. The position of the so-called ionomer peak can used to estimate the characteristic separation distance (d) between the ion-rich phase domains of the AEM, and is given by the scattering maximum (qmax). We have previously shown by SAXS that attaching the QA groups via alkyl spacer units significantly improves the ability of QA groups to form ionic clusters.27,28 This is manifested by a much more pronounced ionomer peak in relation to corresponding QA groups placed directly on the polymer backbone. The efficient ionic clustering leads to high local ionic concentrations which results in significantly higher ion conductivities of AEMs based on the former polymers.27,28 As seen in Fig. 2a, the scattering maximum of the PPO-7QPi-1.7Br membrane with mono-QPi functional side chains occurred at qmax = 1.9 nm−1, which corresponded to d = 3.3 nm (Table 2). These values were quite similar to those of an AEM with IEC = 1.5 mequiv. g−1 based on PPO carrying trimethylammonium groups on heptyl spacers (qmax = 1.8 nm−1, d = 3.6 nm).28 The SAXS profiles of the PPO-7bisQPi-xBr membranes, where each side chain carried two QPi groups, clearly showed ionomer peaks between qmax = 1.28 and 1.35 nm−1 with much increased scattering intensities (Fig. 2a). These peak positions corresponded to d-values between 4.9 and 4.7 nm, respectively, which was considerably higher than for membrane PPO-7QPi-1.7Br. Thus, the introduction of two pendant QPi groups on heptyl side chains seemed to greatly promote the phase separation between the cations and the polymer backbones in the AEMs.Thermal and alkaline stability
TGA was carried out to study the thermal decomposition of the AEMs in the Br− form under nitrogen atmosphere after an initial isotherm at 150 °C for 10 min to remove any residual water. Previously, the insertion of a spacer unit in-between the PPO backbone and QA groups have been shown to increase the thermal stability in relation to QA groups attached in benzylic positions directly on the PPO backbone.28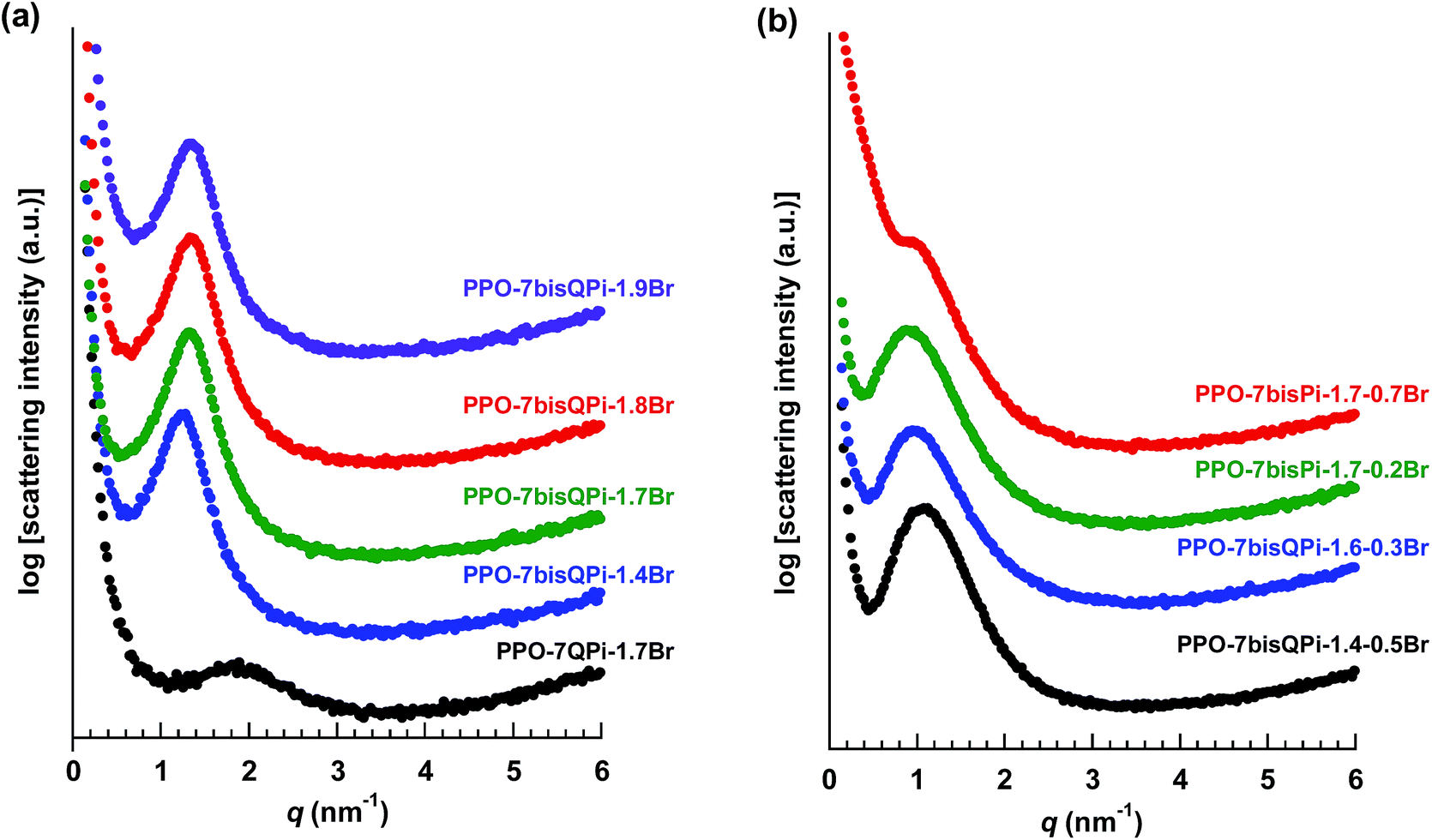 | ||
| Fig. 2 SAXS profiles of dry AEMs based on PPO functionalized with mono- and bis-QPi groups via alkyl spacer units (a), and of dry AEMs crosslinked by bis-QPi units (b). The data were measured with the AEMs in the Br− form and have been shifted vertically for clarity. | ||
As shown in Fig. 3, the current AEMs started to decompose at Td,95 = 225–250 °C depending on the IEC. At an IEC of 1.5 mequiv. g−1, the Td,95 of PPO-7QPi-1.5Br was 247 °C. This can be compared with PPOs functionalized with trimethylammonium groups in benzylic positions (Td,95 = 199 °C) and via heptyl spacers (Td,95 = 213 °C), respectively, at the same IEC.28 Thus, the nature of the QPi group and the presence of the spacer significantly raised the thermal stability of the present AEMs. As seen in Fig. 3b, the TGA traces showed that the degradation and weight loss occurred in two distinct steps. The first step started just above 200 °C and may be attributed to the loss of QPi groups. The second step occurred in the temperature range 400–450 °C and was due to the decomposition of the polymer backbone. Still, the thermal stability was clearly well above the normal operating temperature of most electrochemical devices.
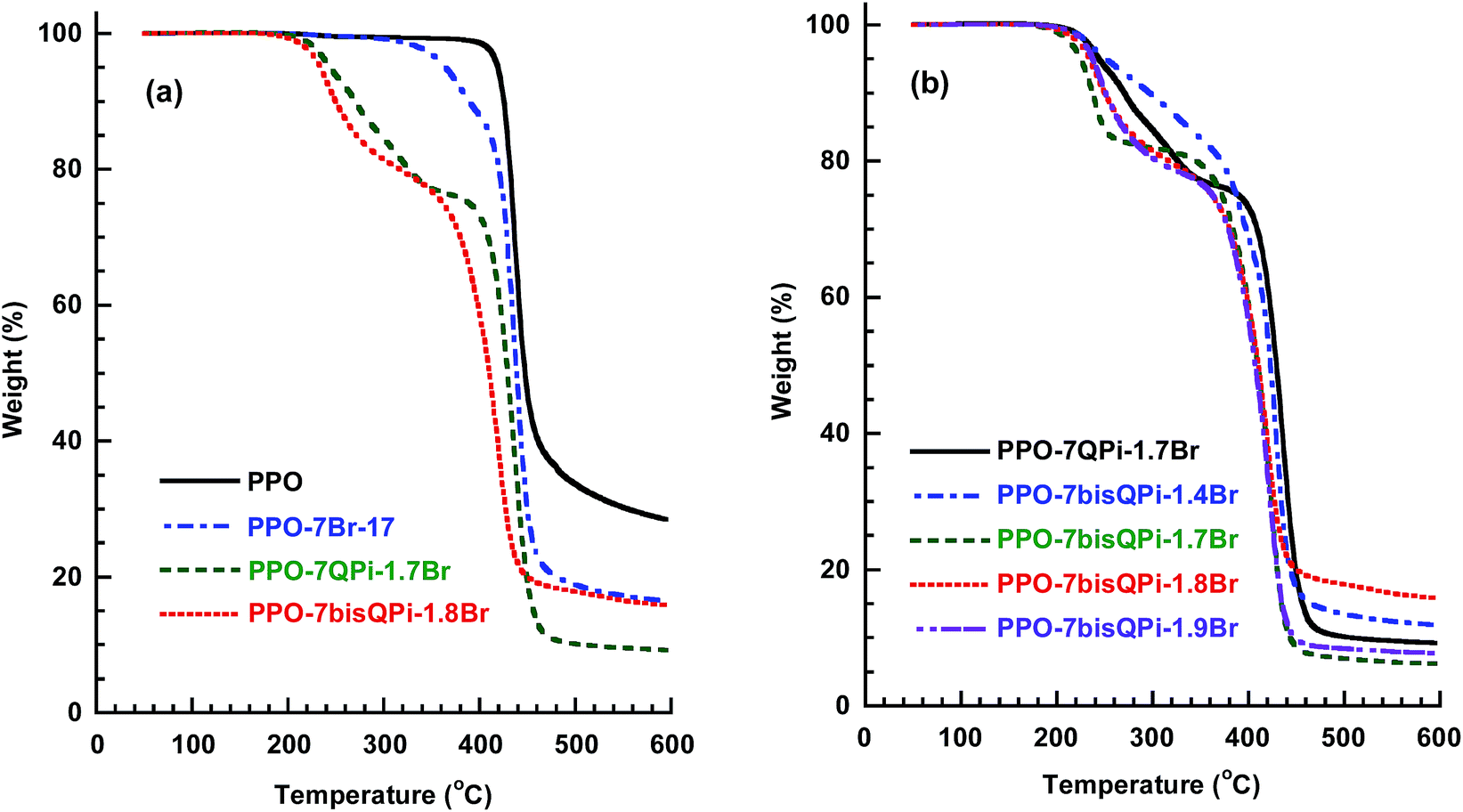 | ||
| Fig. 3 TGA traces of (a) native PPO, a bromoalkylated precursor PPO, a mono-QPi and a bis-QPi functionalized sample, and (b) mono- and bis-QPi functionalized samples with different IEC values. Samples were measured under N2 at 10 °C min−1. | ||
The evaluation of the alkaline stability was carried out by immersing the AEM samples in 1 M aqueous NaOH at 90 °C under nitrogen atmosphere. Samples were removed after 4 and 8 day's storage, ion-exchanged to the Br− form, dissolved in DMSO-d6 and analyzed by 1H NMR spectroscopy to identify possible changes in the molecular structure. Fig. 4 shows representative spectra obtained after the analysis of AEMs based on PPOs with mono- and bis-QPi side chains, respectively. As seen, no new signals and no significant changes in any of the chemical shifts were detected after 8 days. Consequently, no degradation of the molecular structure of the QPi modified PPOs was detected. In addition, the AEMs retained their flexibility and creasability after the alkaline treatment at 90 °C. These results demonstrated an excellent stability of the corresponding AEMs under strongly alkaline conditions. To complement the NMR data, the PPO-7QPi-1.7 and PPO-7bisQPi-1.7 membranes were also analysed by IEC and OH− conductivity measurements after 8 day's storage in 1 M aqueous NaOH at 90 °C. The results showed no detectable change in the IEC values, but a slight decrease in the conductivity of 6 and 9%, respectively. We believe that this minor decline may be due to changes in the AEM morphology rather than degradation. TGA analysis is very sensitive towards loss of cationic groups and the TGA traces of these membranes in the Br− form before and after the alkaline testing were almost identical, again indicating no AEM degradation (ESI, Fig. S4†). The present results on the QPi functional materials are in agreement with theoretical considerations36 and previous studies of model compounds19,49 which predict QPi groups to be very stable.
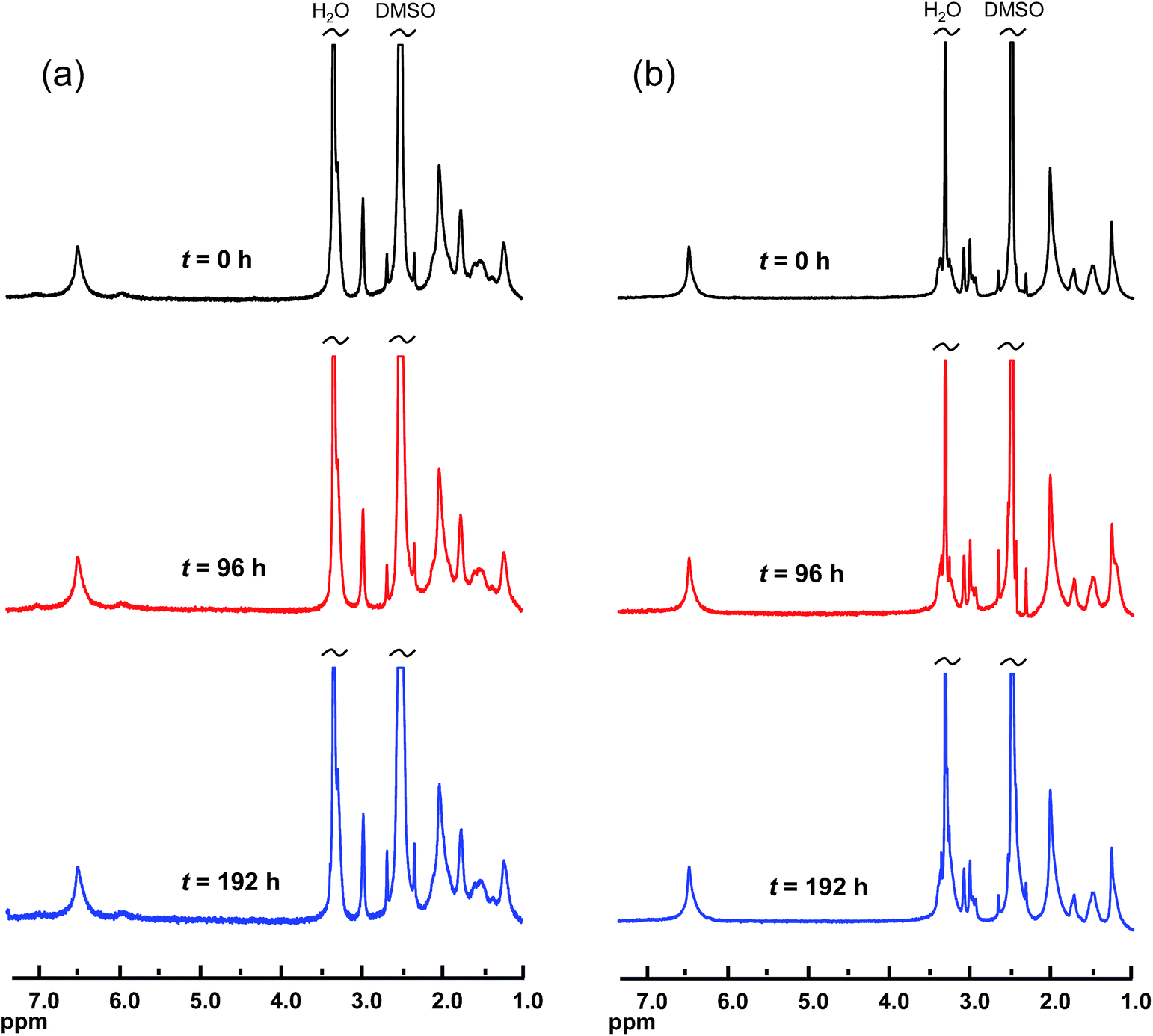 | ||
| Fig. 4 1H NMR spectra of PPOs functionalized with mono- and bis-QPi groups PPO-7QPi-1.7 (a) and PPO-7bisQPi-1.7 (b), respectively, after storage in 1 M aqueous NaOH solution at 90 °C during 0, 96, and 192 h, respectively. | ||
The current findings may be compared with our previous results on PPOs modified with QA groups, which showed extensive loss of QA groups if placed in benzylic positions on the PPO backbone.28 However, if these groups were instead attached via alkyl spacers the alkaline stability was dramatically increased. Hence, no degradation was detected by 1H NMR spectroscopy of PPOs functionalized with QA groups via pentyl or heptyl spacers units after 8 days' storage in 1 M NaOH at 80 °C.28 In fact, by NMR spectroscopy we also found AEMs based on PPOs modified with QA groups to be stable in 1 M NaOH at 90 °C over 8 days. This means that that even harsher conditions and/or longer testing periods are required to distinguish between the alkaline stability of materials functionalized with the QPi and QA groups via alkyl spacers.
Water uptake and OH− conductivity
The water content of AEMs greatly promotes the ionic conductivity but will also influence, e.g., membrane swelling and mechanical properties. Fig. 5a shows the water uptake of the AEMs in the OH− form as a function of temperature under fully hydrated conditions (immersed) in water between 20 and 80 °C. As expected, the water uptake of the AEMs increased with increasing IEC and temperature. The PPO-7bisQPi-x membranes, with IEC values between 1.4 and 1.9 mequiv. g−1, showed a water uptake ranging from 38 to above 82 wt% at 20 °C. This corresponded to λ values ([H2O][QPi]−1) between 15 and 24 (Table 2). As seen, a slight increase in the IEC from 1.8 to 1.9 mequiv. g−1 led to a dramatic increase in the water uptake, and the water uptake of PPO-7bisQPi-1.9 increased progressively with the temperature to reach 120 wt% at 80 °C. This result indicated that an IEC of 1.8–1.9 mequiv. g−1 may be the limit beyond which non-crosslinked AEMs start to absorb excessive amounts of water. Compared at an IEC value of 1.7 mequiv. g−1, the AEM based on PPO with bis-QPi functional side chains (PPO-7bisQPi-1.7) took up more water than the corresponding membrane with mono-QPi functional side chains (PPO-7QPi-1.7). The reason may be the more distinct phase separation of the former AEMs that led to the formation of large water-rich domains upon hydration. The OH− conductivity was measured by EIS with the AEMs fully hydrated (immersed) in a sealed two-probe cell. To avoid uptake of CO2 from air, the membranes in OH− form were carefully immersed, washed and equilibrated with degassed distilled water under nitrogen. After a measurement cycle of heating–cooling–heating between −20 and 80 °C, all the membranes remained flexible and seemingly unaffected by the treatment. The conductivity data collected during the second heating step from −20 to 80 °C are shown in Fig. 5b. As expected, the conductivity followed the same trend as the water uptake and thus increased with increasing IEC and temperature. The conductivity increased significantly between −20 and 20 °C because of the melting of ice in the AEMs. As seen in Fig. 5b, PPO-7bisQPi-1.7 showed a higher conductivity than PPO-7QPi-1.7. Hence, compared at the same IEC, the AEM carrying bis-QPi side chains reached a higher conductivity than the one with mono-QPi side. The most probable reason was the more distinct phase separation, as observed by SAXS, and the higher water uptake of the former AEM. The OH− conductivity of PPO-7QPi-1.7 at 80 °C was 44 mS cm−1 at a water uptake of 50 wt%. This may be compared with the corresponding data of AEMs based on PPO functionalized with single trimethylammonium groups in benzylic positions directly on the backbone (11 mS cm−1 at 28 wt% water uptake) and via heptyl spacers (60 mS cm−1 at 42 wt% water uptake), both at an IEC of 1.5 mequiv. g−1.28 Thus, the QPi functionalized AEM had a slightly lower conductivity compared to the latter AEM carrying trimethylammonium groups on spacers.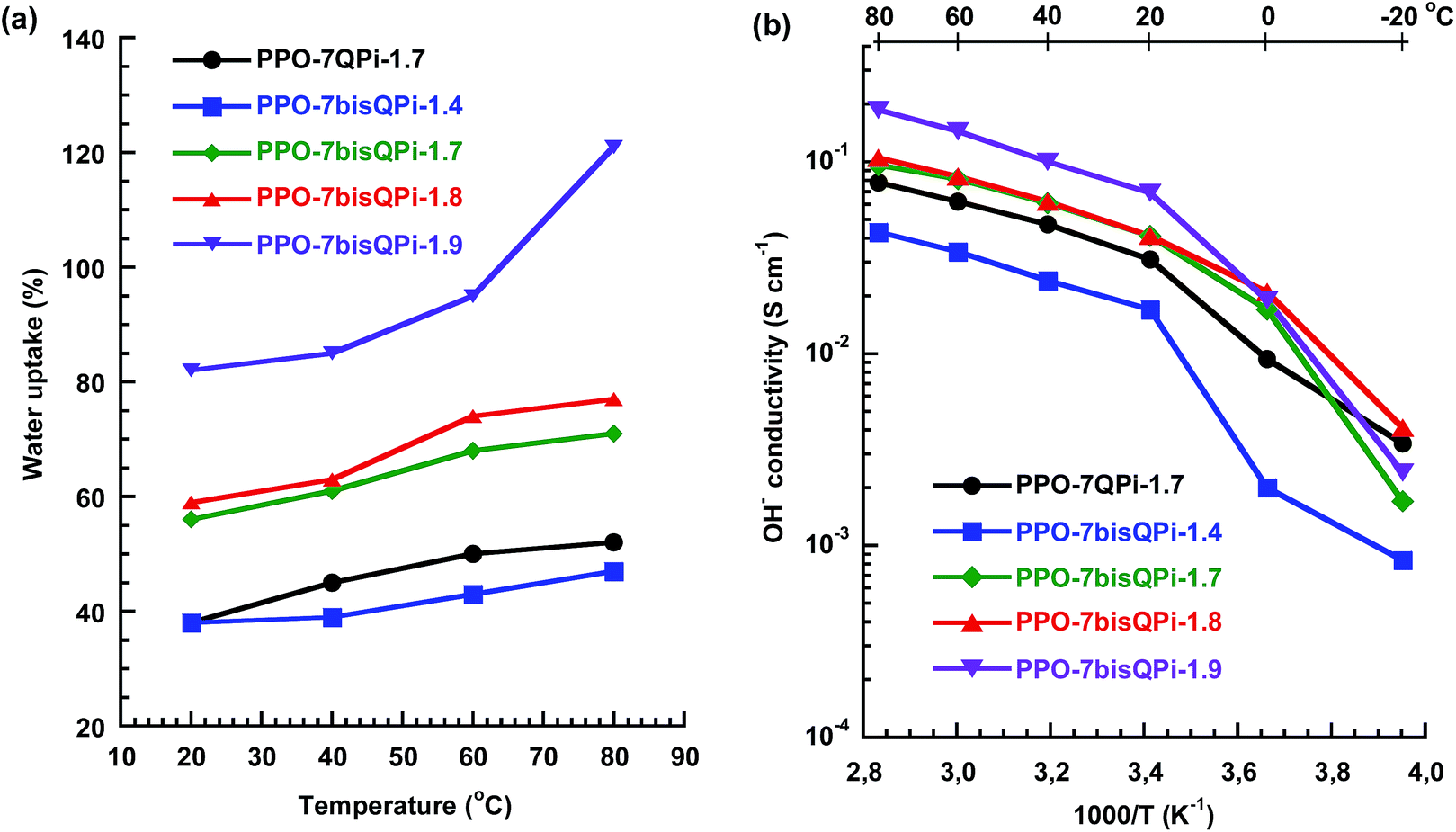 | ||
| Fig. 5 Water uptake of AEMs in the OH− form (a) and Arrhenius plots of the OH− conductivity (b) of mono- and bis-QPi functionalized PPO AEMs measured under fully hydrated (immersed) conditions. | ||
PPO-7bisQPi-1.9 exhibited very high OH− conductivity values, 69 and 186 mS cm−1 at 20 and 80 °C, respectively (Fig. 5b). This can be attributed to the efficient ionic cluster formation and high water content of this AEM. The properties of the present AEMs may be compared with corresponding data of AEMs with alkyl side chains reported previously. For example, Zhuang et al. have reported on polysulfones with long alkyl side chains in addition to benzyl trimethylammonium groups placed directly on the backbone.50 These AEMs reached an OH− conductivity close to 35 mS cm−1 at 20 °C and just above 100 mS cm−1 at 80 °C. The AEMs were however found to degrade in 1 M aq. KOH at 60 °C. In addition, Zhuang and co-workers have reported on polysulfones functionalized with side chains containing two QA groups attached via benzylic positions.51 Fully hydrated membranes (IEC = 2.1 mequiv. g−1) reached 80 mS cm−1 at 80 °C, but degradation was noted in 3 M aq. KOH at 60 °C. Xu et al. prepared PPOs carrying trimethylammonium groups placed on phenylpropyl spacer units via Pd catalyzed Suzuki coupling and quaternization.29 The OH− conductivity was 27 and 63 mS cm−1 at 30 and 70 °C, respectively, for a membrane with IEC = 1.78 mequiv. g−1. Approximately 90% of the OH− conductivity was retained after immersing a membrane in 1 M NaOH at 60 °C over 168 h.29 In another study, Mohanty and Bae have synthesized polyfluorenes with trimethylammonium groups located on hexyl spacers using Pd catalyzed Suzuki coupling of premade bromoalkylated monomers, followed by quaternization.30 At 30 and 80 °C, the OH− conductivity under fully hydrated conditions was measured to be 24 and 85 mS cm−1 at (IEC = 2.9 mequiv. g−1). No significant change in the 1H NMR shifts or in the titrated IEC value was seen for any of the polymers after 720 h at 80 °C in 1 M NaOH.
As seen in Fig. 5b, the temperature dependence of the OH− conductivity of the AEMs followed Arrhenius relationships above the melting point of the water which indicated decoupling from the slow polymer dynamics. The apparent activation energy (Ea) of the conductivity was estimated to be between 12 and 14 kJ mol−1 from the data measured between 20 and 80 °C. This was comparable or slightly below values previously reported for AEMs.28,52Fig. 6a and b shows the OH− conductivity as a function of IEC and water uptake, respectively. The former data of the PPO-7bisQPi-x series showed that the rate of conductivity increase with the IEC value accelerated above 1.8 mequiv. g−1 because of the very high water uptake. The dependence of the ion conductivity on the water uptake can be seen in Fig. 6b. By comparing the data of samples PPO-7QPi-1.7 and PPO-7bisQPi-1.7 (both with IEC = 1.7 mequiv. g−1) it is obvious that the latter sample took up 20 wt% more water than the former in the temperature range 20–80 °C (also seen in Fig. 5a). Concurrently, PPO-7bisQPi-1.7 reached a ∼30% higher conductivity than PPO-7QPi-1.7. In addition to OH−, a large number of alternative anions may be transported by AEMs. The conductivity of Br− is generally significantly lower than the OH− conductivity because of the lower mobility of Br− in dilute solution in combination with the lower water uptake of the AEMs in the Br− form.53 For example, Marino et al. found the OH− conductivity to be up to an order of magnitude higher that the Br− conductivity in a model poly(arylene ether) AEM.53 The water uptake of PPO-7bisQPi-1.9Br at 20 and 80 °C was 16 and 38 wt%, respectively, (ESI, Fig. S5†) which was significantly lower than for the same AEMs in the OH− form.
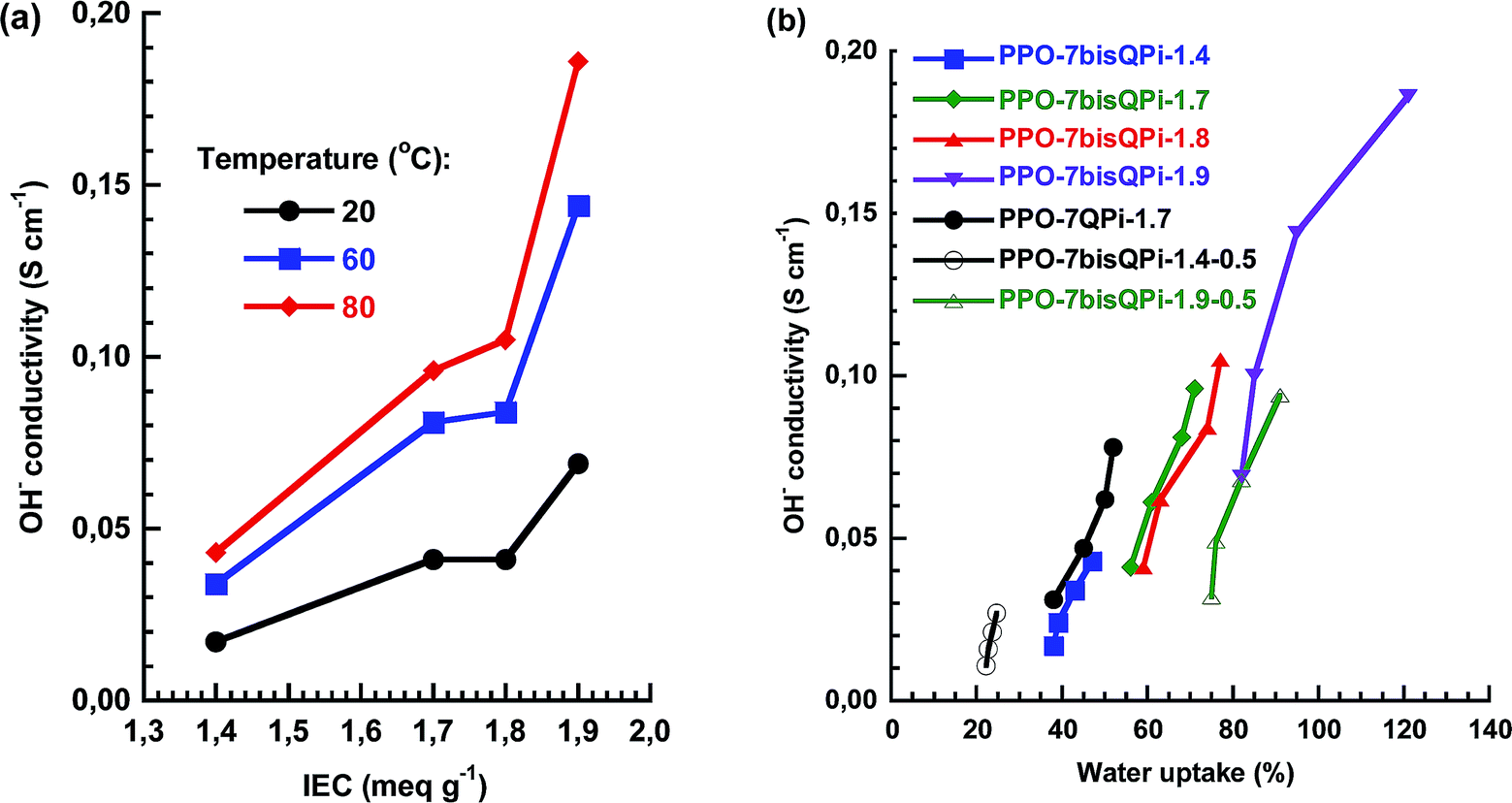 | ||
| Fig. 6 OH− conductivity as a function of (a) IEC and (b) water uptake of the AEMs under fully hydrated (immersed) conditions. Data of crosslinked samples are denoted by open symbols. | ||
As expected, the Br− conductivity of the present AEMs followed the same order among the studied materials as the OH− conductivity (ESI, Fig. S6†). The Br− conductivity of PPO-7bisQPi-1.9Br was 3.8 and 20 mS cm−1 at 20 and 80 °C, respectively.
Preparation and properties of crosslinked AEMs
Normally crosslinking of AEMs is carried out to restrict the water uptake and the degree of swelling, and is thus particularly relevant at high IEC values. This is usually accompanied by a decrease in the ionic conductivity. A number of different crosslinking methods have been reported for AEMs, including the use of reactive thiol,54 aldehyde,55 alkoxysilane,56 allyl57 and epoxy58 groups. In addition, several groups have reported the use of diamines in crosslinking reactions.59–62 For example, Park et al. fabricated crosslinked AEMs by reacting chloromethylated polysulfone with mixtures of TMA and different tetramethyl diamines.61 AEMs quaternized by a mixture of TMA and N,N,N′,N′-tetramethyl-1,6-hexanediamine showed improved OH− conductivity (up to 11 mS cm−1 at 25 °C) and thermal stability than those crosslinked by just the diamine. Using a different approach, Li and co-workers prepared crosslinked AEMs by first functionalizing PPO with benzylic QA groups carrying pendant undecene chains, and in the second step employing Grubbs-catalyzed olefin metathesis for the crosslinking.57 This gave AEMs with good dimensional stability, improved alkaline stability and OH− conductivities reaching 40 mS cm−1 at 20 °C (IEC = 3.20 mequiv. g−1).In all cases known to us where the crosslinks have included QA groups, the crosslinking has been achieved with the QA groups in benzylic positions on the polymer backbones. In the present case, AEMs crosslinked by bis-QPi units were prepared in a reactive casting process where the difunctional BMP reacted with bromoalkylated PPO to form the crosslinks at 85 °C, as shown in Scheme 2.
 | ||
| Scheme 2 Preparation of AEMs crosslinked by bis-QPi units via membrane casting/crosslinking (i), methylation (ii), and ion-exchange (iii) [key: (i) NMP, 85–105 °C, (ii) MeI, NMP, 40 °C, (iii) aqueous NaOH, 20 °C]. | ||
The presence of the spacer can be expected to bring several advantages, including increased accessibility/reactivity during the crosslinking reaction, increased molecular flexibility to reduce membrane brittleness, and increased alkaline stability in relation to systems with benzylic QA groups. The molar ratio of [Br]:[BMP] was kept at 1:1, 1:2 and 1:4, respectively, to ensure the complete displacement of the Br atoms of the bromoalkyl side chains and to vary the degree of crosslinking. In addition, two precursor PPO-7Br-DB samples with DBs of 20 and 30%, respectively, were used to further vary the crosslinking degree. Notably, if solutions of PPO-7Br-DB and BMP were left for more than 1 h at room temperature there was a risk of premature crosslinking and gel formation, which indicated the high reactivity of the system. After the first step the intermediate AEMs contained a mixture of QPi and unreacted tertiary piperidine groups. Mohr titrations of these intermediate AEMs gave IECItitr, i.e., the concentration of QPi groups formed in the first step.
In the second step, the residual tertiary methylpiperidine groups were converted to QPi groups via reaction with an excess of iodomethane with the membranes immersed and swollen in NMP at 40 °C. The fact that the membranes were only moderately swollen by NMP further proved the successful formation of crosslinks in the first step. Mohr titrations of these final AEMs gave IECIItitr, i.e., the concentration of QPi groups formed during both the first and second step.
The crosslinked yellow-brownish AEMs were noticeably stiffer than the non-crosslinked membranes but were still flexible and creasable. As seen in Table 3, the crosslinking density and IEC values of the AEMs were in the range 0.20–0.65 mmol g−1 and 1.3–1.9 mequiv. g−1, respectively, under the conditions employed. As anticipated, both the crosslinking density and the IEC increased when the DB of the PPO-7Br-DB precursor polymer was increased from 20 to 30%.
| AEM | Precursor polymer | [Br]:[BMP] |
IECItitra [mequiv. g−1] | IECIItitrb [mequiv. g−1] | Crosslinking densityc,d [mmol crosslinks per g] | Fraction of bis-QPi units involved in crosslinkingd | T d,95 [°C] | q max [nm−1] | d [nm] | λ |
|---|---|---|---|---|---|---|---|---|---|---|
| a Intermediate IEC value in the Br− form measured by Mohr titration after membrane casting/crosslinking. b Final IEC value in the OH− form measured by Mohr titration after quaternization with iodomethane (values within parenthesis are in the Br− form). c In the OH− form (values within parenthesis are in the Br− form). d Calculated from IECItitr and IECIItitr. e Immersed in the OH− form at 20 °C. | ||||||||||
| PPO-7bisQPi-1.4-0.5 | PPO-7Br-20 | 1:1 |
1.1 | 1.4 (1.3) | 0.50 (0.45) | 0.69 | 260 | 1.08 | 5.8 | 8 |
| PPO-7bisQPi-1.6-0.3 | PPO-7Br-20 | 1:2 |
1.0 | 1.6 (1.4) | 0.33 (0.35) | 0.47 | 258 | 0.95 | 6.6 | 10 |
| PPO-7bisQPi-1.7-0.2 | PPO-7Br-20 | 1:4 |
0.95 | 1.7 (1.5) | 0.22 (0.20) | 0.27 | 256 | 0.87 | 7.2 | 12 |
| PPO-7bisQPi-1.7-0.7 | PPO-7Br-30 | 1:1 |
1.4 | 1.7 (1.5) | 0.72 (0.65) | 0.87 | 268 | 0.95 | 6.6 | 11 |
| PPO-7bisQPi-1.9-0.5 | PPO-7Br-30 | 1:2 |
1.3 | 1.9 (1.7) | 0.50 (0.45) | 0.53 | 264 | — | — | 22 |
Equally expected, an increase in the molar BMP concentration in relation to that of the bromoalkyl side chains decreased the crosslinking density, as well as the fraction of bis-QPi units involved in the crosslinking, but increased the IEC value (Table 3). Thus, both the DB and the [Br]:[BMP] ratio were efficiently employed to tune and optimize the AEM properties.
The SAXS profiles of the PPO-7bisQPi-x-yBr membranes showed clear scattering peaks between qmax = 0.87–1.08 nm−1, which corresponded to d values in the range 5.8–7.2 nm (Fig. 2b). As seen in Table 3, the d spacing was found to increase with increasing IEC and decreasing crosslink density. The data of PPO-7bisQPi-1.7-0.7Br indicated a much smaller scattering intensity. This may be due to the considerably higher degree of crosslinking compared to the other samples, which may restrict the clustering of the ions. A comparison with the data of the non-crosslinked PPO-7bisQPi-xBr membranes, the crosslinked membranes displayed higher d spacings at comparable IECs (Tables 2 and 3). This may be explained by the difference in the membrane formation process where the latter membranes were swollen with NMP during the second quaternization step which possibly favoured the formation of larger ionic clusters.
TGA traces of the PPO-7bisQPi-x-yBr membranes recorded under nitrogen revealed a two-step decomposition process (as seen in Fig. S7 and S8†). The value of Td,95 increased significantly with the degree of crosslinking at a given IEC (Table 3). Compared at an IEC of 1.7 mequiv. g−1, the non-crosslinked PPO-7bisQPi-1.7 showed Td,95 = 225 °C, while PPO-7bisQPi-1.7-0.2Br and PPO-7bisQPi-1.7-0.7Br reached Td,95 = 256 and 268 °C at degrees of crosslinking of 0.2 and 0.7 mmol g−1, respectively (Tables 2 and 3).
In order to probe the alkaline stability of the crosslinked AEMs, the IEC and OH− conductivity of PPO-7bisQPi-1.7-0.2 were measured after 8 days in 1 M aqueous NaOH at 90 °C. The results were very similar to those of the non-crosslinked AEMs. Thus, the IEC was found to be unaffected but the conductivity decreased by 7%. As mentioned above, this decrease may be due to, e.g., changes in morphology rather than degradation. TGA measurements of the AEM in the Br− form before and after the alkaline treatment showed identical traces, which indicated no degradation (ESI, Fig. S4†). Consequently, no macromolecular degradation was detected.
Fig. 7a shows the water uptake of the crosslinked AEMs as a function of temperature and indicate that the water content was efficiently controlled by the IEC and crosslinking density with the predicted trends. At 80 °C, membrane PPO-7QPi-1.4-0.5 and PPO-7QPi-1.9-0.5 (same crosslink density but different IEC) took up 25 and 91 wt% water, respectively, and PPO-7QPi-1.7-0.2 and PPO-7QPi-1.7-0.7 (same IEC value but different crosslink density) took up 46 and 42 wt% water, respectively. In particular, the water uptake was markedly reduced in relation to the non-crosslinked AEMs. For example, at the same IEC and 80 °C, the non-crosslinked PPO-7bisQPi-1.7 took up 71 wt% water, while only 46 wt% was taken up by the crosslinked PPO-7bisQPi-1.7-0.2 (Fig. 7a).
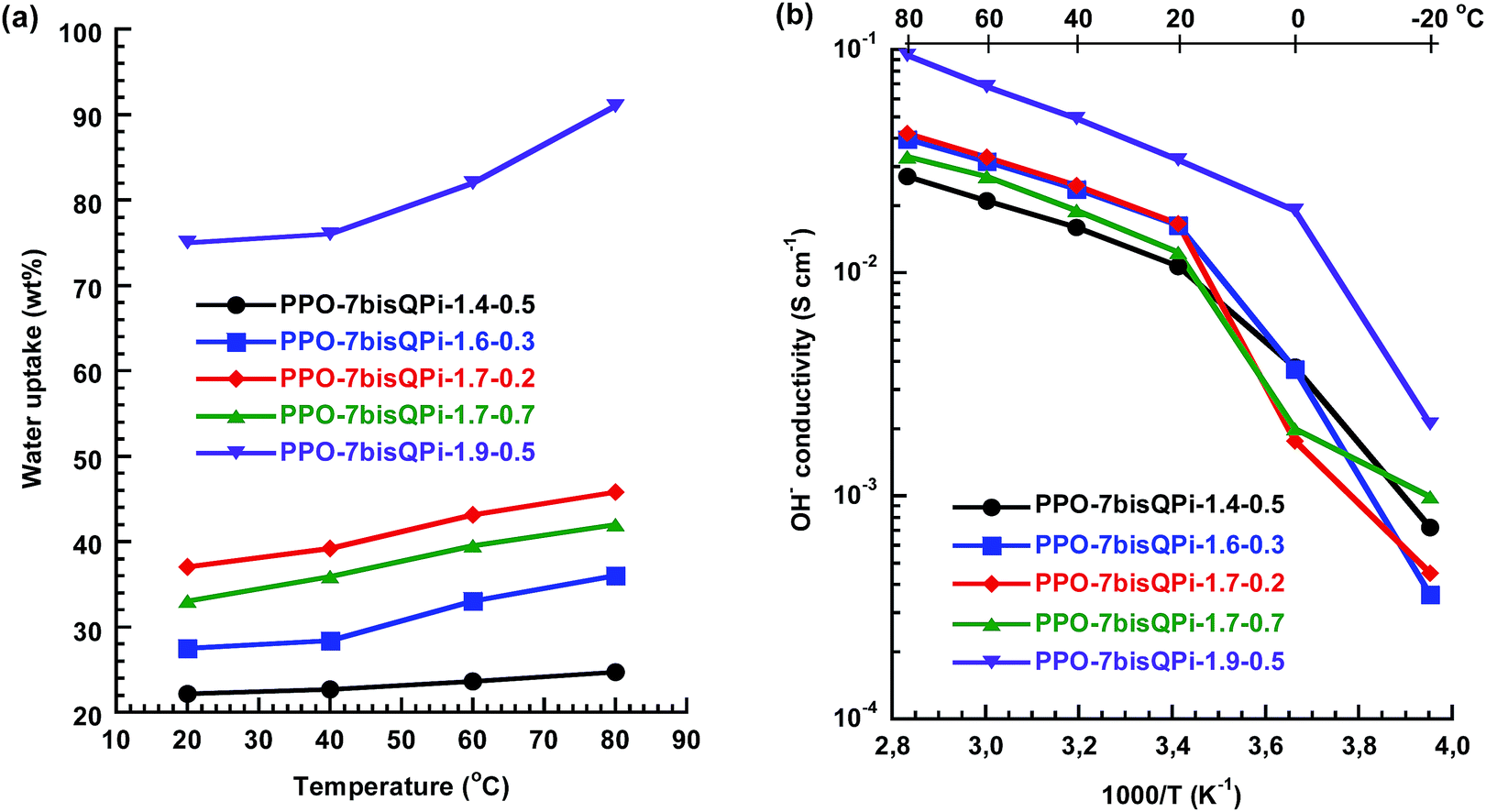 | ||
| Fig. 7 Water uptake (a) and OH− conductivity (b) of the bis-QPi crosslinked AEMs under fully hydrated (immersed) conditions. | ||
Just as observed for the non-crosslinked AEMs, the temperature dependence of the OH− conductivity of the crosslinked AEMs followed Arrhenius relationships above the melting point of the water (Fig. 7b). The Ea value of the conductivity between 20 and 80 °C was equal to the non-crosslinked AEMs and thus indicated a similar conduction mechanism. At a given IEC value, the crosslinking lowered the OH− conductivity of the AEM, primarily because of the reduced water content. For example, the conductivity of PPO-7bisQPi-1.7 and PPO-7bisQPi-1.7-0.2 was 96 and 42 mS cm−1, respectively, at 80 °C. The highest OH− conductivity of the crosslinked AEMs, 32 and 94 mS cm−1 at 20 and 80 °C, respectively, was reached by PPO-7bisQPi-1.9-0.5. This was in level with the highest conductivity reported by Li and co-workers for AEMs with crosslinked via Grubbs-catalyzed olefin metathesis.57 However, the latter result was achieved at a much higher IEC (3.2 mequiv. g−1).
Conclusions
PPO was successfully functionalized with mono- and bis-QPi groups via flexible heptyl spacers, thus combining a proven alkali-stable cycloaliphatic QA cation with the spacer concept to produce high-performance membranes. Flexible, transparent and tough AEMs functionalized with QPi showed a significantly higher thermal stability than corresponding AEMs carrying trimethylammonium groups via benzylic positions or via heptyl spacer units. Furthermore, no degradation was detected after storage in 1 M NaOH at 90 °C during 8 days, demonstrating an excellent alkaline stability. SAXS results showed an efficient clustering of the QPi cations, especially when placed in pairs on the spacer units. Consequently, AEMs functionalized with mono-QPi groups showed high OH− conductivity. Still, the ones functionalized with bis-QPi groups showed even higher values, up to 69 and 186 mS cm−1 in the fully hydrated state at 20 and 80 °C, respectively. With the aim to tune the water uptake, crosslinked AEMs were effectively prepared in a reactive casting process were bromoalkylated PPO reacted with BMP to form bis-QPi crosslinks with heptyl spacers in-between the QPi cations and the PPO backbones, thus avoiding QA groups in labile benzylic positions. The joint results of this work showed that AEMs with excellent chemical and thermal stability and OH− conductivity can be prepared by placing cycloaliphatic QA cations such as QPi on flexible alkyl side chains. Furthermore, by introducing QPi functional crosslinks the properties can be further tuned and optimized for use in alkaline fuel cells and other electrochemical energy devices.Acknowledgements
We thank the Swedish Energy Agency and the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning for financial support. We are also grateful to Tommy Garting for assistance with SAXS measurements and data treatment.Notes and references
- U. Lucia, Renewable Sustainable Energy Rev., 2014, 30, 164–169 CrossRef CAS.
- T. Yoshida and K. Kojima, Electrochem. Soc. Interface, 2015, 24, 45–49 CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 CAS.
- N. W. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727–1735 CrossRef CAS.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521–1557 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- Y. J. Wang, J. L. Qiao, R. Baker and J. J. Zhang, Chem. Soc. Rev., 2013, 42, 5768–5787 RSC.
- C. Vogel and J. Meier-Haack, Desalination, 2014, 342, 156–174 CrossRef CAS.
- G. W. He, Z. Li, J. Zhao, S. F. Wang, H. Wu, M. D. Guiver and Z. Y. Jiang, Adv. Mater., 2015, 27, 5280–5295 CrossRef CAS PubMed.
- K. D. Kreuer, Chem. Mater., 2014, 26, 361–380 CrossRef CAS.
- J. Ran, L. Wu, Y. F. Ru, M. Hu, L. Din and T. W. Xu, Polym. Chem., 2015, 6, 5809–5826 RSC.
- C. G. Arges, L. Wang, J. Parrondo and V. Ramani, J. Electrochem. Soc., 2013, 160, F1258–F1274 CrossRef CAS.
- T. Xu, D. Wu and L. Wu, Prog. Polym. Sci., 2008, 33, 894–915 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- C. G. Arges, J. Parrondo, G. Johnson, A. E. Nadhan and V. Ramani, J. Mater. Chem., 2012, 22, 3733–3744 RSC.
- S. A. Nuñez and M. A. Hickner, ACS Macro Lett., 2012, 2, 49–52 CrossRef.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- E. A. Weiber and P. Jannasch, Macromol. Chem. Phys., 2016, 217, 1108–1118 CrossRef.
- L. Zhu, J. Pan, C. M. Christensen, B. Lin and M. A. Hickner, Macromolecules, 2016, 49, 3300–3309 CrossRef CAS.
- J. J. Han, Q. Liu, X. Q. Li, J. Pan, L. Wei, Y. Wu, H. Q. Peng, Y. Wang, G. W. Li, C. Chen, L. Xiao, J. T. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2015, 7, 2809–2816 CAS.
- L. Wang and M. A. Hickner, Soft Matter, 2016, 12, 5359–5371 RSC.
- M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161–1167 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- J. Wang, H. B. Wei, S. Z. Yang, H. G. Fang, P. Xu and Y. S. Ding, RSC Adv., 2015, 5, 93415–93422 RSC.
- H.-S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 CAS.
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS.
- Z. J. Yang, J. H. Zhou, S. W. Wang, J. Q. Hou, L. Wu and T. W. Xu, J. Mater. Chem. A, 2015, 3, 15015–15019 CAS.
- W. H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS.
- B. C. Lin, L. H. Qiu, B. Qiu, Y. Peng and F. Yan, Macromolecules, 2011, 44, 9642–9649 CrossRef CAS.
- Y. Z. Zhuo, A. L. Lai, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2015, 3, 18105–18114 CAS.
- M. Zhang, J. L. Liu, Y. G. Wang, L. A. An, M. D. Guiver and N. W. Li, J. Mater. Chem. A, 2015, 3, 12284–12296 CAS.
- W. H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CAS.
- A. Alaaeddine, F. Boschet and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1721–1729 CrossRef CAS.
- M. L. Di Vona, R. Narducci, L. Pasquini, K. Pelzer and P. Knauth, Int. J. Hydrogen Energy, 2014, 39, 14039–14049 CrossRef CAS.
- A. Katzfuss, V. Gogel, L. Jorissen and J. Kerres, J. Membr. Sci., 2013, 425, 131–140 CrossRef.
- C. G. Morandi, R. Peach, H. M. Krieg and J. Kerres, J. Mater. Chem. A, 2015, 3, 1110–1120 CAS.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61–71 CrossRef CAS.
- A. Wohlfarth, J. Smiatek, K. D. Kreuer, S. Takamuku, P. Jannasch and J. Maier, Macromolecules, 2015, 48, 1134–1143 CrossRef CAS.
- E. A. Weiber and P. Jannasch, ChemSusChem, 2014, 7, 2621–2630 CrossRef CAS PubMed.
- H. L. Hsieh, ACS Symp. Ser., 2009, 696, 28–33 CrossRef.
- M. Ingratta, M. Elomaa and P. Jannasch, Polym. Chem., 2010, 1, 739–746 RSC.
- A. J. Chalk and T. J. Hoogeboom, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 1359–1369 CrossRef CAS.
- A. J. Chalk and A. S. Hay, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 691–705 CrossRef CAS.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 CAS.
- J. Pan, C. Chen, Y. Li, L. Wang, L. S. Tan, G. W. Li, X. Tang, L. Xiao, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354–360 CAS.
- J. Pan, Y. Li, J. J. Han, G. W. Li, L. S. Tan, C. Chen, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912–2915 CAS.
- J. Pan, S. F. Lu, Y. Li, A. B. Huang, L. Zhuang and J. T. Lu, Adv. Funct. Mater., 2010, 20, 312–319 CrossRef CAS.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61–71 CrossRef CAS.
- D. Stoica, L. Ogier, L. Akrour, F. Alloin and J. F. Fauvarque, Electrochim. Acta, 2007, 53, 1596–1603 CrossRef CAS.
- C. C. Yang, S. J. Chiu, W. C. Chien and S. S. Chiu, J. Power Sources, 2010, 195, 2212–2219 CrossRef CAS.
- Y. H. Wu, C. M. Wu, T. W. Xu, F. Yu and Y. X. Fu, J. Membr. Sci., 2008, 321, 299–308 CrossRef CAS.
- N. W. Li, L. Z. Wang and M. Hickner, Chem. Commun., 2014, 50, 4092–4095 RSC.
- J. F. Zhou, M. Unlu, I. Anestis-Richard and P. A. Kohl, J. Membr. Sci., 2010, 350, 286–292 CrossRef CAS.
- E. N. Komkova, D. F. Stamatialis, H. Strathmann and M. Wessling, J. Membr. Sci., 2004, 244, 25–34 CrossRef CAS.
- J. J. Wang, G. H. He, X. M. Wu, X. M. Yan, Y. P. Zhang, Y. D. Wang and L. Du, J. Membr. Sci., 2014, 459, 86–95 CrossRef CAS.
- J. S. Park, S. H. Park, S. D. Yim, Y. G. Yoon, W. Y. Lee and C. S. Kim, J. Power Sources, 2008, 178, 620–626 CrossRef CAS.
- S. M. Zhang, C. P. Li, X. F. Xie and F. S. Zhang, Int. J. Hydrogen Energy, 2014, 39, 13718–13724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta01905f |
| This journal is © The Royal Society of Chemistry 2016 |