
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent status and challenges of biohydrogels for applications as supercapacitors and secondary batteries
Elaine
Armelin
*ab,
Maria M.
Pérez-Madrigal
ab,
Carlos
Alemán
ab and
David Díaz
Díaz
*cd
aDepartament d'Enginyeria Química, ETSEIB, Universitat Politècnica de Catalunya, Avda. Diagonal 647, E-08028, Barcelona, Spain. E-mail: elaine.armelin@upc.edu
bCenter for Research in Nano-Engineering, Universitat Politeècnica de Catalunya, Campus Sud, Edifici C', C/Pasqual i Vila s/n, E-08028, Barcelona, Spain
cInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de
dIQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain
First published on 3rd May 2016
Abstract
Progress in the chemical sciences has formed the world we live in, both on a macroscopic and on a nanoscopic scale. The last decade has witnessed the development of high performance materials that store charge in many ways: from solar cells to fuel cells, and from batteries to supercapacitor devices. One could argue that inorganic hybrid materials have played a central, starring role for the assembly of various electrochemical energy conversion systems. However, energy conversion systems fabricated from biopolymers has just emerged as a new prospect. Here, we summarize the main research results on the attractive use of biohydrogels for the fabrication of either conductive electrolytes or electrodes for battery science and technology.
 Elaine Armelin | Elaine Armelin is Associate Professor in the Chemical Engineering Department and in the Center for Research in NanoEngineering, at the Universitat Politècnica de Catalunya (UPC-Spain). She received her Bachelor's degree in Chemistry in 1995 and Master Degree in Organic Chemistry in 1997 at the Universidade de São Paulo (USP-Brazil). In 1997, she moved to Barcelona to perform her PhD studies in Polymer Science at UPC. Her current work focuses on the developing of hybrid materials with applications mainly within the life sciences and for energy storage, and this includes the development of new organic–inorganic coatings to protect metals and alloys against corrosion. |
 Maria M. Pérez-Madrigal | Maria M. Pérez-Madrigal received her BSc degree in Chemical Engineering (2010) and her MSc degree in Polymers and Biopolymers (2011) at the Universitat Politècnica de Catalunya (UPC), Barcelona, Spain. In 2010, she joined the “Innovation in Materials and Molecular Engineering” (IMEM) group in the Chemical Engineering Department at UPC. Under the supervision of Prof. Carlos Alemán and Dr Elaine Armelin, she conducted her PhD and graduated in 2015. Her research focused on combining conducting polymers (CPs) with conventional polymers to obtain biointerfaces at the nanoscale level for bioapplications. Currently, she is a postdoctoral member in the IMEM group working on developing supercapacitor devices based on CPs and hydrogels. |
 Carlos Alemán | Carlos Alemán graduated in Chemistry from the University of Barcelona (Spain). He received his PhD from UPC in 1994 wherein he was promoted to the position of Full Professor of Physical Chemistry. He was a postdoctoral researcher at ETH in Zürich (Switzerland) and visiting professor at the Università di Napoli Federico II (Italy), University of Twente (Holland) and Universidade Federal do Rio Grande do Sul (Brazil). Since 2003, he has been the leader of the IMEM group in the Chemical Engineering Department and in the Centre of Research on Nano-Engineering. His main research interests focus on cps and biopolymers with biomedical and technological (energy) applications. |
 David Díaz Díaz | David Díaz Díaz received his PhD in Chemistry from the University of La Laguna (Spain). In 2002, he joined Prof. Finn's group as a postdoctoral fellow at The Scripps Research Institute (USA). Since 2006, he has held various positions in academia and industry (‘Ramón y Cajal’ Researcher, UAM, Spain, 2006; Sr. Chemist, Dow Chemical, Switzerland, 2007; Tenured Scientist, CSIC, Spain, 2009; Alexander von Humboldt Experienced Researcher, University of Regensburg, Germany, 2010). In 2013, he was awarded with the DFG Heisenberg Professorship and appointed an Associate Professor at the University of Regensburg. He has received the Young Investigator Award from the Polymer Network Group and is the Editor-in-Chief of Gels. His main research interest focuses on functional soft materials. |
1. Introduction
Nature offers abundant examples of polymeric biomolecules from the aspects of both simplicity and complexity. Biopolymers are naturally occurring materials that have aroused tremendous interest for their application as useful engineered technical systems.As a matter of fact, obtaining and processing biopolymers are the key steps for the development of product from start to finish, which is the essence of all known current applications. However, the basic strategies followed to reach such steps are not related with biology, but more specifically related with polymer chemistry and physics. Essentially, three core aspects need to be explored for this purpose: (i) to elucidate structure–function relationships from the study of biomimetic model systems and to apply them to easily scale-up fabrication processes; (ii) to correlate ordered primary structures with the properties required for further engineering applications; and (iii) to discover new pathways for biopolymer processing that are expected to extend the scope of the natural system to design and develop new materials.
In fact, new approaches are almost solving all questions. Soft materials, like hydrogels, exhibit a number of unique properties. Among those properties, multi-functionality, biocompatibility, adaptability, responsiveness to stimuli, tunable tensile strength and elastic modulus deserve special attention.1 Hydrogels are not only model systems suitable for investigating natural phenomena, such as shape transformation, but also very promising components of hybrid materials that can acquire a pre-programmed shape and electroconductive properties for electrode and electrolyte production.
Such soft materials have been extensively explored and are widely used in diverse biomedical applications, for example: tissue engineering,2,3 drug delivery4–6 and molecular actuators.7–9 Furthermore, their applicability has been extended due to their self-healing10,11 response. In addition, they have new and ongoing recent applications in soft robotics12 and electronic devices like fuel cells, optoelectronic devices,13 organic batteries and supercapacitors.14–17 Indeed, they can be scaled-up using sustainable processes (i.e. using non-toxic solvents and low or moderate temperatures) based on their ability to be deposited as a liquid, enhancing production viability.
Regarding sustainable processes, Billiet et al.18 produced 3D gels based on gelatin methacrylamide activated with photoinitiators that were print-laden with HepG2 cells using a 3D-bioplotter. 3D printing has grown in sophistication since the late 1970s, otherwise 4D printing is the next fabrication process revolution that converts the 3D shape of one object to another ordered shape. The fourth dimension can be activated by time-, temperature-, mechanical- or water-stimulation that happens to hydrogels when they absorb water. Recently, Spinks and co-workers19 fabricated a smart valve by 4D printing of thermally actuating and mechanically robust hydrogels. These 4D structures, which involved an interpenetrating network of alginate and poly(N-isopropylacrylamide), were obtained by printing that “dynamic” hydrogel ink alongside other static materials. The result is a smart hydrogel actuator able to control the water flow dependent on the water temperature. Both methods, 3D and 4D, can be scaled-up for the preparation of supercapacitors and batteries based on biopolymer hydrogels (also called biohydrogels).
Supercapacitors (SCs), also called ultracapacitors, are electrochemical energy storage devices capable of providing higher power density and longer cycling life compared to batteries. Supercapacitors are formed by two non-reactive porous electrodes that are immersed in an electrolytic medium and electrically isolated by a membrane that is porous enough to enable the migration of ions (Fig. 1A). According to differences in the charge storage mechanism, SCs can be categorized into two families: electrical double layer capacitors (EDLCs) and redox capacitors. Both types present an interface (electrical double layer) between an electronic conductor and an ionic conductor, an electrolyte.16
 | ||
| Fig. 1 Schematic of: (A) a charged EDLC (AC: activated carbon, EDL: Electrical Double Layer, S: separator/porous membrane), and (B) example of the first-generation rechargeable lithium-ion cell. During charging, lithium ions flow to the negative electrode through the electrolyte and electrons flow from the external circuit. During discharge, the directions are reversed. Adapted with permission from ref. 23. Copyright © 2014 Royal Chemical Society. | ||
In a battery one or more electrochemical cells are electrically connected to supply energy. Depending on the energy consumption, batteries can be classified into two families. Primary batteries are intended to be used until exhausted and then discarded. Accordingly, discharge is the primary process during operation of such batteries and they are assembled in the charged state. In contrast, secondary batteries (also named rechargeable batteries or accumulators) can be restored to their original charged status after discharge. The original charged condition is recovered through an electric current flowing in the direction opposite to the flow of current when the cell was discharged (Fig. 1B).20 Hall et al.21 reviewed the application of the most studied materials used for electrical energy storage through batteries and SCs and all them were related to synthetic polymers and ceramic compounds.
Inganäs and Admassie22 recently reported the need to implement renewable energy technologies based on solar energy conversion and storage to facilitate the development of regions near to the equator with rapid economic growth and large populations. More specifically, these authors proposed novel SCs and batteries using organic materials. They emphasized that these technologies would have an enormous impact on poverty eradication.
Storing charge in SCs, or in secondary batteries, constructed using renewable materials is very attractive from an environmental point of view. Hence, replacement of organic solvents by water-containing gel-polymer electrolytes is highly desirable. The use of porous hydrogel nanostructures may provide the advantages of compactness, freedom of liquid leakage, excellent solid–liquid interface, ease of processability, larger electrochemically active surface area, shortened pathways for charge/mass transport and effective accommodation of the strain caused by intensive cycling electrochemical processes. In addition, this strategy results in a significant reduction of the device cost. Unfortunately, water-containing gel-polymer electrolytes, called hydrogel-polymer electrolytes, limit the operating potential window, which is of only ∼1.23 V. However, supramolecular biohydrogels are currently considered as an important part of the next-generation of materials to enter the era of SCs and batteries.
These investigations provide indication of the current relevance of biohydrogels in the field of energy storage devices. Accordingly, this review aims to establish the state-of-the-art for this field, paying special attention to areas where further research is urgently needed. Table 1 summarizes the main biopolymers reported in this review and the related references.












2. Polysaccharide-based hydrogels
Natural polysaccharides are being developed as alternatives to synthetic inorganic materials for electrochemical devices. Major attention has been recently paid to the fabrication of compact organic capacitors combining solid polymers with gel polymer electrolytes (GPE). For this purpose, hydroxylated, carboxylated, or sulfated-polysaccharides have been extensively used because of their high ionic conductivity and advantageous mechanical properties.242.1. Chitin and chitosan
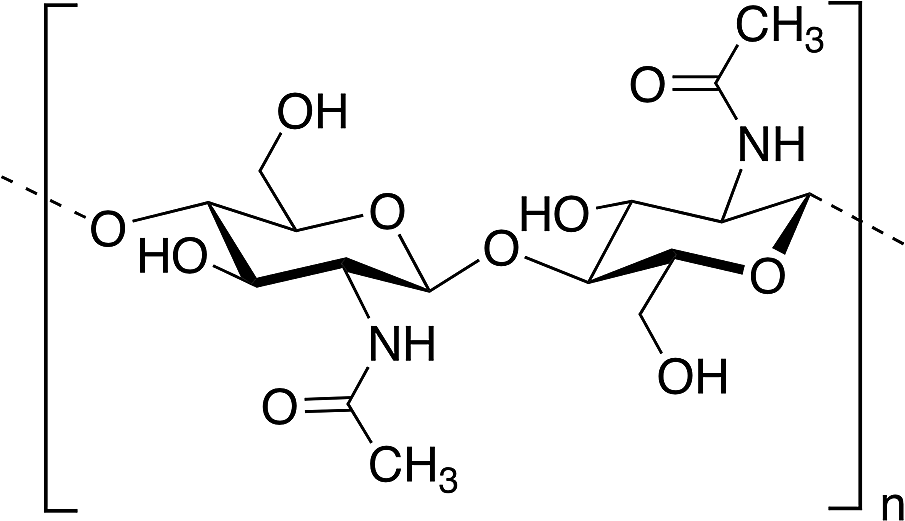
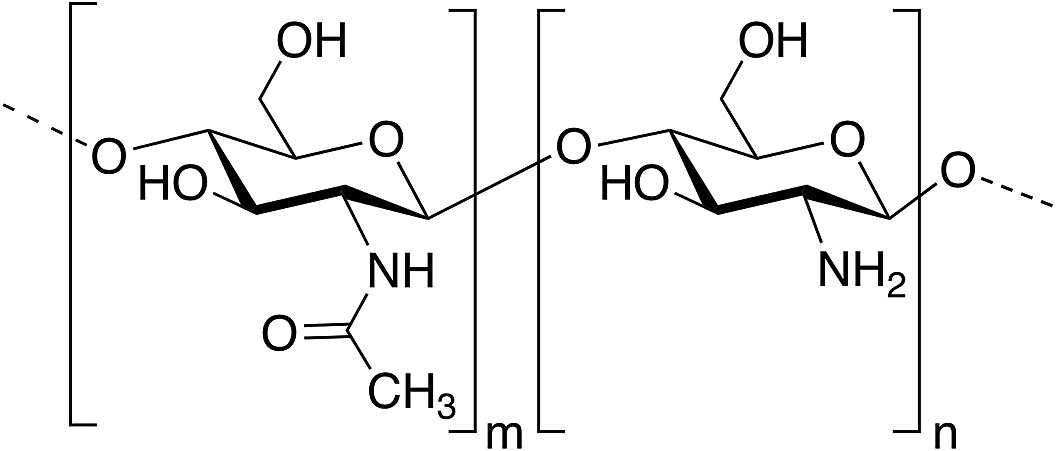
Among polysaccharides, chitin and chitosan (CS) have been explored for developing electrolytic mediums and conductive electrodes. Chitin, which is the second most abundant biopolymer in the biosphere after cellulose,25 is a linear polysaccharide of α- or β-(1-4)-2-acetamido-2-deoxy-D-glucopyranose where the repeating unit is N-acetylglucosamine.26 It is found as a structural material in, for example, fungal cell walls, insect and crustacean exoskeletons, mollusk radula, and cephalopod beaks.27 Man-made chitin waste coming from seafood processing is used to produce commercial chitin by harsh chemical treatments. Furthermore, biowaste from other sources, for example, mushroom, silk and honey-harvesting industries, has been recommended as a substitute for obtaining chitin. On the other hand, CS is derived from controlled chitin deacetylation. This material can be produced with a wide range of physical properties simply by varying the chain length of the precursor material, the degree of chemical depolymerization or the extent of deacetylation. Both chitin and CS bear nitrogen- and oxygen-containing functional groups that can be chemically modified, exhibit good biocompatibility and form uniform films and hydrogels. However, CS is, by far, more widely used as a biomaterial than chitin, including in hydrogel-based applications.27–29CS has been used for the preparation of solid-state proton-conducting polymer batteries. However, due to its insolubility in water, CS is dissolved in acetic acid to provoke ionic conductivity. The transport of protons from acetic acid occurs through microvoids inside the biopolymer, which was probed through piezoelectric studies.30 Different strategies have been explicitly developed for biopolymer processing and could be applied for the generation of chitin- or CS-based electrolytes or electrodes.27Fig. 2 depicts an example of such strategies.
 | ||
| Fig. 2 (A) Instrumental setup for electrodeposition of thin CS films on substrate electrodes and (B) the mechanism of CS precipitation onto the cathode. Both illustrate easy methods for obtaining biopolymer electrodes. Adapted with permission from ref. 27. Copyright © 2013 American Chemical Society. | ||
Among the pioneers in the preparation of either electrolytes or electrodes using biohydrogels for electrochemical energy-storage cells, Subramanian and co-workers31 deserve special mention. These researchers designed a solid-state EDLC combining a novel, cost-effective and eco-friendly electrode binder made of CS chemical hydrogel (CCH) and a separator consisting of an ionically cross-linked CS hydrogel membrane electrolyte (ICCSHME). The EDLC, which was prepared using 40% loading of CCH electrode binder and ICCSHME with 1 M NaOH dopant, was assembled with black carbon pearls as the electrode. The EDLC was cycled between charged and discharged states at constant current until failure. Fig. 3 reflects that such a capacitor is stable during operation between 20 and 200 cycles. However, the capacity decreased with increasing number of cycles, even though it was not definitely exhausted until a large number of cycles (i.e. from 500 to 1400). This evidenced that the EDLC model works well, but the charging/discharging stability needs improvement.
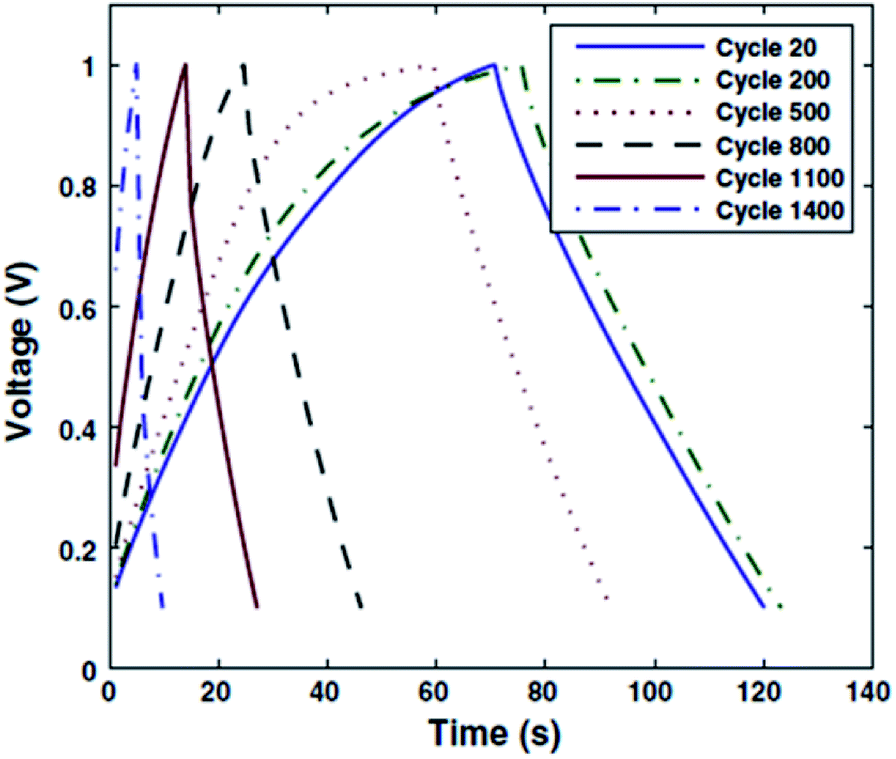 | ||
| Fig. 3 Charge/discharge plots after a determined number of cycles for an EDLC capacitor prepared as reported in ref. 31. Reprinted with permission from ref. 31. Copyright © 2012 Springer. | ||
The same authors studied the design of polymer electrolyte membranes based on an ionically cross-linked CS hydrogel prepared in ref. 31 to fabricate fuel cells.31 In this case, CS was chemically doped by the addition of sodium sulfate and sodium hydrogen phosphate. The operating cell voltage decreased only 100 mV, from about 0.8 to 0.7 V during the 100 h period of continuous operation, thus implying a good fuel cell performance and durability.32 The application of CS to other energy storage devices needs to be explored.
2.2. Cellulose and derivatives
Cellulose is another powerful candidate for the design of new electrolytes for electronic devices. This is the most abundant biopolymer in nature. Cellulose was discovered in 1838 by the French chemist Anselme Payen, who isolated this biopolymer from plants and determined its chemical formula. Cellulose is a good candidate for electrode and electrolyte preparation since it strongly binds to organic and inorganic materials such as conducting polymers (CPs),33 graphene oxide (GO)34 and carbon nanotubes (CNTs).33Development of environmentally friendly methods for “green” cellulose extraction, and approaches for suitable cellulose dissolution, are requirements for the green use of this biopolymer. Within this context, several researchers have used ionic liquids, such as 1-butyl-3-methylimidazolium chloride and 1-allyl-3-methylimidazolium chloride, to dissolve cellulose. Another interesting approach is microwave heating, which significantly accelerates the cellulose dissolution process and allows the separation of cellulose from other lignocellulosic materials like lignin and hemicellulose. The main “green” approaches were reviewed early on by Zhu et al.35
In addition, several cellulose derivatives, such as hemicellulose and hydroxyethylcellulose (HEC), have also been explored in the field of capacitors. Hemicellulose consists of a branched polymer bearing 500–3000 sugar units per polymer chain, whereas cellulose is unbranched and involves 7000–15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 glucose units per polymer chain. Recently, electrically conductive hemicellulose hydrogels (ECHHs) have been prepared by a one-pot reaction between biocompatible O-acetyl-galactoglucomannan and an aniline pentamer (AP) using epichlorohydrin as a crosslinking agent. This simple and green approach, which occurs in water and at ambient temperature, is illustrated in Fig. 4.36
000 glucose units per polymer chain. Recently, electrically conductive hemicellulose hydrogels (ECHHs) have been prepared by a one-pot reaction between biocompatible O-acetyl-galactoglucomannan and an aniline pentamer (AP) using epichlorohydrin as a crosslinking agent. This simple and green approach, which occurs in water and at ambient temperature, is illustrated in Fig. 4.36
 | ||
| Fig. 4 Schematic of ECHHs using epichlorohydrin as a cross-linker in basic media. Reprinted with permission from ref. 36. Copyright © 2014 American Chemical Society. | ||
Albertsson and co-workers36 found that the presence of AP inside the biohydrogel allows the preparation of a p-type semiconductor. The presence of holes and its concentration was found to increase with the AP, thus improving the conductivity. Those results represented an important achievement for the development of new biohydrogel electrolytes from hemicellulose materials.
Rosi et al.37 combined hydrogel-polymer electrolytes based on polyvinyl alcohol (PVA) with HEC for the preparation of capacitors. HEC products are water-soluble polymers with particular abilities. They are thick, retain water, form films, exhibit pseudoplastic solution behavior, and tolerate salts. The content of HEC in the hydrogel polymer electrolyte was 1, 2.5, and 5 wt%. The highest capacitance (160 F g−1) was obtained using the hydrogel with 1 wt% of HEC and corresponded to the electrolyte with the lowest ohmic resistance, which was attributed to the rapid ionic transport offered by the O–H groups throughout the chain of HEC in the electrolyte. However, the specific capacitance decreased with increasing concentration of HEC. This behavior was related to the reduction of free-volume in the hydrogel-polymer electrolyte with 2.5 and 5 wt% HEC. Those observations were in accordance with the results of Sampath et al.,38 who used PVA-based membranes doped with HClO4 as electrolytes for electrochemical capacitors. It should be noted that the specific capacitance reported by Rosi et al.37 was similar to that obtained with synthetic organic gels (about 150–250 F g−1), even though such values are not yet competitive with those described for metal oxides (about 700–1500 F g−1).17,39
On the other hand, bacterial cellulose (BC) is an interesting eco-friendly biomaterial produced by various microorganisms (e.g. Acetobacter xylinum, Pseudomonas, Rhizobium, and E. coli) that can be produced at the industrial scale via the microbial fermentation process.40,41 BC is made of ultrafine nanofibers, with a diameter of less than 100 nm, arranged forming an ultrafine network that confers unique properties (i.e. porosity, high biodegradability, high tensile strength, and high water retention capabilities). Renewable, biodegradable and electro-conducting composites have been obtained by combining BC with CPs (e.g. polyaniline (PAni) and polypyrrole (PPy)). The mass specific capacitance of these materials, which were obtained by in situ polymerization of the corresponding monomers, ranges from 273 F g−1 to 316 F g−1 at 0.2 A g−1.42,43 These interesting results obtained with BC will be addressed in detail in Section 4.
Overall, the research developed up to now demonstrates that natural cellulose, and its derivatives, can be employed to avoid the presence of toxic and volatile organic solvents in the battery electrode fabrication process while being compatible with the aqueous processing of hydrogel electrodes.
2.3. Alginate
Alginates are a family of water-soluble polysaccharides (carbohydrate polymers) extracted from brown seaweed. These materials were commercialized over 50 years after they were first investigated in the late 19th century by the British chemist E.C. Standford. Alginates have been mainly used by the pharmaceutical industry for specific gelling, thickening, and stabilizing applications.From the structural point of view, alginates are heteropolysaccharides with a block-copolymer structure that involves building blocks made of mannuronic acid (M) and guluronic acid (G) monomers. Thus, the block-copolymer is composed of longer homopolymeric blocks of M and G alternating with shorter blocks with a sequential M–G distribution. The proportion, distribution, and length of such blocks are crucial because of their implications in the material properties. While G-blocks are responsible for the gel-forming capacity, MM and MG units provide flexibility to the polymer chains. The composition of the blocks depends on the species used for extraction and the part of the thallus from which extraction is made. The acid that is extracted from seaweed is converted into the sodium salt, which is then extracted from the thick slurry, heavily diluted, and filtered to remove the unwanted inorganic residues. Sodium alginate (SA) obtained using this procedure can then be used to produce other useful salts such as potassium and calcium alginates. Nowadays, the main alginate producers are Scotland, Norway, China and the USA, followed by Japan, Chile, and France.
The number of studies associated with the application of alginate hydrogels for energy-storage devices is very scarce. These studies are mainly devoted to the development of new electrodes and the improvement of the capacity cycle-life in Li-ion batteries. Taking into account the electrode materials used for the fabrication of lithium-ion batteries, there is enormous interest in solving the failure mechanisms, such as electrode pulverization and cracking, associated with charge/discharge cycles. Although the specific capacity of silicon is ten times higher than that of graphite, anodes made of the former material undergo fast capacity decay and short cycle-lives. This has been attributed to the volumetric expansion experienced by silicon upon full lithium insertion (lithiation) and the significant contraction upon lithium extraction (delithiation). These drastic volumetric variations can provoke cracking and pulverization in the electrode, leading to both loss of electrical contact and excessive solid–electrolyte interphase growth.44 Accordingly, the development of more resistant electrode materials has become necessary. Different materials have been proposed as polymeric binders for Si-based anodes: poly(divinylidene fluoride) (PVDF), carboxymethyl cellulose (CMC), poly(acrylic acid) (PAA), polyamide imide, polyamide amine,44 SA, etc. Kovalenko et al.45 provided evidence that Si nanoparticles coated with a SA binder result in a stable silicon electrode with a volumetric capacity (∼1520 mA h cm−3) higher than that obtained using other binders, such as PVDF, PAA, and CMC.
Thus, SA had a positive impact on the cycling performance of the Li-ion battery. Fig. 5 compares the electrochemical behavior of SA with that of the binder-free electrode as well as with those obtained using other polymeric binders. The electrochemical stability of silicon nanoparticles coated with SA does not decrease after the first cycle, behaving as electroactive as the binder-free electrode (Fig. 5d).
 | ||
| Fig. 5 Electrochemical performance of alginate-based nano-Si electrodes (electrode density = 0.50 g cm−3, weight ratio of Si:C = 3:1). (A) Reversible Li extraction capacity and coulombic efficiency (CE) of the nano-Si electrodes versus cycle number for Li insertion level fixed to 1200 mA h g−1 Si. (B) Reversible Li extraction capacity of nano-Si electrodes with alginate, CMC, and PVDF binders versus cycle number collected for the current density of 4200 mA g−1 for cells cycled in the potential window from 0.01 to 1 V versus Li/Li+. (C) Galvanostatic discharge profiles of the nano-Si electrode at different current densities between 0 and 1 V. (D) Differential capacity curves of the nano-Si electrode in the potential window from 0 to 1 V versus Li/Li+ collected at a rate of 0.025 mV s−1 after the first galvanostatic charge–discharge cycle. All electrochemical measurements were performed at room temperature in a two-electrode coin-type half-cell. In (A), the capacity is reported for the Si contribution only. In (B) to (D), the capacity is normalized by the total weight of Si and C additives. Adapted with permission from ref. 45. Copyright © 2011 American Association for the Advancement of Science. | ||
In general, conventional hydrogels cannot be used as electrode materials because of their inherent poor mechanical integrity. However, the chemical structure of SA, which contains a large number of hydroxyl groups, facilitates its application not only as polyelectrolyte, but also as an electrode material in the presence of other conductive particles (e.g. CPs, carbon black, and metal particles). Thus, the favorable mechanical strength of SA hydrogels with conductive particles has been attributed to (i) the inter- and intramolecular hydrogen bonding interactions between SA chains and (ii) the attractive electrostatic interactions inside the entanglement among SA chains and the conductive particles, the latter being also responsible for the conductive properties. The fabrication of CP-SA hydrogel electrodes for supercapacitors and the synergistic interaction between SA and CP will be discussed in Section 4.
2.4. Other polysaccharides
The poor mechanical properties of commonly used hydrogels, discussed before, represent a drawback for the fabrication of stable electrolyte and electrode systems for SCs and secondary batteries devices. However, Moon et al.46 recently reported a simple procedure to prepare a mechanically robust NaCl–agarose-gel (AGAR)-mediated MnO2 hybrid electrolyte. This system displayed high potential for large scale fabrication (scale-up) of cost-effective, safe, nontoxic, and flexible SCs (Fig. 6). An agarose-based hydrogel that is composed of agar with an agarobiose monomeric unit,47 features submicrometer-sized pores (sizes of 400–500 nm), high elasticity (elastic modulus of 116 kPa), low-cost production, and environmental benignity. | ||
| Fig. 6 (A) Schematic showing the fabrication of an agarose-gel-mediated MnO2 symmetric cell assembly. (B) (a) Optical image and (b) SEM surface image of agarose gel; (c) optical image and (d) SEM surface image of MnO2 thin film deposited onto ITO–PET substrate. (C) Charge/discharge profile of NaCl–AGAR supercapacitor assembled between two electrodeposited MnO2 electrodes. Adapted with permission from ref. 46. Copyright © 2015 American Chemical Society. | ||
Among the advantages of NaCl–AGAR, its transparency deserves special mention. In order to characterize the implications of its flexibility, a one-planar interdigitated SC prototype was designed to check the electrochemical response upon compressive and tensile bending.46 Analyses, which were performed combining cyclic voltammetry and electrochemical impedance measurements, did not show significant deviations in the electrochemical behavior. The excellent supercapacitive behavior of the flexible NaCl–AGAR-based supercapacitor as a full cell was attributed to the porous structure of the material that provided several advantages for ion transport (Fig. 6b). Charge and discharge galvanostatic curves displayed symmetric triangular forms with a small internal resistance drop in a broad range of current densities, demonstrating excellent rate capabilities (Fig. 6c).46 Additionally, the authors fabricated NaCl–AGAR supercapacitors enveloped with poly(ethylene terephthalate) (PET) sealant tape and sticky polypropylene tape. These devices revealed an enhanced ability to maintain normalized capacitances, as revealed by the very slight reduction observed after 600 cycles, as well as retaining 80% of the maximum capacity after 1200 cycles. In contrast to the high stability of enveloped SCs, open systems presented a continuous decay after only 400 cycles (Fig. 7). Accordingly, Moon et al.46 proved that the fabrication of almost 100% biohydrogel SCs is feasible and needs further study.
 | ||
| Fig. 7 (A) Long-term cycling stability test profile of enveloped and non-enveloped NaCl–AGAR SCs performed at a scan rate of 100 mV s−1 and (B) digital image of an LED activated by the NaCl–AGAR SC. Reprinted with permission from ref. 46. Copyright © 2015 American Chemical Society. | ||
Sudhakar et al.24 employed guar gum (GG) as the gel polymer electrolyte (GPE) for the fabrication of SCs. GG is a non-ionic, hydrophilic, galactose- and mannose containing polysaccharide and belongs to the galactomannans family. The linear backbone involves β 1,4-linked mannose residues while galactose residues form short side-branches through 1,6-links to every second mannose. GG forms highly viscous solutions when hydrated in cold water. This unique property is crucial for the use of GG as GPE in SCs cells.
For construction of the SC cell, Sudhakar et al.24 used GG as the gel and binder, glycerol as the plasticizer, lithium perchlorate (LiClO4) as the dopant salt, and AC derived from areca fibers (an abundantly available fiber extracted from Palmecea plants) as the electrode material. The GPE was sandwiched between two prepared AC coated electrodes. The unit cell was sealed in a plastic coated aluminum pouch keeping the two wires outside. Amazingly, the GG gel exhibited an unusual tubular array-like morphology without crystalline and/or phase separation, and showed a great stability in aqueous medium. Unfortunately, the capacitor charge–discharge profile, measured by the galvanostatic method, showed evidence that the energy density and power density were not remarkable in comparison to other biohydrogels.
Channel type mesoporous architecture represents an adequate morphology for high power SC electrode applications. This has promoted the exploration of sophisticated materials, especially hybrid organic–inorganic compounds. Wahid et al.48 reported the template based synthesis of supercapacitive carbon by infiltrating a natural hydrogel, pectin, into a mesoporous silica template and, subsequently, applying pyrolysis in an inert atmosphere (SBA-15). Pectin is an indigestible soluble fiber that, combined with water, forms colloidal systems and gels and are frequently used as ingredients in food technology (e.g. it provides smooth and sticky texture to jellies and jams). From a chemical point of view, pectin is a biopolymer rich in 1,4-linked α-glucuronic acid units, having its carboxylic group esterified. This biomaterial exhibits higher performance than other gels (e.g. resorcinol, PVA, polyvinylpyrrolidone, agarose, or alginate) as a precursor for carbon synthesis because the concentration of hydroxyl groups is adequate to interact with water and with the template walls via hydrogen bonds. Indeed, pectin hydrogel gives place to unbreakable columns inside silica channels with less sideways diffusion, fulfilling all the necessary requirements for the transformation into one dimensional carbon threads. Wahid et al.48 doped the carbon-pectin activated electrodes with urea molecules, which also increased the viscosity due to the formation of urea-pectin chain interactions. Indeed, the compactness of the electrode increased with the viscosity since the sideways flow decreased, even at higher temperatures. This study clearly proved that the electrode morphology, with nanometer-sized mesoporous channels, exerts a crucial role in the performance of the capacitor (Fig. 8). On the other hand, Wahid et al.48 also compared the stability of pectin-containing electrodes in terms of capacitance by applying 2000 consecutive charge–discharge cycles. Si-carbon-pectin electrodes (PU-SBA11) retained 96% of their capacitance at a current density of 10 A g−1 (Fig. 9). This cycling stability was higher than that observed for other activated carbon electrodes prepared from agarose and alginate gels (AgU-SBA11 and AlU-SBA11, respectively). Similarly, electrodes prepared using pectin gels had better electrochemical behavior than those derived from other gels. This superiority was attributed to the particular chemical structure of pectin, which presents additional hydrogen bonding centers in comparison with agarose and alginate.
 | ||
| Fig. 8 TEM micrographs, with increasing magnification from (A) to (C), of pure SBA-15 template-prepared with pectin-urea. Adapted with permission from ref. 48. Copyright © 2015 Royal Chemical Society. | ||
 | ||
| Fig. 9 Electrochemical performance comparison of three natural gels, namely, agarose (AgU-SAB11), alginate (AlU-SBA) and pectin (PU-SBA11), on a three assembly in 1 M H2SO4 with calomel as the reference electrode: (A) compares the CV behavior of the three systems at a scan rate of 50 mV s−1; and (B) compares charge–discharge characteristics at 10 A g−1 for the three biohydrogel systems studied in this work. Adapted with permission from ref. 48. Copyright © 2015 Royal Chemical Society. | ||
Overall, the results recently reported in the literature suggest that, in general, polysaccharides represent a versatile alternative to conventional metal-oxide materials currently employed to fabricate energy-storage devices.
3. Lignin and lignin-derived quinones
Lignin, which is composed of cross-linked phenolic polymeric chains, is the major byproduct of the paper and pulp industry because it is present in the cell walls of woody plant matter. For instance, the content of lignin in wood varies between 15% and 35% depending on the wood species. Within the field of environmentally friendly energy storage applications, there has been a great interest in developing sustainable organic electrodes using lignin as a low-cost carbon source, which entails high scalable and economic production processes. However, in the research found in the literature, lignin is not applied as a hydrogel in the design of SC and battery devices, but mainly as the carbon precursor to obtain carbonized fibers mats49–53 or aerogels (of lignin alone54,55 or combined with BC56)that act as the storing charge electrode through EDLC. These renewable lignin-based electrodes are highly porous with interconnected 3D architectures, thus exhibiting an electrochemical response similar to that displayed by commercial carbon-based anodes (i.e. high power and energy densities). Therefore, because the evolution towards a green energy storage technology is of vital importance, the above cited studies on lignin-based electrodes have been included in this review even though they are not hydrogel-like. Besides, to increase the specific capacitance of lignin-based electrodes by faradic reactions, they are modified with aniline57 or CPs, such as PPy,58 and PEDOT.59Finally, lignin derivatives, which are rich in phenol groups, can be further modified by oxidation processes to render polymers with a high content of quinone groups.60 These conjugated cyclic dione structures can store and exchange charge during redox processes; thus, they can further enhance the charge-storage capability of the biopolymer when also combined with PPy.61–63 In fact, there are plenty of works in the literature that use the redox function of quinone derivatives, such as hydroquinone and benzoquinone, to contribute to the pseudocapacitance of the energy storage device after molecule immobilization, or coating onto the electrode surface, or its dissolution in the electrolyte. Nevertheless, this line of investigation is out of the scope of this review.
4. Protein-based hydrogels
Gelatin is a combination of randomly coiled peptides and proteins that result from the hydrolysis of the collagen triple helix. Consequently, the chemical structure of gelatin resembles that of collagen, presenting a high amount of glycine, proline and hydroxyproline residues. Gelatin dissolves in hot water, forming a pale yellow, semi-transparent and viscous solution, while a hydrogel is obtained by cooling down below 308 K (34.8 °C). This biopolymer exhibits high stability against electrolytes, effective adhesion and easy dispersion into positive electrode materials. In spite of such advantageous characteristics, gelatin is frequently limited by its intrinsically poor mechanical strength with improvements being required through chemical cross-linking, as an example.Gelatin has been recently developed as a binder for graphite electrodes in Li-ion batteries.64 Cathodes using gelatin present better capacity and cyclability than those obtained using synthetic polymeric binders. On the other hand, Choudhury et al.65 developed gelatin hydrogel electrolytes (GHEs) by cross-linking an aqueous solution of gelatin with aqueous glutaraldehyde for application as the electrolyte in SC cells. These authors obtained light brown, semi-transparent, and mechanically strong free-standing films with a large volume of water absorbed in the polymer hydrogel matrix. The morphology of GHE films was found to change when current was applied, their charge–discharge capacity improving upon the addition of NaCl as a dopant. The ionic conductivity of pristine GHE was explained by considering that protons associated to amino, hydroxyl, and thiol groups, as well as readily removable small chain fragments, migrate through the hydrogel under the influence of a small electric field. On the other hand, the ionic conductivity in NaCl-doped GHEs is predominantly due to the free diffusion of hydrated Na+ and Cl− ions inside the biopolymer matrix. This effect is illustrated through the variation of the specific capacitance, which increased from 37.6 to 81.2 F g−1 when the concentration of NaCl in the GHEs was modified from 0 to 3 N.65 A comprehensive review of hydrogel-polymer electrolytes for electrochemical capacitors was reported by the same authors.17
Despite the tremendous improvements in the development of batteries and SCs, materials of different chemical nature are still necessary to assemble an electronic device for energy storage. Within this context, researchers have used both hard materials, such as metals, and very soft materials, such as hydrogels. However, the use of the most important biological molecule in life, DNA, exceeds even science fiction.
Luo and co-workers,66 from Cornell University, were pioneers in the preparation of mechanically stable DNA-hydrogels. This was achieved by modifying the biomolecule with a polymerase enzyme that provoked the elongation of DNA chains, weaving them non-covalently into the hydrogel. The new hydrogel, which was termed meta-hydrogel (recalling the concept of metamaterials), exhibited liquid-like properties when it was taken out of water and solid-like properties, when it was in water. DNA-based meta-hydrogels presented a hierarchical internal structure that was useful for different potential applications. Among them, the authors explored the behavior of the biohydrogel doped with gold nanoparticles in an electrical circuit, employing water as a switch (Fig. 10).
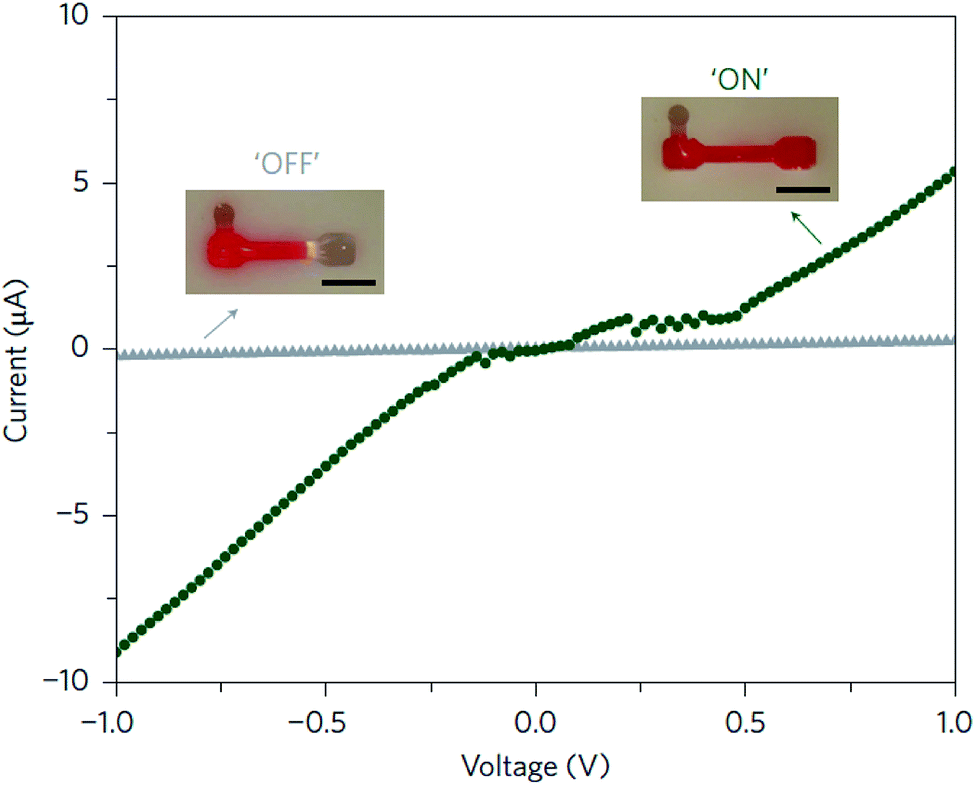 | ||
| Fig. 10 Electric circuit switch formed using the liquid- and solid-like properties of a DNA meta-hydrogel. When the DNA meta-hydrogel (containing 10 nm gold nanoparticles) has liquid-like properties, the circuit can be covered by the gel (green circles, ‘ON’). By simply adding water, the gel metamorphoses to its original shape, which is shorter, resulting in the gel rapidly moving away (within seconds) from the electrode and completely shutting off the current (grey triangles, ‘OFF’). Scale bars (inset), 5 mm. Reprinted with permission from ref. 66. Copyright © 2012 Macmillan Publishers Limited. | ||
Regarding the innovative research developed by Luo and co-workers,66 Hur et al.67,68 used the same concept to create SCs with layer-by-layer (LBL)-deposited polyelectrolyte multilayers (PEMs). These were prepared combining DNA hydrogel (Dgel) with either poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) or PAni (Fig. 11 and 12). These capacitors could be directly dipped in physiological fluids in a very small package form and be operated with great performances, which represent important advantages. Furthermore, these devices showed very stable galvanostatic charge–discharge cycling responses in artificial urine and PBS solutions. When the number of cycles reached 1000, the initial capacitance of PEM1.0-, PEM3.0-, and PEM5.0-DNA hydrogel (subscript numbers represent the repeated LBL PEM depositions) decreased by 71, 84, and 89%, respectively. It should be remarked that, although studies in this field are still at the initial stages, one can anticipate that successful applications in living organisms will be achieved in the near future.
 | ||
| Fig. 11 Schematic of: (A) PEM-Dgel SC operation in physiological environment (stacked features underneath the device represent cells); (B) enlarged detailed structure of PEM-Dgel where porous layered Dgel (grey) is covered by conductive PEM (indigo); (C) magnified view of Dgel where X-shaped double stranded DNAs are crosslinked by an enzymatic reaction; (D) construction of PEM-Dgel electrodes by coating the Dgel with the polyelectrolyte pair polydiallydimethyl-ammonium chloride and poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PDADMAC/PEDOT:PSS) using LBL deposition. Reprinted with permission from ref. 67. Copyright © 2015 Nature Publishing Group. | ||
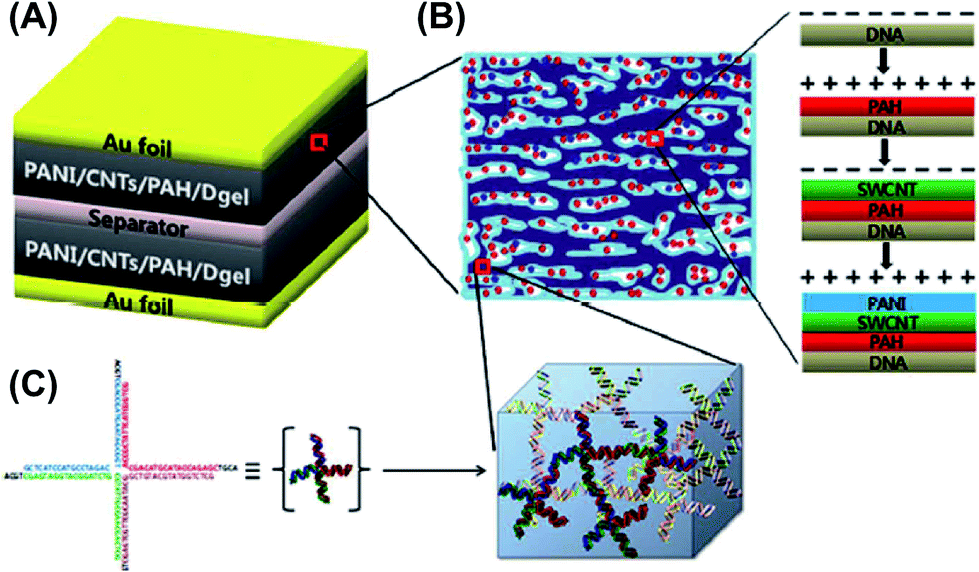 | ||
| Fig. 12 Schematic of the sophisticated DNA gel-based SC developed by Hur and co-workers. (A) Capacitor assembly was carried out by immersing the dried Dgel into the positively charged PAH solution. Negatively charged CNTs and positively charged PAni were electrostatically assembled to form a porous conductive and pseudocapacitance layer, respectively. Each film was performed by a conventional LBL deposition process on top of Au foils. (B) Details of the conversion of insulating Dgel into a conductive Dgel by electrostatic deposition of PAH, CNTs, and PAni explained in (A). (C) Representation of the DNA building block (X-shaped DNA) consisting of a Dgel phase. Adapted with permission from ref. 68. Copyright © 2013 Royal Chemical Society. | ||
5. Hydrogels made from biopolymers and electroactive conducting polymers
The main advantages associated with the incorporation of CPs into non-conducting hydrogels matrices are the achievement of large conducting surface areas and fast electron-ion transfers. Furthermore, CP-based biohydrogels fulfill the requirements of 3D-self-supported structures for anode and cathode manufacturing (i.e. they eliminate the need of binders).As it has been reported, PAni, PPy, and PEDOT improve the power density of SCs. Accordingly, a large number of works available in the literature describe the combination of CP with isolating synthetic polymers.69–72 Nevertheless, studies related to the synergistic relationship between biopolymers and CPs are very scarce.
In a recent work, we have reported a comprehensive study of the use of κ-carrageenan (κC), SA, CS and gelatin hydrogels as the electrolytic media, and PEDOT-supported stainless steel for the active electrodes.73 Among these green electrolytes, κ-carrageenan was the biohydrogel with the best electrochemical behavior, with good specific capacitance, small leakage current, and high cycling stability. The maximum energy density, the amount of energy stored per unit of mass, and the power density, which represents the speed at which the energy stored can be delivered, were 6.1 W h kg−1 and 1700 W kg−1 for the κC-PEDOT system, respectively. Fig. 13 shows the schematic representation of the complete SC system (A) as well as the four organic electronic SC OESC-κC devices connected in series to power a light emitting diode (LED) (B).
 | ||
| Fig. 13 (A) One electrochemical cell fabricated with κ-carrageenan as electrolyte and both counter-electrode (CE) and work-electrode (WE) made of PEDOT CP. The reference electrode (RE) was in direct contact with the WE in the charge–discharge experiment. (B) Four-assembled electrochemical cells, similar to that shown in (A), connected in series with electrical cables.73 | ||
As mentioned in Section 2.2, bacterial cellulose (BC) is one promising polysaccharide for use in energy storage devices. Within this context, highly conductive BC composites were prepared with PPy43 and PAni42,74 for SC applications. For this purpose, a simple procedure based on the in situ synthesis of the conductive electrodes by oxidative polymerization using BC as a template was employed. The resulting CP-coated BC nanofibrils formed uniform and flexible membranes due to the structure of BC, which consists on ribbon-shaped ultrafine nanofiber networks (Fig. 14). These studies provide a straightforward method to prepare conducting thin films with good mechanical properties that could be successfully used for the fabrication of flexible electrodes.
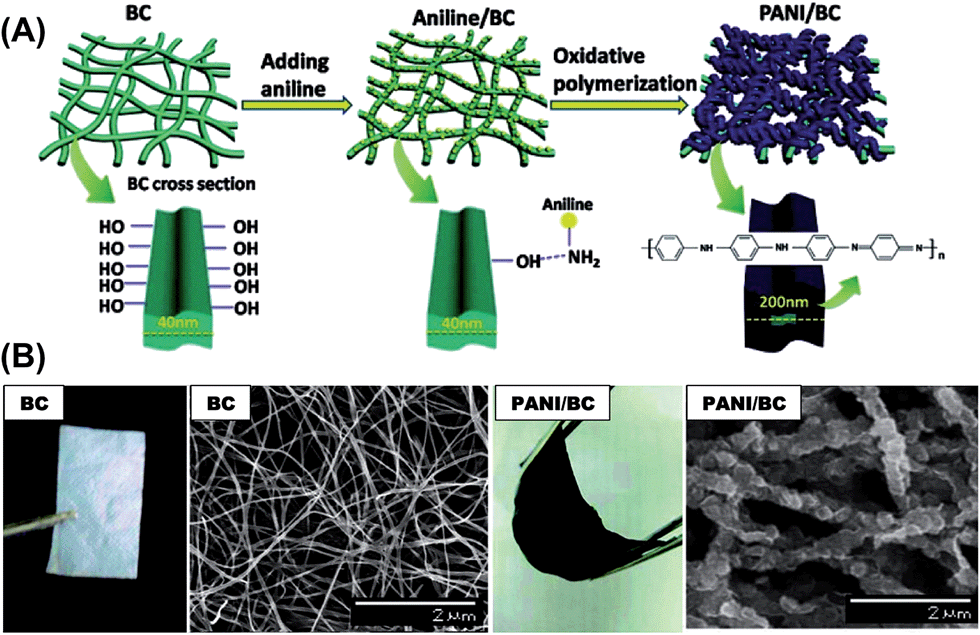 | ||
| Fig. 14 (A) Schematic showing the fabrication of a flexible BC/PAni nanocomposite membrane. (B) Optical images and SEM images of pure BC and the BC/PAni composites after 90 min of oxidative polymerization. Reprinted with permission from ref. 74. Copyright © 2011 American Chemical Society. | ||
Flexible films prepared with BC/PPy showed higher conductivity than BC/PAni (77 and 5.1 S cm−1 for the former and the latter, respectively).42,43 However, both the BC/PPy and BC/PAni hybrid electrodes were found to exhibit very high stabilities, retaining about 88.2% (278.7 F g−1) and 94.3% (234.7 F g−1) of the initial capacitance after 1000 cycles, respectively. This behavior was attributed to both the ordered structure of the composites and the strong interactions between the BC core and the CP shell. The high electrochemical stability of the BC/PAni and BC/PPy nanocomposites is illustrated in Fig. 15 by means of galvanostatic charge/discharge curves. The profiles registered during the charge and discharge processes are almost symmetrical, indicating good pseudocapacitance behavior for the two BC/CP electrodes.
 | ||
| Fig. 15 Galvanostatic charge/discharge plots of BC/CP composites at varied current density: (A) BC/PAni in 1 M H2SO4 solution and (B) BC/PPy in 0.5 M KCl solution. Reprinted with permission from ref. 42 and 43. Copyrights © 2012 American Chemical Society and 2013 Royal Chemical Society, respectively. | ||
SCs based on free-standing and flexible electrodes derived from BC and PAni and activated with multiwalled carbon nanotubes (MWCNTs) have also been prepared using an easy and scalable synthetic method.33 In this case, PAni deposition was performed by anodic polymerization.33 On the other hand, growth of PPy in cellulose paper by chemical polymerization also produced conducting materials. Such materials were successfully used to build SCs and batteries.20 Unfortunately, research using other CPs has been much less explored.
Among polysaccharides, alginate has been recently investigated with CPs, specifically SA. Li et al.75 described the electrochemical properties of PAni–SA nanofibers that were prepared by oxidative polymerization of aniline and ammonium persulfate previously introduced into a dilute solution of SA (0.025–0.2 wt%). The new conducting biohydrogel was used as an electrode material for SC assemblies. These PAni/SA electrodes exhibited an excellent specific capacitance (as high as 2093 F g−1), long cycle life, and fast reversible oxidation/reduction on high current charges. Such remarkable electrochemical characteristics should be attributed to the nanostructure of the electrodes, which presented high electrode/electrolyte contact areas and short path lengths for electronic transport and electrolyte ions. Fig. 16 displays the electrochemical response of the PAni/SA biohydrogels. This simple approach could be easily extended to fabricate nanostructured composites for supercapacitor electrodes or electrolyte materials.
 | ||
| Fig. 16 Galvanostatic charge/discharge curves of the (A) pure PAni electrode and the (B) PAni/SA electrode at various current densities in 1 M H2SO4; (C) cycling stability of PAni and PAni/SA electrodes. Inset shows the CV curves at 100 mV s−1 for PAni (left; black) and PAni/SA (right; red) after the first (solid line) and the 1000th (dashed line) charge/discharge cycle. Reprinted with permission from ref. 75. Copyright © 2011 American Chemical Society. | ||
Yang and co-workers61,76 used the same procedure (in situ polymerization of aniline in an aqueous solution of SA) to prepare reinforced PAni/SA conducting hydrogels. The main advantage of their system, which was based on the use of highly concentrated solutions of SA and aniline (2 wt%) in comparison to the dilute solution employed by Li et al.,75 was the achievement of mechanically stable gels (Fig. 17). After 1000 consecutive charge–discharge cycles, the retained specific capacitances and the compressive tablets of PAni/SA hydrogels with high content of SA were found to be 71% and 76%, respectively. The good cycling stability of PAni/SA hydrogels was due to their substantial mechanical strength. As discussed above, both the interactions between the components of the system and the 3D nanofibers network efficiently overcome the tendencies to expand and shrink during the charge–discharge processes, providing high stability to the capacitor device.76
 | ||
| Fig. 17 (A) Optical images of PAni/SA hydrogel formation and mechanical integrity. PAni/SA hydrogels showed excellent mechanical strength, samples maintained intact shapes even under a pressure of 200 g weight. (B) Schematic illustration of hydrogen bonds and van der Waals interactions within the PAni-SA polymeric chains. The synergy of the two interactions leaded to an increase in the hydrogel strength. Reprinted from ref. 76. Copyright © 2014 Royal Chemical Society. | ||
CP-based flexible SCs are among the most potential materials for energy storage devices due to their high redox performance, specific capacitance, and inherent elastic polymeric nature. There are several papers devoted to the study of flexible solid-state SCs with enhanced electrochemical performance made of CPs combined with synthetic hydrogels, like PVA and phytic acid,77,78 or graphene oxide.79
Within this context, Kaskel and co-workers80 recently reported an extraordinary stretchable PAni-containing SC with high capacitance retention (up to 92%) and remarkable cycling stability (up to 35000 cycles at a high current density of 8 A g−1). It is worth noting that the capacitance of previously reported PAni-based SCs decreased by 70–50% over 1000 cycles, which was attributed to swelling and shrinking of the CP during charge and discharge processes, respectively. Although the innovative system described by Kaskel80 was not fully bio-based, it represented an important advance in the combination of biohydrogel and synthetic polymer for the assembly of highly capacitive polymer electrodes. The biopolymer chosen for the hydrogel was α-cyclodextrin (αCD), a cyclic polysaccharide that contains six glucose units attached end-to-end via α-1,4 linkages. αCD presents a characteristic “micro heterogeneous environment” with a hydrophobic cavity and a hydrophilic exterior. In order to improve the mechanical integrity, these authors used polyacrylamide (PAAm) that was crosslinked with N,N′-methylenebis(acrylamide) and PAni. The whole system, called αCD-PAAm-PAni, was directly employed as formed, without the need of any binders (Fig. 18).
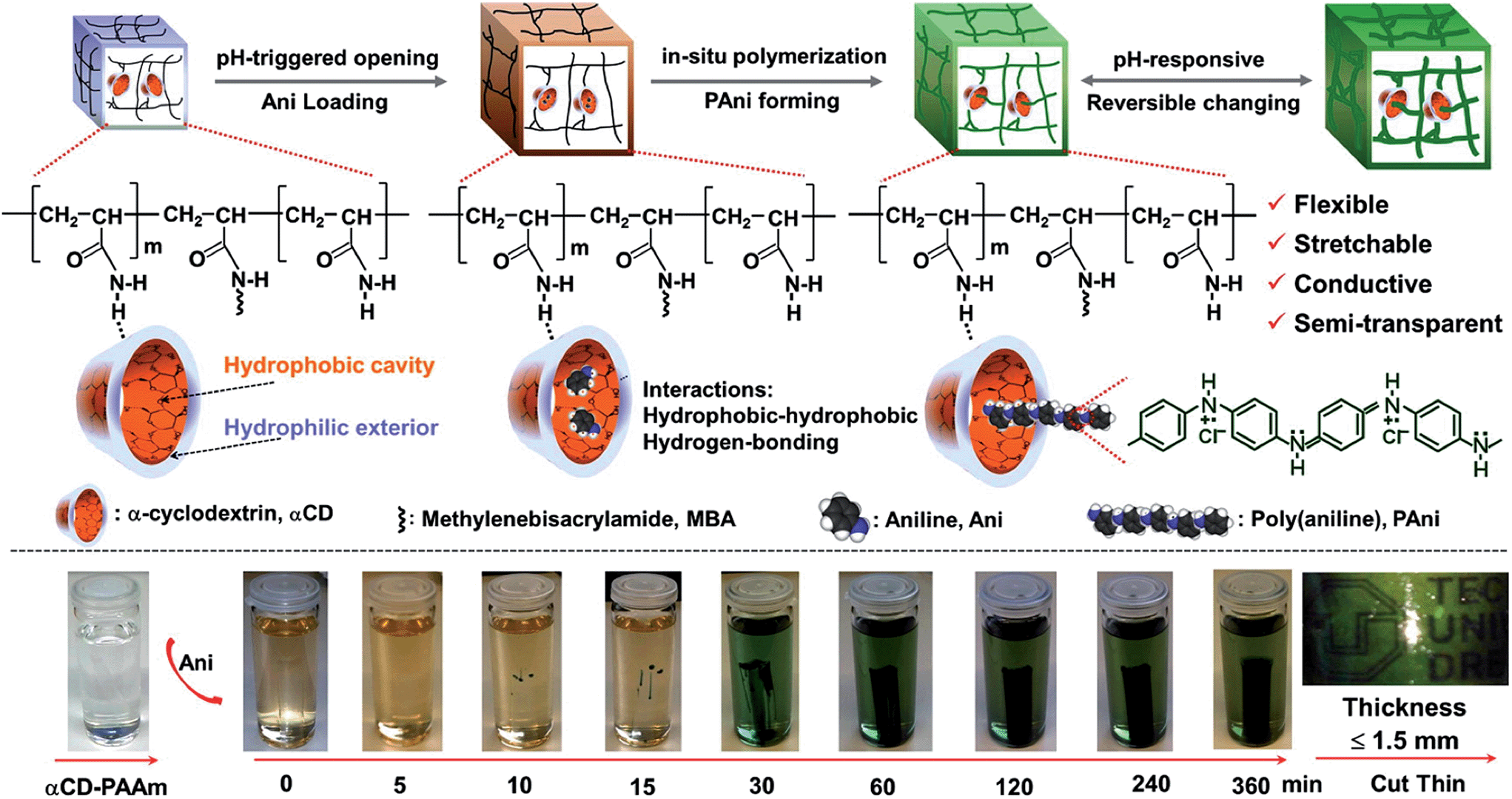 | ||
| Fig. 18 Schematic of the synthetic steps (top) and hydrogel formation (bottom) of the structurally flexible and stretchable electrically conductive αCD-PAAm-PAni. The synthesis is based on preorganized αCD-containing polyacrylamide hydrogels that are amphiphilic and pH responsive. The conductive polymer networks become semi-transparent for films with thickness ≤1.5 mm (bottom right). Reprinted with permission from ref. 80. Copyright © 2014 American Chemical Society. | ||
6. Structure-property relationships
The selection of an appropriate electrolyte and electrode material is fundamental to improving the performance of energy storage devices. Considering electrode fabrication, the most common form of electrode material for EDLC is activated carbon and its variety of forms.21 The electrode must have high surface area, low electrical resistance, good polarizability, and controllable pore size. With polymer-derived carbon electrodes, the porosity is one of the most critical parameters to be controlled and the activation process is expensive. With hydrogels, the porosity can be achieved by simply choosing a suitable biopolymer or modulating its processing. Additionally, larges surface areas are better achieved with hydrogels than with carbon-activated materials due to the easier preparation step. However, hydrogels must be converted into conducting materials with semiconducting particles, like oxide metals,37,42,43 or with the incorporation of CPs.73,80Besides ion diffusion, oxidative stability also affects the electrode's performance. Control of the hydrogel porous density and ion transport inside, either with conductive nanoparticles or with electroactive polymers, is particularly important because it influences the fraction of electrode material that is electrochemically accessible; and subsequently, the capacity, energy, and power of the EDLC devices. In this way, the dispersion of the conductive biopolymer part and the micro-morphology obtained play important roles.
Several works have provided evidence that 3D porous structures are better than 3D compact biopolymer films. Comparing electrochemical performance results obtained from cellulose derivatives and SA, the later present more ordered structure, regular distribution of porosity, and is able to accommodate conductive particles with good dispersability inside the polymer matrix. The dispersability is highly dependent on the chemical interactions in the hybrid material. Yang and co-workers76 specially described the beneficial intra/inter hydrogen bond interactions between the polar groups of SA with –NH groups from PAni monomer units. The same observation was done by Kaskel and co-workers80 when they employed α-cyclodextrin as hydrogel and PAni as the conductive network for flexible SCs. Controlled porosity and excellent hydrogel-conducting intramolecular interactions can increase the electrode's specific capacitance (which has a basis of mass), and the energy density (which has a basis of volume). Therefore, it is important to design an electrode's porosity to suit a particular application's needs.
With respect to the electrolyte media in SCs and batteries cells, the importance of micro- and meso-porosity obtained with biopolymers is still to be resolved. Major research is necessary to understand the relationship between electrode porosity and electrolyte interfaces.
In our recent work,73 we compared the efficiency of SA, κC, CS, and gelatin hydrogels as electrolytic media. The κC hydrogel exhibited a good SC response in terms of specific capacitance and cycling stability. Both SA and κC presented fibrillar structures, homogenously distributed pores, and charge transfer capability. Nevertheless, κC fibers were more robust than SA fibers,73 resulting in a 3D porous structure perfectly interconnected among fibers. Low crystallinity and a more brittle structure in the SA hydrogel compared to the flexible and mechanically stable electrolyte obtained with the κC hydrogel led to a better electrochemical response with the later than the former, despite the oxidative stability being similar in both cases.
Thus, in addition to morphology and complex intra/intermolecular interactions, anisotropy and crystallinity of biopolymers will strongly affect properties like charge mobility and charge–discharge efficiency in SCs and secondary battery devices.
7. Conclusion and outlook
This review compiles the recent advances in SC, battery science, and technology based on biohydrogel materials. The selection of articles reflects a combination of what is seen as impactful, thus opening up new directions and providing deep and important insights.Conductive, flexible and mechanically stable biohydrogels are the most promising candidates to replace metal inorganic and ceramic electrical storage devices. From the viewpoint of material design, there are various possible combinations of gel electrolytes and electrodes to improve energy storage and power density performances. Among the different types of biopolymers, cellulose derivatives (HEC, BC, cellulose nanocrystals and cellulose nanofibrils) seem to combine the ideal features for SCs and secondary battery fabrication, which include compactness, lightweight nature, high mechanical strength, and biocompatibility. Nowadays, it represents a sustainable route towards the large-scale use of energy storage devices (portable batteries for mobile phone, power for household appliances, electric cars, etc.) because it can be extracted directly, or eventually synthesized from biomass, or recycled through paper waste, simplifying the cycle life of rechargeable batteries and SCs.
Another interesting approach is the combination of biohydrogels with inherently CPs. Both materials retain their unique responsive properties in the final product. This perspective has described the synchronization of various remarkable characteristics of 3D nanostructured CP-biohydrogels, such as excellent conductivity, biocompatibility, processability, and mechanical flexibility for obtaining electrolyte and electrode materials.
However, considering that a conventional inorganic battery has a typical discharge time of 0.3–3 h, specific energy density of up to 230 W h kg−1, specific power density up to 1500 W kg−1, and cycle life of 500–2000; the effort to improve the overall energy and storage parameters using biopolymer systems is still ongoing. Specifically, one strategy recently being followed to enhance the energy density of biopolymer-based SCs and batteries relies on modifying the electrode material. Increased specific surface area and porosity, high conductivity, and well-connected channels all favor ion diffusion during the charging and discharging steps. Similarly, adding redox fillers in the electrolyte that contribute to the pseudocapacitance of the system also represents an attractive approach to improve the overall electrochemical performance of the energy storage device. Finally, superior cycling stability can be achieved if appropriate electrodes are prepared that are able to withstand the volumetric expansions and contractions experienced during cycling. Hence, flexible materials with suitable mechanical properties and less brittleness may undergo less drastic structural changes upon cycling, thus extending the long-term use of the device.
Fortunately, the research community at large clearly recognizes the need to push new advances in these topics by building on current research. It is an exciting opportunity to continue investigating in this direction: towards a more sustainable energy storage technology.
Acknowledgements
This work has been supported by MICINN and FEDER funds (MAT2015-69367-R) and by DFG (DI 1748/3-1). E. A. acknowledges the Ministry of Education, Culture and Sports of Spain for a research mobility grant (PRX14/00627). Support for the research of C. A. was received through the award “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. D. D. D. thanks the DFG for the Heisenberg professorship award.References
- A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed., 2011, 50, 9026 CrossRef CAS PubMed.
- A. Sivashanmugam, R. Arun Kumar, M. Vishnu Priya, S. V. Nair and R. Jayakumar, Eur. Polym. J., 2015, 72, 543 CrossRef CAS.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, J. Mater. Chem. B, 2014, 2, 5319 RSC.
- F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883 RSC.
- J. Li, Y. Kuang, Y. Gao, X. Du, J. Shi and B. Xu, J. Am. Chem. Soc., 2013, 135, 542 CrossRef CAS PubMed.
- A. Vashist, A. Vashist, Y. K. Gupta and S. Ahmad, J. Mater. Chem. B, 2014, 2, 147 RSC.
- H. J. Schneider and R. M. Strongin, Acc. Chem. Res., 2009, 42, 1489 CrossRef CAS PubMed.
- L. Ionov, Adv. Funct. Mater., 2013, 23, 4555 CrossRef CAS.
- E. Ye, P. L. Chee, A. Prasad, X. Fang, C. Owh, V. Jing, J. Yeo and X. J. Loh, Mater. Today, 2014, 17, 194 CrossRef CAS.
- H.-P. Cong, P. Wang and S.-H. Yu, Chem. Mater., 2013, 25, 3357 CrossRef CAS.
- M. D. Golinska, M. K. Wlodarczyk-Biegun, M. W. T. Werten, M. A. C. Stuart, F. A. de Wolf and R. de Vries, Biomacromolecules, 2014, 15, 699 CrossRef CAS PubMed.
- D. Morales, E. Palleau, M. D. Dickey and O. D. Velev, Soft Matter, 2014, 10, 1337 RSC.
- H. A. Al-Mohsin, K. P. Mineart and R. J. Spontak, Adv. Energy Mater., 2015, 5, 1401941 Search PubMed.
- L. Li, Z. Wu, S. Yuan and X.-B. Zhang, Energy Environ. Sci., 2014, 7, 2101 CAS.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 Search PubMed.
- X.-L. Wu and A.-W. Xu, J. Mater. Chem. A, 2014, 2, 4852 CAS.
- N. A. Choudhury, S. Sampath and A. K. Shukla, Energy Environ. Sci., 2009, 2, 55 CAS.
- T. Billiet, E. Gevaert, T. De Schryver, M. Cornelissen and P. Dubruel, Biomaterials, 2014, 35, 49 CrossRef CAS PubMed.
- S. E. Bakarich, R. Gorkin III, M. in het Panhuis and G. M. Spinks, Macromol. Rapid Commun., 2015, 36, 1211 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed.
- P. J. Hall, M. Mirzaeian, I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238 CAS.
- O. Inganäs and S. Admassie, Adv. Mater., 2014, 26, 830 CrossRef PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185 RSC.
- Y. N. Sudhakar, M. Selvakumar and D. K. Bhat, Mater. Sci. Eng., B, 2014, 180, 12 CrossRef CAS.
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1 CrossRef CAS.
- J. L. Arias and M. S. Fernández, Chem. Rev., 2008, 108, 4475 CrossRef CAS PubMed.
- W. Suginta, P. Khunkaewla and A. Schulte, Chem. Rev., 2013, 113, 5458 CrossRef CAS PubMed.
- Chitosan-based hydrogels: functions and applications, ed. K. Yao, J. Li, F. Yao and Y. Yin, CRC Press, Taylor & Francis Group, Boca Raton, USA, 2011 Search PubMed.
- Chitin and chitosan derivatives: advances in drug discovery and developments, ed. S. K. Kim, CRC Press, Boca Raton, FL, 2013 Search PubMed.
- L. Arof, R. H. Y. Subban and S. Radhakrishna, in Polymer and Other Advanced Materials: Emerging Technologies and Business, ed. P. N. Prasad, Plenum Press, New York, 1995, p. 539 Search PubMed.
- N. A. Choudhury, P. W. C. Northrop, A. C. Crothers, S. Jain and V. R. Subramanian, J. Appl. Electrochem., 2012, 42, 935 CrossRef CAS.
- N. A. Choudhury, J. Ma and Y. Sahai, J. Power Sources, 2012, 210, 358 CrossRef CAS.
- S. Li, D. Huang, B. Zhang, X. Xu, M. Wang, G. Yang and Y. Shen, Adv. Energy Mater., 2014, 4, 1 Search PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042 CrossRef CAS PubMed.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Dinga and G. Wu, Green Chem., 2006, 8, 325 RSC.
- W. Zhao, L. Glavas, K. Odelius, U. Edlund and A.-C. Albertsson, Chem. Mater., 2014, 26, 4265 CrossRef CAS.
- M. Rosi, F. Iskandar, M. Abdullah and K. Khairurrijal, Int. J. Electrochem. Sci., 2014, 9, 4251 Search PubMed.
- S. Sampath, N. A. Choudhury and A. K. Shukla, J. Chem. Sci., 2009, 121, 727 CrossRef CAS.
- W. Dmowski, T. Egami, K. E. Swider-Lyons, C. T. Love and D. R. Rolison, J. Phys. Chem. B, 2002, 106, 12677 CrossRef CAS.
- W. Czaja, D. Romanovicz and R. M. Brown, Cellulose, 2004, 11, 403 CrossRef CAS.
- Z. Schnepp, Angew. Chem., Int. Ed., 2013, 52, 1096 CrossRef CAS PubMed.
- H. H. Wang, E. W. Zhu, J. Z. Yang, P. P. Zhou, D. P. Sun and W. H. Tang, J. Phys. Chem. C, 2012, 116, 13013 CAS.
- H. H. Wang, L. Y. Bian, P. P. Zhou, J. Tang and W. H. Tang, J. Mater. Chem. A, 2013, 1, 578 CAS.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042 CrossRef CAS PubMed.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75 CrossRef CAS PubMed.
- W. G. Moon, G.-P. Kim, M. Lee, H. D. Song and J. Yi, ACS Appl. Mater. Interfaces, 2015, 7, 3503 CAS.
- M. Tako and S. Nakamura, Carbohydr. Res., 1988, 180, 277 CrossRef CAS.
- M. Wahid, G. Parte, R. Fernandes, D. Kothari and S. Ogale, RSC Adv., 2015, 5, 51382 RSC.
- R. Berenguer, F. J. García-Mateos, R. Ruiz-Rosas, D. Cazorla-Amorós, E. Morallón, J. Rodríguez-Mirasol and T. Cordero, Green Chem., 2016, 18, 1506 RSC.
- S.-X. Wang, L. Yang, L. P. Stubbs, X. Li and C. He, ACS Appl. Mater. Interfaces, 2013, 5, 12275 CAS.
- S. Hu, S. Zhang, N. Pan and Y.-L. Hsieh, J. Power Sources, 2014, 270, 106 CrossRef CAS.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X.-F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134 CrossRef CAS.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86 CrossRef CAS.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer III, S. Dai, M. P. Paranthaman and A. K. Naskar, Langmuir, 2014, 30, 900 CrossRef CAS PubMed.
- P. Hao, Z. Zhao, J. Tian, H. Li, Y. Sang, G. Yu, H. Cai, H. Liu, C. P. Wong and A. Umar, Nanoscale, 2014, 6, 12120 RSC.
- X. Xu, J. Zhou, D. H. Nagaraju, L. Jiang, V. R. Marinov, G. Lubineau, H. N. Alshareef and M. Oh, Adv. Funct. Mater., 2015, 25, 3193 CrossRef CAS.
- K. Wang, Y. Cao, X. Wang, M. A. Castro, B. Luo, Z. Gu, J. Liu, J. D. Hoefelmeyer and Q. Fan, J. Power Sources, 2016, 307, 462 CrossRef CAS.
- S. Admassie, A. Elfwing, E. W. H. Jager, Q. Bao and O. Inganäs, J. Mater. Chem. A, 2014, 2, 1974 CAS.
- F. N. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838 CAS.
- T. Y. Nilsson, M. Wagner and O. Inganäs, ChemSusChem, 2015, 8, 4081 CrossRef CAS PubMed.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468 CrossRef CAS PubMed.
- S. Admassie, T. Y. Nilsson and O. Inganäs, Phys. Chem. Chem. Phys., 2014, 16, 24681 RSC.
- T. Rębiś, T. Y. Nilsson and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1931 Search PubMed.
- Handbook of battery materials, ed. C. Daniel and J. O. Besenhard, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed.
- N. A. Choudhury, S. Sampath and A. K. Shukla, J. Electrochem. Soc., 2008, 155, A74–A81 CrossRef CAS.
- J. B. Lee, S. Peng, D. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. Chen, R. Long, M. Wu and D. Luo, Nat. Nanotechnol., 2012, 7, 816 CrossRef CAS PubMed.
- J. Hur, K. Im, S. Hwang, B. Choi, S. Kim, S. Hwang, N. Park and K. Kim, Sci. Rep., 2013, 3, 1282 Search PubMed.
- J. Hur, K. Im, S. W. Kim, U. J. Kim, J. Lee, S. Hwang, J. Song, S. Kim, S. Hwanga and N. Park, J. Mater. Chem. A, 2013, 1, 14460 CAS.
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C.-K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287 CrossRef CAS PubMed.
- Y. Zhao, B. Liu, L. Pan and G. Yu, Energy Environ. Sci., 2013, 6, 2856 CAS.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- L. Chen, Y. Chen, J. Wu, J. Wang, H. Bai and L. Li, J. Mater. Chem. A, 2014, 2, 10526 CAS.
- M. M. Pérez-Madrigal, F. Estrany, E. Armelin, D. Díaz Díaz and C. Alemán, J. Mater. Chem. A, 2016, 4, 1792 Search PubMed.
- W. Hu, S. Chen, Z. Yang, L. Liu and H. Wang, J. Phys. Chem. B, 2011, 115, 8453 CrossRef CAS PubMed.
- Y. Li, X. Zhao, Q. Xu, Q. Zhang and D. Chen, Langmuir, 2011, 27, 6458 CrossRef CAS PubMed.
- H. Huang, X. Zeng, W. Li, H. Wang, Q. Wang and Y. Yang, J. Mater. Chem. A, 2014, 2, 16516 CAS.
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086 CAS.
- K. Wang, X. Zhang, C. Li, H. Zhang, X. Sun, N. Xu and Y. Ma, J. Mater. Chem. A, 2014, 2, 19726 CAS.
- I. Shown, A. Ganguly, L.-C. Chen and K.-H. Chen, Energy Sci. Eng., 2015, 3, 2 CrossRef CAS.
- G.-P. Hao, F. Hippauf, M. Oschatz, F. M. Wisser, A. Leifert, W. Nickel, N. Mohamed-Noriega, Z. Zheng and S. Kaskel, ACS Nano, 2014, 8, 7138 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |