
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct photocatalytic hydrogen evolution from water splitting using nanostructures of hydrate organic small molecule as photocatalysts†
Huihui
Li
ab,
Liulun
Jie
ab,
Jiannan
Pan
ab,
Longtian
Kang
*ab and
Jiannian
Yao
*c
aKey Laboratory of Design and Assembly of Functional Nanostructures, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China
bFujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: longtiank@fjirsm.ac.cn
cBeijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: jnyao@iccas.ac.cn
First published on 30th March 2016
Abstract
Organic small molecules with a suitable energy level have usually been considered as photosensitizers rather than catalysts for photocatalytic hydrogen evolution (PHE). Herein, we achieved direct PHE using hydrate zinc tetraphenylporphyrin (ZnP, ZnTPP·H2O) nanostructures synthesized by a liquid-phase chemical reaction as photocatalysts. The shape-dependent photocatalysis revealed that the ZnP nanosheets (ZnP-NS) exhibit higher PHE activity (∼0.16 mmol g−1 h−1) than the ZnP octahedron nanoparticles (ZnP-NPs) (∼0.06 mmol g−1 h−1). After in situ construction of the rubrene/ZnP-NS heterostructure, more efficient PHE of this pure organic nanostructure was obtained due to the occurrence of photoinduced electron transfer and Förster resonance energy transfer (FRET). The optimal PHE rate is ∼0.56 mmol g−1 h−1. Furthermore, with the addition of 3.0 mM methyl viologen (MV) and 3.8 wt% platinum, a PHE rate of ∼9.3 mmol g−1 h−1 can be achieved at pH = 7. This study offers a new route to design organic small molecules as photocatalysts.
Introduction
Over the past half century, PHE from water splitting has attracted more and more attention because of the increasing worldwide energy shortage and environmental issues.1–7 With the rapid development of nanoscience and nanotechnology, obvious progress has been made in the photocatalysis of nano-semiconductors due to their abundant active sites and efficient absorption of solar light.1–5 To date, a variety of inorganic nanomaterials have been synthesized as photocatalysts such as transition metal oxides and metal sulfides.3–5 Although significant development has been achieved in the field of inorganic systems, there are few reports on PHE when organic small molecules nanostructures were used directly as photocatalysts rather than photosensitizers over the past two decades.6,7In recent years, π-conjugated organic semiconductors, particularly organic dye molecules with high light-harvesting properties, have attracted considerable attention as a result of their relatively low-cost and various stable modifiable structures.8–10 To take metal porphyrin (MP) for example, theoretically, the porphyrin macrocycle can not only offer a unique platform for the coordination of different metal atoms to tune the spectroscopic and/or electrochemical properties of the chromophore, but also stabilize the redox-active metal centre. These advantages indicate their potential applications in photoelectric transfer. In fact, compared to the normal hydrogen electrode (NHE), the reduction potential of many MPs from MP+ to MP excited stage (MP*), such as MgTPP, ZnTPP and AlTPP, is more negative and more positive than that from MP* to MP−.11 This means that these MPs may give photogenerated electron to reduce a proton. With the help of an appropriate sacrificial electron donor (SED), PHE may be achieved via the application of a Z-scheme.7,9,12,13 Although the porphyrin-based PHE has been widely studied, to the best of our knowledge, there are few reports on directly splitting water for PHE using MPs as photocatalysts rather than photosensitizers.9,14 Now, more efforts are seemingly focused on constructing a molecule system for PHE than nanostructures.
The effect of organic nanostructures with different shapes, size, crystal types and self-assembly on the optoelectronic properties have been studied widely.15–20 In particular, the recent study of the self-assembly of naturally-occurring molecules provides a new opportunity to investigate and simulate the function of biological systems. For example, Yan's group successfully adjusted the optical properties of porphyrin based on peptide-tuning self-assembly method.18–20 This inspired us to explore the role of self-assembling porphyrin-based nanostructures analogous to natural photocatalytic system. At present, few reports on the PHE properties of organic small molecule nanostructures can be attributed to three factors. The first is the difficult production of large-scale monodisperse organic nanostrucutures.21 The second is their hydrophobic essence of main body, which means difficult diffusion of water or protons to the reaction interface and high overpotential.10 The third is the higher Coulomb binding energy of Frenkel exciton existed in an organic system than Wannier exciton in inorganic semiconductors, which prevents photoinduced electrons and holes from effectively separating.2,6,22 To solve these tough problems, an entirely new strategy must be proposed. In this regard, we wondered whether these difficulties can be overcome by preparing hydrate organic nanostructures via a liquid-phase chemical reaction.16,23 In this way, a variety of monodisperse ZnP nanostructures can be synthesized on a large scale. It is very exciting that ZnP-NS with fully exposed crystal water on the crystal face can also be synthesized.23 Due to the existence of a hydrogen bond between ZnP and body-phase water via crystal water, in theory, ZnP-NS not only have well hydrophilicity, but also contribute to the transfer of a photoinduced electron from the metal atom on the porphyrin ring to proton/H2O.24 As far as the third problem was concerned, it was proved that the construction of a heterostructure is an effective route.25,26
Compared to hydrogen, ZnP has a more negative reduction potential from ZnP+ to ZnP* (whether single or triplet excited state) as well as a long triplet state lifetime, which implies that it may be a potential photocatalyst.11,12,27 For organic semiconductor rubrene, its far long diffusion distance of triplet excitons, more positive LUMO (−2.10 eV) than ZnP (−2.39 eV) and excellent optical properties make it suitable as a photosensitizer when recombined with ZnP.27–30 In this study, large amounts of ZnP-NS were synthesized via a liquid-phase chemical reaction and achieved direct PHE from water splitting when ZnP-NS and triethanolamine (TEOA) were used as the photocatalyst and sacrificial electron donor (SED), respectively. Through the in situ construction of rubrene/ZnP-NS heterostructure in the aqueous phase with ZnP-NS as templates, the efficiency of PHE was further improved. Moreover, it was also found that more efficient PHE can be observed when the system contains methyl viologen (MV) and H2PtCl6·6H2O.
Results and discussions
Fig. 1A shows the synthesis process of ZnP-NS and rubrene/ZnP-NS nanostructures. In a typical experiment, 1 mL of 1 mM ZnP perchlorate (10−6 mol) in acetonitrile was injected rapidly into 5 mL of an aqueous solution, including 0.5 M poly(vinyl pyrrolidone) (PVP) and 10 mM sodium nitrite (NaNO2), at room temperature.23 As shown in Fig. 1B by the scanning electron microscopy (SEM) images, the square nanoflakes with an edge length of ∼900 nm and a thickness of <100 nm can be well synthesized. After centrifuging and washing, ZnP-NS were redispersed in ultrapure water to form a 1.0 mg/30 mL colloid dispersion system after mild sonication. To fabricate the rubrene/ZnP-NS heterostructure, the precursor of rubrene perchlorate was first synthesized according to the reported method.31 After it was added to water, MALDI-TOF mass spectroscopy in Fig. S1† proved the formation of the rubrene NPs, as shown in Fig. S2.† Similarly, to obtain the composites of rubrene/ZnP with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6, 1:3 and 2:3, 1 mL of 1 mM rubrene perchlorate in acetonitrile was, respectively, added to 120 mL, 60 mL and 30 mL of a 1.0 mg/30 mL ZnP-NS aqueous dispersion and aged for 12 hours. The as-prepared samples were characterized by SEM, transmission electron microscopy (TEM), selected area and elemental mapping. Fig. 1C–E shows that with increasing molar ratio of rubrene/ZnP from 1:6, 1:3 to 2:3, the electron diffraction (SAED) average size of rubrene NPs increases from ∼100 nm, ∼150 nm to ∼200 nm and more detached rubrene NPs can be found, as marked by the red frames. This should result from the increase in rubrene monomer chemical potential after a rapid reaction with the decreasing volume of dispersion agent.23 Interestingly, Fig. 1F and inset maps of Fig. 1D and E show that the rubrene NPs prefer to form at the edge of two smooth faces of ZnP-NS. This was attributed to the existence of rough faces on the edge of the ZnP-NS and higher reduction potential of rubrene+/rubrene (0.9 eV vs. SCE) than ZnP+/ZnP (0.71 eV vs. SCE), which means not only the reaction between the rubrene cation and ZnP, but also the preferred nucleation of rubrene at these rough faces with high surface energy.11,31 In addition, TEM image in Fig. 1F shows the close contact between the rubrene NPs and ZnP-NS, indicating the in situ formation of rubrene NPs on the surface of the ZnP-NS. The corresponding ED pattern in Fig. 1G confirms that a smooth crystal face of ZnP-NS can be indexed to (020) with fully exposed crystal water (see Fig. 1A). The nanosheet in Fig. 1H and relevant Zn element mapping in Fig. 1I further verify the synthesis of a high-quality rubrene/ZnP-NS nano-heterostructure and the morphology of ZnP remains unchanged after the formation of rubrene via chemical reaction.
:6, 1:3 and 2:3, 1 mL of 1 mM rubrene perchlorate in acetonitrile was, respectively, added to 120 mL, 60 mL and 30 mL of a 1.0 mg/30 mL ZnP-NS aqueous dispersion and aged for 12 hours. The as-prepared samples were characterized by SEM, transmission electron microscopy (TEM), selected area and elemental mapping. Fig. 1C–E shows that with increasing molar ratio of rubrene/ZnP from 1:6, 1:3 to 2:3, the electron diffraction (SAED) average size of rubrene NPs increases from ∼100 nm, ∼150 nm to ∼200 nm and more detached rubrene NPs can be found, as marked by the red frames. This should result from the increase in rubrene monomer chemical potential after a rapid reaction with the decreasing volume of dispersion agent.23 Interestingly, Fig. 1F and inset maps of Fig. 1D and E show that the rubrene NPs prefer to form at the edge of two smooth faces of ZnP-NS. This was attributed to the existence of rough faces on the edge of the ZnP-NS and higher reduction potential of rubrene+/rubrene (0.9 eV vs. SCE) than ZnP+/ZnP (0.71 eV vs. SCE), which means not only the reaction between the rubrene cation and ZnP, but also the preferred nucleation of rubrene at these rough faces with high surface energy.11,31 In addition, TEM image in Fig. 1F shows the close contact between the rubrene NPs and ZnP-NS, indicating the in situ formation of rubrene NPs on the surface of the ZnP-NS. The corresponding ED pattern in Fig. 1G confirms that a smooth crystal face of ZnP-NS can be indexed to (020) with fully exposed crystal water (see Fig. 1A). The nanosheet in Fig. 1H and relevant Zn element mapping in Fig. 1I further verify the synthesis of a high-quality rubrene/ZnP-NS nano-heterostructure and the morphology of ZnP remains unchanged after the formation of rubrene via chemical reaction.
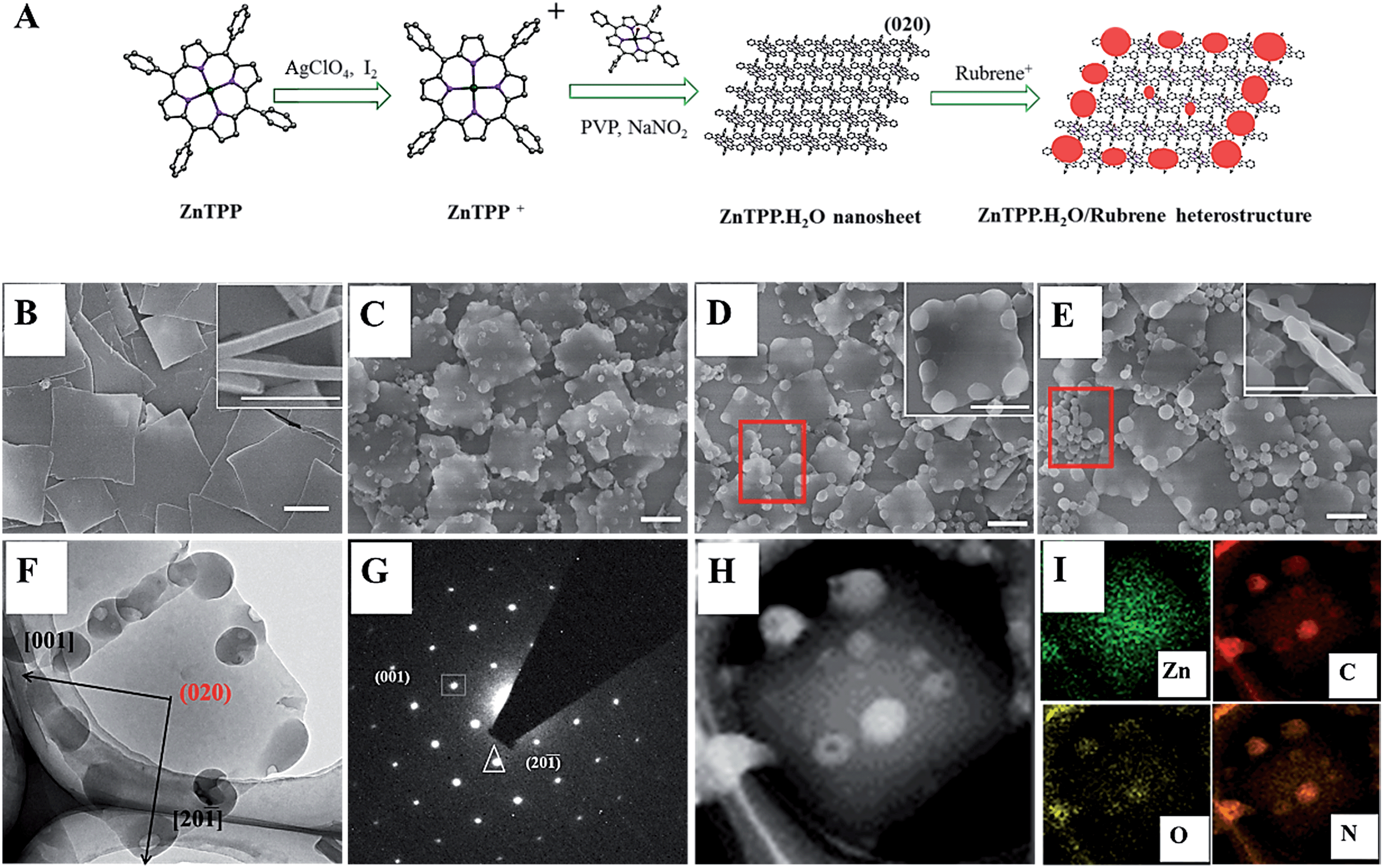 | ||
| Fig. 1 (A) Illustration of the formation of the ZnP-NS and rubrene/ZnP-NS heterostructure in an aqueous solution; SEM images of (B) sole ZnP-NS and rubrene/ZnP-NS heterostructure with different molar ratio of rubrene/ZnP (C) 1:6, (D) 1:3 and (E) 2:3. (F and H) TEM images and (G) relevant SAED pattern as well as (I) Zn, C, O and N mapping of rubrene/ZnP-NS heterostructure when the molar ratio of rubrene/ZnP is 1:3. All scale bars are 500 nm. | ||
To gain insight into the interaction between the ZnP-NS and rubrene, the optical properties of ZnP-NS, rubrene NPs and the composite of rubrene/ZnP-NS in the aqueous phase were studied via an analysis of their UV-vis absorption and steady-state photoluminescent (PL) spectra as well as time-resolved fluorescent lifetime (TRFL), as illustrated in Fig. 2. In Fig. 2A, the peak at 305 nm of the pristine rubrene NPs originates from n–π* transitions, whereas the shoulder peak at 432 nm, the two peaks at 450 and 515 nm as well as the two peaks in the range of 600–800 nm of ZnP-NS belong to the Soret-band, J-aggregate and Q-band from a1μ(π) and a2μ(π) to eg(π*) electron transition, respectively.32 With increasing rubrene content, the UV-vis spectra (a–c) of the composites present two obvious phenomena compared to pristine ZnP-NS (d) and rubrene NPs (f). One is the enhancement of both rubrene absorption peaks and ZnP (J-aggregate and Q-band) absorption peaks, another is the red-shift of rubrene and ZnP Q-band absorption peaks. It can be found that the absorption of rubrene NPs is shifted from 307 nm to 309 nm, whereas the ZnP Q-band is shifted from 558 nm to 563 nm when the ZnP Soret-band remains constant. These phenomena strongly suggest a π–π interaction between ZnP and rubrene in the composite, which in turn decreases the free energy of surface molecules of ZnP and rubrene and thus promotes the formation of more aggregates and intermolecular charge transfer.33
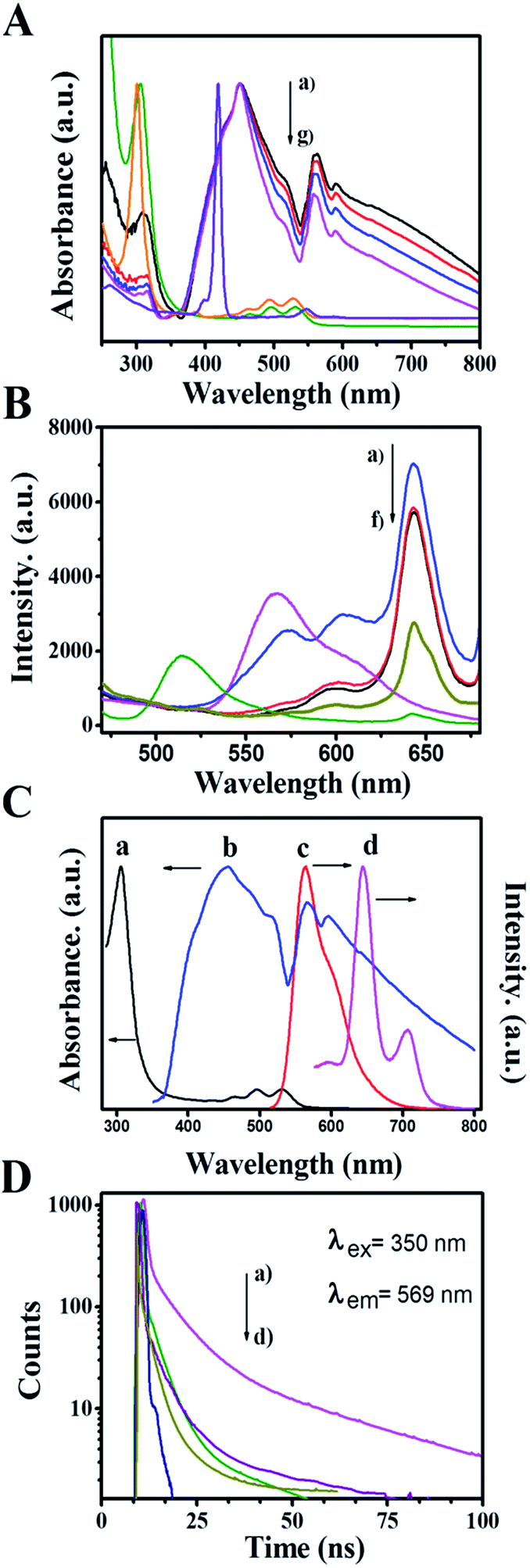 | ||
| Fig. 2 In an aqueous dispersion, (A) UV-vis absorption spectra of rubrene/ZnP-NS NPs with molar ratio of rubrene/ZnP (a) 2:3, (b) 1:3 and (c) 1:6, (d) sole ZnP-NS and (f) rubrene NPs as well as (e) rubrene and (g) ZnP solution in CH2Cl2; (B) PL spectra of rubrene/ZnP-NS with molar of (a) 2:3, (b) 1:3 and (c) 1:6, (d) sole ZnP-NS and (e) rubrene NPs as well as (f) rubrene/ZnP-NS + MV; (C) the UV-vis absorption and PL spectra of (a and c) rubrene NPs and (b and d) ZnP-NS in aqueous dispersion. (D) Fluorescence decay profiles of (a) sole rubrene NPs and rubrene in rubrene/ZnP-NS composite with molar of (b) 2:3, (c) 1:3 and (d) 1:6. The nanodispersed system was monitored at 569 nm and the excitation wavelength was 350 nm. | ||
To further study the interaction between ZnP-NS and rubrene in the heterostructure, the fluorescence of the as-prepared samples was carried out, as shown in Fig. 2B. The figure shows that the PL spectrum of the rubrene NPs has an emission peak at 569 nm upon excitation at 350 nm. However, the emission peak intensity of rubrene NPs decreases drastically and is red shifted to 577 nm after rubrene NPs formed on the surface of the ZnP-NS, which indicates the formation of a new excited state in the composites such as (rubrene·rubrene)* exciplex and (rubrene·ZnP)* excimer.34 In this case, the fluorescence of rubrene NPs (c) at ∼570 nm can be quenched totally by a factor of ten compared to the pristine rubrene NPs (e). The sudden increase of rubrene fluorescence (a) in Fig. 2B when the molar ratio of rubrene/ZnP is 2:3 should result from the existence of sole rubrene NPs, as shown in Fig. 1E. In addition, Fig. 2B displays that ZnP-NS has an emission peak at 643 nm upon excitation at 350 nm. It can be found that the peak intensity obviously increases in the composites and the emission wavelength remains constant. Fig. 2C shows that the UV-vis absorption spectrum of ZnP Q-band (c) overlaps very well with the steady-state emission spectrum of the rubrene NPs (b) in the wavelength range of 600–700 nm. This is essential for the FRET from rubrene to ZnP-NS. The existence of a π–π interaction between ZnP and rubrene at the heterojunction also strongly indicates that the relative orientation of transition dipoles for the rubrene donor to the ZnP acceptor is coincident and the distance between rubrene and ZnP molecules is close,35 which meets requirements for the occurrence of FRET. Thus, FRET from rubrene to ZnP is responsible for the enhanced emission of ZnP in the heterostructure and indicates the formation of the excimer of (rubrene·ZnP)* at the heterojunction.
The fluorescence decays of rubrene at 569 nm were measured using an excitation wavelength of 350 nm, as shown in Fig. 2D. The average lifetime of rubrene NPs (a) in the aqueous phase is 12.5 ns. In the case of rubrene/ZnP-NS heterostructure, the TRFLs of rubrene (b–d) obviously become shorter as the content of rubrene decreased. As listed in Table 1, the decay curves of rubrene in all samples can be well fitted by three exponents, which means three relaxation processes of rubrene excited state (S*). According to the corresponding excitation spectra (see Fig. S3†), it might be related to the transition from S* to the exciplex or excimer (S*e), aggregate (S*a) and ground state (S0). The longest TRFL (τ3) should arise from S* → S0, as reported earlier.36 For rubrene, the formation of heterojunction means the appearance of more excimers due to π–π interaction between ZnP and rubrene. When the molar ratio of rubrene/ZnP is >1:3 in the composite, there are more excimers with the increased content of rubrene. Based on this, τ1 should be from the transition of S* → S*e as referred to the percentage change of TRFL in Table 1. Therefore, τ2 represents the lifetime of S* → S*a. The corresponding TRFLs are τ(S* → S0) > τ(S* → S*a) > τ(S* → S*e). The data in Table 1 show that the TRFLs of τ1 and τ2 become longer as the content of rubrene increased. This may involve the size-effect of rubrene NPs. Smaller size means higher energy level of S*e and/or S*a. In a word, compared to pristine rubrene NPs, the formation of more rubrene-related excimers and exciplex at the heterojunction is responsible for the quenching of rubrene PL and the enhancement of ZnP PL. Under the excitation of light, it will result in the appearance of more photoinduced electrons on the surface of ZnP-NS and achieve more efficient utilization of light.35
| Molar ratio of rubrene/ZnP | τ 1 | τ 2 | τ 3 | τ a/ns | |||
|---|---|---|---|---|---|---|---|
| ns | %1 | ns | %2 | ns | %3 | ||
| a Average fluorescence lifetime, τa = (τ1%1 + τ2%2 + τ3%3)/100. | |||||||
| 1:0 |
0.55 | 37.9 | 6.96 | 36.4 | 37.86 | 25.7 | 12.47 |
| 2:3 |
0.33 | 76.7 | 4.98 | 15.1 | 40.43 | 8.3 | 4.36 |
| 1:3 |
0.32 | 69.5 | 3.69 | 22.1 | 36.78 | 8.4 | 4.13 |
| 1:6 |
0.28 | 64.0 | 2.30 | 29.9 | 32.13 | 6.1 | 2.83 |
To further investigate the effect of the formation of rubrene/ZnP-NS heterojunction on the photoelectric properties, PHE activity of different samples was studied when they were used as photocatalysts with TEOA as SED. PHE was carried out under a 300 W Xe lamp after 2 mg of sample was dispersed in 90 mL ultrapure water by sonication and 10 mL of TEOA was added to it. To survey the shape-dependent photocatalysis of the ZnPs, ZnP-NPs with an average size of ∼80 nm were synthesized (see Fig. S4†). Fig. 3A shows the time course of PHE over the ZnP-NPs, ZnP-NS and rubrene/ZnP-NS heterostructures. The ZnP-NS presents higher photocatalytic activity than ZnP-NPs. The PHE rate of ZnP-NS is ∼0.16 mmol g−1 h−1, whereas that of ZnP-NPs is ∼0.06 mmol g−1 h−1. This indicates that the catalytic activity of the ZnP nanostructure may be closely associated with its shape. Combined with the crystal structure of ZnP-NS in Fig. 1A, it is believed that the active sites of ZnP were concentrated on the (020) crystal face. Compared to pristine ZnP-NS and rubrene NPs, the heterostructures of rubrene/ZnP-NS constructed on the ±(020) crystal faces of ZnP-NS reveal more efficient PHE activity.
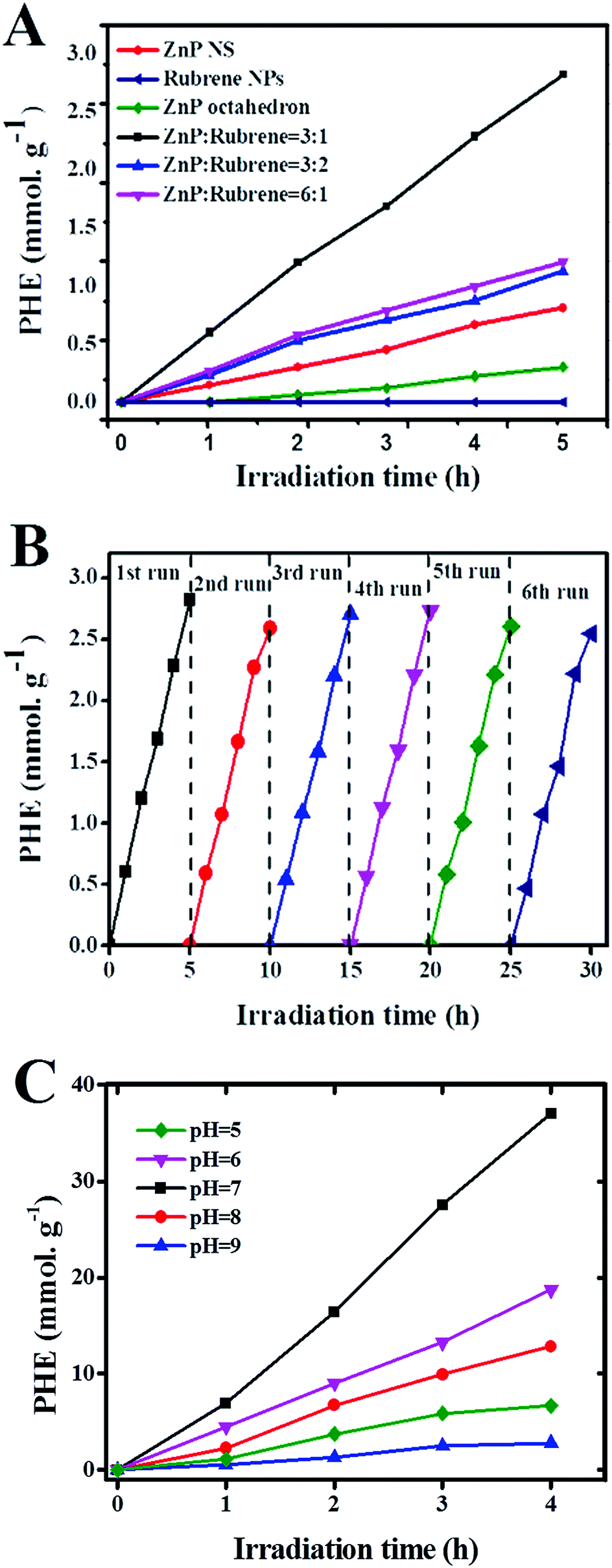 | ||
| Fig. 3 (A) Time course of PHE of ZnP-NPs, ZnP-NS, rubrene NPs and rubrene/ZnP-NS heterostructure with different molar ratios (6:1, 3:1 and 3:2); (B) stability test of PHE (every 5 h) and (C) time course of PHE of rubrene/ZnP-NS (3:1) at different pH in the presence of 3.0 mM MV, 10 mL TEOA and 3.8 wt% Pt in 100 mL aqueous system. | ||
The experimental results in Fig. 3A show that the loading of rubrene plays an important role in adjusting the photocatalytic activity of the heterostructure. With increasing content of rubrene, the amount of hydrogen first increases and then decreases when the molar ratio of rubrene/ZnP is changed from 1:6, 1:3 to 2:3. As we know, the photocatalytic activity of a nanocomposite is closely related to two factors. One is the ability to harvest light, another is the amount of photocatalytic active sites at the surface. The increase in heterojunction usually means a decrease in the number of active sites. In our system, the PL spectra in Fig. 2A clearly display that the intensity of UV-vis absorption enhances as the content of rubrene increased. However, the amount of heterojunction, in fact, did not increase when the molar ratio of rubrene/ZnP was increased to 2:3. As observed in Fig. 1E, there are a large number of independent rubrene NPs, which have little photocatalytic activity in our experiment. In nature, the PL spectra of curves (b) and (c) in Fig. 2B also imply that increasing the loading of rubrene to ZnP-NS will not improve the energy and/or electron transfer from rubrene to ZnP. The optimal molar ratio of rubrene/ZnP for PHE is 1:3. Its PHE rate is 0.56 mmol g−1 h−1 and it is more than three times than that of pure ZnP-NS, which confirms that the formation of the rubrene/ZnP-NS heterojunction has a positive effect on the PHE from water splitting. Actually, the result is better than the PHE rate of ZnP-NS with ∼3.8 wt% Pt loaded, as illustrated in Fig. S5.† In this system, the turnover numbers (TONZnP) and the turnover frequency (TOFZnP) for the ZnP are ∼5 and 2.72 × 10−4 s−1 after 5 hours irradiation, respectively. Furthermore, the PHE durability of the rubrene/ZnP-NS (1:3) heterostructure was performed. The results can be seen in Fig. 3B. For each cycle, there is no evident decay of PHE activity, indicating the excellent photocatalytic stability of the composite.
As a matter of fact, zinc porphyrin and its derivatives are usually used for PHE in a three component system, including MV and Pt.14 As shown in Fig. 2B, the PL spectrum (f) of the heterostructure at 643 nm was quenched by a factor of nineteen after the addition of MV. In view of this, we tried constructing the system of TEOA/rubrene/ZnP-NS/MV/Pt. The results are displayed in Fig. 3C. Taking the effect of the pH on PHE into account, we achieved a PHE rate of ∼9.3 mmol g−1 h−1 at pH = 7. After 4 hours irradiation, the TONPt and TOFPt were 385 and 2.68 × 10−2 s−1, respectively. The TONZnP was ∼65 and the TOFZnP was 4.51 × 10−3 s−1. In addition, at a wavelength of 365 ± 10 nm, a quantum yield (Ø) of 1.65% was obtained. In this type of system, MV is usually used as an electron relay to transfer photoinduced electrons from a photosensitizer to the Pt surface, whereas Pt is often seen as a catalyst.9 The existence of Pt further improves the efficiency of photocatalytic hydrogen evolution (PHE) by limiting the recombination of photoinduced electrons and holes due to the Schottky effect and providing more and better activity sites for the reaction from the proton to hydrogen and the desorption of hydrogen. In the photocatalytic system of TEOA/rubrene/ZnP-NS/MV/Pt, the effect of pH on the PHE results from two factors. (1) A higher pH value will decrease the concentration of protons in water and in turn reduce the reaction rate from protons to hydrogen. (2) When hydrochloric acid (HCl) was used to adjust the pH, a lower pH value may influence the activity of TEOA as a sacrificial electron donor. Therefore, there is an optimal pH in our experiment.
Based on our experiments, we established the schematic of PHE of rubrene/ZnP-NS, as illustrated in Scheme 1. Under solar light illumination, both ZnP and rubrene are pumped to the excited state due to an electron transition and then form excitons (the pair of electron and hole), as shown in eqn (1) and (2).
 | (1) |
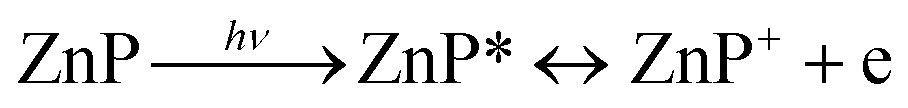 | (2) |
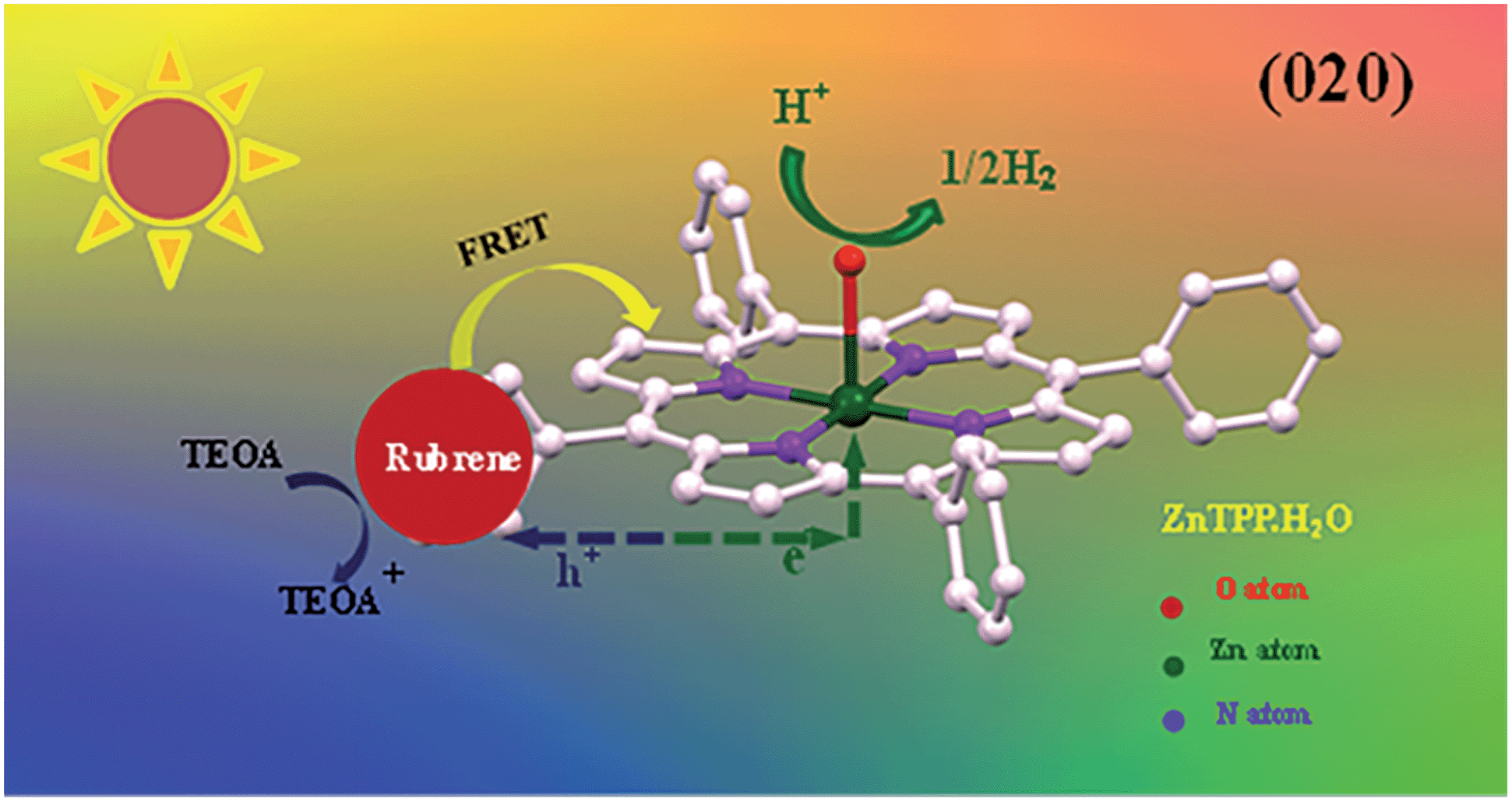 | ||
| Scheme 1 Schematic photoexcited electron transfer and FRET as well as PHE over the rubrene/ZnP-NS photocatalyst under light irradiation. | ||
In addition, FRET between the excited-state rubrene and ground-state ZnP occurs and generates an excimer, finally forms ground-state rubrene and excited-state ZnP because of the π–π stacking interaction and close contact between ZnP and rubrene at the heterojunction, as referred in eqn (3). This process promotes the occurrence of reaction (2).
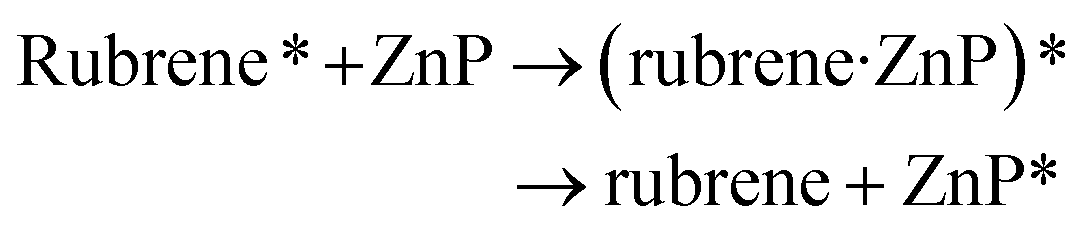 | (3) |
Because the LUMO and HOMO of ZnP and rubrene are −2.39 eV and −5.13 eV and −2.10 eV and −4.62 eV, respectively, higher energy photogenerated electrons of rubrene should transfer to ZnP, and higher energy photogenerated holes of ZnP should transfer to rubrene, as shown in eqn (4) and (5).27,30 In this process, the TRFLs of both rubrene and ZnP are obviously lowered (see Tables 1, S1 and Fig. S6†).
| Rubrene− + ZnP → rubrene + ZnP− | (4) |
| Rubrene + ZnP+ → rubrene+ + ZnP | (5) |
On the (020) crystal face of ZnP-NS, the photoinduced electrons from ZnP and rubrene first transfer from ligand TPP to the central atom of Zn(II), then to crystal water, finally to water and/or protons in body phase.24 Subsequently, as listed in eqn (6) and (7) and Scheme 1, holes on the surface of rubrene were reduced by TEOA and hydrogen is generated at the active sites.
| Rubrene+ + TEOA → rubrene + TEOA+ | (6) |
| H+ + e → H2 | (7) |
According to the abovementioned results and analysis for the photocatalytic system of TEOA/rubrene/ZnP-NS, we believe that in the entire process of PHE from water, (1) ZnP-NS is the photocatalyst, (2) the crystal water of the ZnP-NS (020) crystal face may not only play a bridge role in the transfer of photogenerated electron to protons, but also be a reaction site of proton to hydrogen, and (3) rubrene only acts as a photosensitizer.
Conclusions
In summary, we achieved direct PHE using the nanostructures of organic small molecule as photocatalysts. In this study, a series of nanostructures, including ZnP-NS, ZnP-NPs, rubrene NPs and heterostructures of rubrene/ZnP-NS with different molar ratios, were synthesized via a liquid-phase chemical reaction for PHE from water splitting. The shape-dependent PHE activity showed that ZnP-NS presented higher PHE activity than ZnP-NPs because of more exposed crystal water on the surface. The PHE rate of ZnP-NS was ∼0.16 mmol g−1 h−1. After in situ constructing the heterostructure of rubrene/ZnP-NS in an aqueous solution, FRET from rubrene to ZnP occurs, and in turn markedly promotes the capability of light absorption of ZnP and limits the recombination of photoinduced electrons and holes. The PHE rate of the optimal heterostructure was ∼0.56 mmol g−1 h−1. Moreover, the value of ∼9.3 mmol g−1 h−1 can be obtained at pH = 7 when the optimal composite of rubrene/ZnP-NS was added to the system including MV and colloid Pt. Our study not only demonstrates that the nanostructures of organic small molecules can be used directly as a photocatalyst, but also paves a new way for PHE from water splitting.Experimental
Materials
Zinc tetraphenylporphyrin (ZnTPP) (95%, J&K Chemicals Co.), rubrene (98.5%, J&K Chemicals Co.), silver perchlorate (97%, J&K Chemicals Co.), iodine (99.5%, Sinopharm Chemical Reagent Co.), anhydrous dichloromethane (J&K Chemicals Co.) and acetonitrile (J&K Chemicals Co., water <30 ppm) were used in this study. Ultrapure water with a resistivity of 18.2 MΩ cm−1 was produced using a Water Purifier apparatus (WP-UP-IV-20). Triethanolamine (99%, Sinopharm Chemical Reagent Co.) and methyl viologen (98%, J&K Chemicals Co.) were used for photocatalysis. Hydrochloric acid (HCl, 36–38%, Sinopharm Chemical Reagent Co.) was used to adjust the pH. All chemicals were used without further purification.Preparation of ZnP-NS
ZnP-NS were synthesized by a liquid phase chemical reaction method. First, the precursor of ZnTPP perchlorate was produced via reaction (8):23| 2ZnTPP + 2AgClO4 + I2 → 2ZnTPPP·ClO4 + 2AgI↓ | (8) |
In a typical synthesis, a solution of dry silver perchlorate in anhydrous acetonitrile (2 mL, 100 mM) and a solution of I2 in anhydrous dichloromethane (4 mL, 25 mM) were added to a 10 mL solution of 20 mM ZnTPP in anhydrous dichloromethane with stirring for 3 hours and aging for one hour. Silver chloride was filtered by centrifuging at 10000 rpm for 10 min. The solution of ZnTPP·ClO4 was then poured into 80 mL petroleum ether and sealed at room temperature for 3 h. Finally, the green-black precipitate was obtained by centrifuging at 10000 rpm for 10 min and further washed twice with petroleum ether. The as-obtained ZnTPP·ClO4 was dried under vacuum for further use. Second, ZnTPP·ClO4 (1 mL, 1 mM) in anhydrous acetonitrile was injected rapidly into 5 mL ultrapure water solution containing 10 mM sodium nitrite and 500 mM polyvinyl pyrrolidone (PVP). The nanosheets can be obtained according to reaction (9):
| 2ZnTPP·ClO4 + NaNO2 + 3H2O → 2ZnTPP·H2O + NaNO3 + 2HClO4 | (9) |
After injection of the precursor and aging 3 hours, the colloidal suspension of ZnP-NS was found. The sample was separated and washed three times with pure water to remove the excess PVP. The final product was dried for further use.
Preparation of rubrene nanostructure
First, the rubrene precursor was produced via reaction (10):31| 2Rubrene + 2AgClO4 + I2 → 2rubrene·ClO4 + 2AgI↓ | (10) |
An olive-drab rubrene·ClO4 solution was obtained. The solution was then treated by decompressed distillation to remove the solvent. The as-obtained rubrene·ClO4 was dried under vacuum at room temperature. Second, rubrene·ClO4 (1 mL, 1 mM) in anhydrous acetonitrile was injected rapidly into a 3 mL pure water solution. After injection and aging for 3 hours, the rubrene nanostructure was obtained. The sample was then washed twice with pure water and dried for further use. Rubrene was prepared via reaction (11):
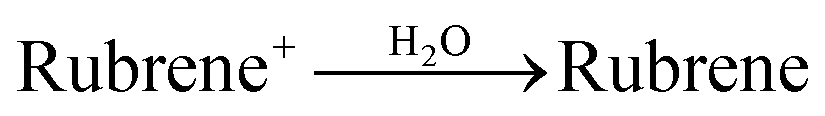 | (11) |
Characterization
The morphologies and elemental composition were measured by a field emission scanning electron microscope (FESEM, Hitachi SU8010) and a transmission electron microscope (TEM, F20). The UV-vis absorption spectra were obtained using a UV-vis spectrophotometer (Shimadzu, UV2550). The photoluminescence (PL) spectra were acquired on a Hitachi F-4600 fluorescence spectrophotometer. The fluorescence lifetime decay spectra were performed with a transient state fluorescence spectrometer (Edinburgh Instrument, FLS980). The mass spectrum was measured using MALDI-TOF-MS spectrometer (Bruker, Autoflex III).Photocatalysis experiments
The photocatalytic water splitting reactions were performed with a Pyrex top-irradiation reaction vessel connected to a closed glass gas circulation system (Labsolar III AG, Beijing Perfect-light Technology Co. Ltd). In a typical experiment, 2 mg of ZnP-NS or rubrene/ZnP-NS (2 mg) was added to 90 mL ultrapure water, and 10 mL TEOA was then added. The irradiation light source was a 300 W Xe lamp (PLS-SXE 300). In the case of H2PtCl6·6H2O (3.86 × 10−7 mol) and MV2+ (2.95 × 10−4 mol), after all regents were added to 100 mL reaction agents, the 300 W Xe lamp was applied for 2 hours so that H2PtCl6 could be reduced to Pt nanoparticles and to deposit on the surface of the ZnP-NS via reaction37(12):| [PtCl6]2− + 4e− → 6Cl− + Pt | (12) |
The pH was adjusted with HCl. The evolved gas was analyzed by gas chromatography (GC 7900, Shanghai Techcomp Instrument Ltd). A flow of cooling water was used to maintain the reactant solution at room temperature.
The estimated turnover numbers (TON), turnover frequency (TOF) and quantum yield of hydrogen production (Ø) were obtained using these equations:
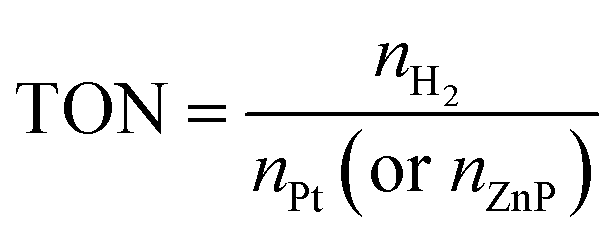 | (13) |
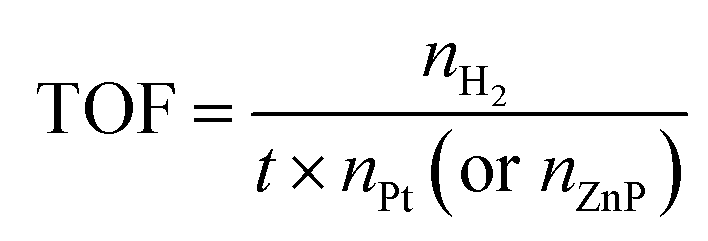 | (14) |
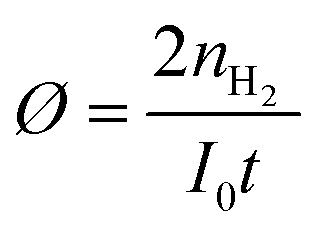 | (15) |
n H2 represents the moles of hydrogen production. nPt is the moles of Pt, whereas nZnP is the moles of ZnPt is the irradiation time. I0 is the number of photons per second. The quantum efficiency (Ø) was obtained by applying a Xe lamp (300 W) with a 365 nm bandpass filter.
Acknowledgements
The authors gratefully acknowledge the financial from the National Natural Science Foundation of China (No. 21252001, 21473204), the Special Project of National Major Scientific Equipment Development of China (No. 2012YQ120060), the Natural Science Foundation of Fujian Province (2015J01070), and the Science and Technology Planning Project of Fujian Province, Grant No. 2014H2008.Notes and references
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa and M. Oshikiri, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- F. X. Xiao, J. Miao, H. B. Tao, S. F. Hung, H. Y. Wang, H. B. Yang, J. Chen, R. Chen and B. Liu, Small, 2015, 11, 2115–2131 CrossRef CAS PubMed.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed.
- T. Y. Zhai, X. S. Fang, L. Li, Y. Bando and D. Golberg, Nanoscale, 2010, 2, 168–187 RSC.
- Z. J. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- D. H. Wang, J. N. Pan, H. H. Li, L. J. Liu, Y. B. Wang and L. T. Kang, J. Mater. Chem. A, 2015, 4, 290–296 Search PubMed.
- Y. M. Tian, K. E. Martin, J. Y. Shelnutt, L. Evans, T. Busani, J. E. Miller, C. J. Medforth and J. A. Shelnutt, Chem. Commun., 2011, 47, 6069–6071 RSC.
- A. Mishra and P. Bauerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38–54 CrossRef CAS.
- J. L. Brédas, B. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5003 CrossRef PubMed.
- J. P. Darwent, P. Douglas, A. Harriman, G. Porter and M. C. Richoux, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS.
- I. Okura and N. K. Thuan, J. Mol. Catal., 1979, 6, 227–230 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- J. W. Lee, K. Kim, J. S. Jung, S. G. Jo, H. M. Kim, H. S. Lee, J. Y. Kim and J. S. Joo, Org. Electron., 2012, 13, 2047–2055 CrossRef CAS.
- L. T. Kang, Z. C. Wang, Z. W. Cao, Y. Ma, H. B. Fu and J. N. Yao, J. Am. Chem. Soc., 2007, 129, 7305–7312 CrossRef CAS PubMed.
- L. W. Huang, Q. Liao, Q. Shi, H. B. Fu, J. S. Ma and J. N. Yao, J. Mater. Chem., 2010, 20, 159–166 RSC.
- Q. L. Zou, L. Zhang, X. H. Yan, A. H. Wang, G. H. Ma, J. B. Li, H. Mohwald and S. Mann, Angew. Chem., Int. Ed., 2014, 53, 2366–2370 CrossRef CAS PubMed.
- K. Liu, R. R. Xing, C. J. Chen, G. Z. Shen, L. Y. Yan, Q. L. Zou, G. H. Ma, H. Mohwald and X. H. Yan, Angew. Chem., Int. Ed., 2015, 54, 500–505 CAS.
- Q. L. Zou, K. Liu, M. Abbas and X. H. Yan, Adv. Mater., 2016, 28, 1031–1043 CrossRef CAS PubMed.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okda, H. Matsuda, N. Minami, A. Kakuta, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., Part 2, 1992, 31, 1132–1134 CrossRef.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. X. Guo and J. W. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- L. T. Kang, H. B. Fu, X. Q. Cao, Q. Shi and J. N. Yao, J. Am. Chem. Soc., 2011, 133, 1895–1901 CrossRef CAS PubMed.
- J. H. Yang, Y. J. Gao, W. Zhang, P. Tang, J. Tan, A. H. Lu and D. Ma, J. Phys. Chem. C, 2013, 117, 3785–3788 CAS.
- Z. Zhao, Y. J. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- A. Irfan, N. Hina, A. G. Al-Sehemi and A. M. Asiri, J. Mol. Model., 2012, 18, 4199–4207 CrossRef CAS PubMed.
- H. Najafov, B. Lee, Q. Zhou, L. C. Feldman and V. Podzorov, Nat. Mater., 2010, 9, 938–943 CrossRef CAS PubMed.
- S. Tavazzi, A. Borghesi, A. Papagni, P. Spearman, L. Silvestri, A. Yassar, A. Camposeo, M. Polo and D. Pisignano, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(1–5), 245416 CrossRef.
- F. Anger, R. Scholz, E. Adamski, K. Broch, A. Gerlach, Y. Sakamoto and T. Suzuki, Appl. Phys. Lett., 2013, 102(1–5), 013308 CrossRef.
- L. Luo, G. Liu, L. W. Huang, X. Q. Cao, M. Liu, H. B. Fu and J. N. Yao, Appl. Phys. Lett., 2009, 95(1–3), 263312 CrossRef.
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1977, 100, 5075–5080 CrossRef.
- H. B. Fu and J. N. Yao, J. Am. Chem. Soc., 2001, 123, 1434–1439 CrossRef CAS.
- T. M. R. Viseu, G. Hungerford, A. F. Coelho and M. I. C. Ferreira, J. Phys. Chem. B, 2003, 107, 13300–13312 CrossRef CAS.
- S. Sardar, P. Kar, H. Remita, B. Liu, P. Lemmens, S. K. Pal and S. Ghosh, Sci. Rep., 2015, 5(1–14), 17313 CrossRef CAS PubMed.
- L. W. Huang, Q. Liao, Q. Shi, H. B. Fu, J. S. Ma and J. N. Yao, J. Mater. Chem., 2010, 20, 159–166 RSC.
- S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. ASai, Z. Li, S. S. K. Ma, F. X. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta01582d |
| This journal is © The Royal Society of Chemistry 2016 |