
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–property relationships for bis-diketopyrrolopyrrole molecules in organic photovoltaics†
Qiang (Mike)
Wang
ab,
Jacobus J.
van Franeker
ab,
Bardo J.
Bruijnaers
a,
Martijn M.
Wienk
ac and
René A. J.
Janssen
*ac
aMolecular Materials and Nanosystems, Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: r.a.j.janssen@tue.nl
bDutch Polymer Institute (DPI), P. O. Box 902, 5600 AX Eindhoven, The Netherlands
cDutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ Eindhoven, The Netherlands
First published on 14th June 2016
Abstract
The design of small organic molecules for efficient solution-processed organic solar cells is hampered by the absence of relationships that connect molecular structure via processing to blend morphology and power conversion efficiency. Here we study a series of bis-diketopyrrolopyrrole molecules in which we systematically vary the aromatic core, the solubilizing side chains, and the end groups to achieve power conversion efficiencies of 4.4%. By comparing the morphology and performance we attempt to identify and rationalize the structure–property relationships. We find that the tendency to aggregate or crystallize are important factors to control and that these require a subtle balance.
1. Introduction
The surge in power conversion efficiency (PCE) of organic photovoltaic (OPV) cells in the last decade has primarily been achieved by developing new organic semiconductors that feature high charge carrier mobilities and carefully tuned frontier molecular orbital (HOMO and LUMO) levels to maximize the open-circuit voltage and short-circuit current. The progress further relied on optimizing the phase separation between the donor and acceptor materials via judicious processing to achieve high current densities and fill factors. The highest reported efficiencies for single junction polymer solar cells exceed 10%.1–6 One drawback of synthetic semiconducting polymers is their statistically determined nature, reflected – amongst others – in their molecular weight distribution and the occurrence of undesired homo-coupling reactions during the polymerization.7–10Small conjugated molecules and well-defined oligomers of intermediate dimensions with distinct molecular structures and controllable purity alleviate the lack of chemical and structural definition of conjugated polymers.11,12 Using well-defined oligomers and reproducible processing methods, improved understanding of structure–property relationships can be anticipated.13 Solution-processed small-molecule organic solar cells were first described for oligothiophenes,14 merocyanines,15 and squaraine dyes.16 Presently the highest PCEs of solution-processed small-molecule organic solar cells rival those of the most efficient polymer solar cells.17–19 A common design element of these efficient molecules is that they are based on complementary electron rich (donor, D) (mostly thiophene based) and electron deficient (acceptor, A) units in D–A–D or A–D–A motifs.
To extend the spectral coverage to the near infrared, organic semiconductors based on diketopyrrolopyrrole (DPP) have been applied in organic solar cells.20–23 DPP polymers exhibit high charge carrier mobilities, which add to their suitability for application in solar cells.24,25 Also small molecules incorporating a DPP unit have extensively been investigated for use in organic solar cells.26–42 Among the many DPP molecules that have been considered for use in organic solar cells, those incorporating the A–D–A or D–A–D–A–D motifs with DPP as acceptor unit have been very popular.43–104 By introducing extended planar aromatic donor bridges between the two DPP acceptor units it is possible to obtain high charge mobility and to manipulate the energies of the frontier orbitals to control voltage and charge transfer to the fullerene acceptor. Several bis-DPP molecules afford PCEs up to 6%, and some even higher.72,87,90 The record PCE of 8.08% for a solution-processed small-molecule solar cell based on DPP units has been obtained for a compound comprising two DPP units linked to a central porphyrin via acetylene bonds.90
In all bulk-heterojunction organic solar cells the morphology of the blend and the nanoscale phase separation is crucial to achieve efficient exciton dissociation into charge carriers and to provide suitable percolating pathways to transport charges to the electrodes. Undoubtedly, achieving an optimized morphology is the key to unlock the full potential of the small-molecule organic donors. While this is well recognized in the field, there is little general knowledge on how the molecular structure directs the blend morphology and how this is influenced by processing conditions such as solvent, co-solvent, and thermal annealing. In this paper we present a series of bis-DPP molecules with A–D–A and D–A–D–A–D motifs with different central cores, solubilizing side chains, and end groups connected to the two DPP units (Fig. 1). The series of molecules was designed to provide similar optical band gaps and HOMO and LUMO levels, to focus on effects of the structural changes in the different parts of the bis-DPP molecules on performance and morphology. The results contribute to establishing structure–performance relationships for solution-processed small-molecule organic solar cells.
 | ||
| Fig. 1 Structures of bis-DPP molecules. | ||
2. Results and discussion
2.1. Synthesis
The bis-DPP molecules shown in Fig. 1 were synthesized via Stille reactions using the appropriate bis(trimethyltin)-substituted aromatic core and 3-(5-bromothiophen-2-yl)-2,5-bis(dialkyl)-6-(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione in toluene at 115 °C using tris(dibenzylideneacetone)dipalladium(0) as a catalyst (Scheme 1). For BDT-EH-S-BT, 3-(5-(benzo[b]thiophen-2-yl)thiophen-2-yl)-6-(5-bromo-thiophen-2-yl)-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione was used instead. All products were purified via column chromatography or preparative recycling gel permeation chromatography and characterized with NMR and MALDI-TOF mass spectrometry. The details of the synthesis and characterization of the molecules shown in Fig. 1 can be found in the ESI†. | ||
| Scheme 1 Generalized synthetic procedure of the bis-DPP molecules. | ||
2.2. Optical absorption
The UV-vis absorption spectra of the bis-DPP molecules in solution are similar (Fig. 2a). The optical absorption of bis-DPP molecules with a benzo[1,2-b:4,5-b′]dithiophene (BDT) core is slightly hypsochromically shifted with respect to those of the thieno[3,2-b]thiophene (TT) and dithieno[3,2-b:2′,3′-d]thiophene (DTT) cores. The five bis-DPP derivatives with a BDT core show a vibronic transition at lower wavelengths, whereas the other bis-DPPs show a featureless absorption band. The two benzo[b]thiophen-2-yl (BT) end groups in BDT-EH-S-BT cause a substantial bathochromic shift, compared to BDT-EH-S. On the other hand, the two 2-thienyl substituents on the 4 and 8 positions of BDT have a negligible effects on the optical absorption in solution.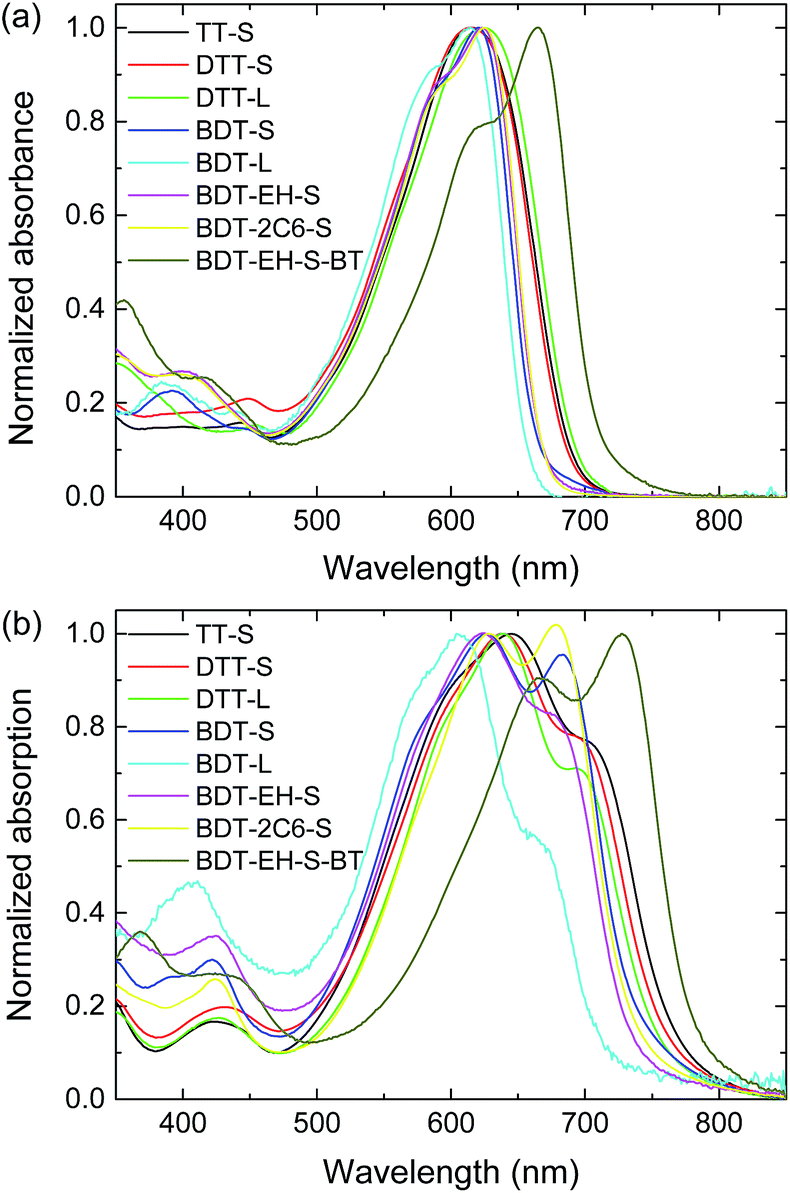 | ||
| Fig. 2 UV-vis absorption spectra of bis-DPP molecules shown in Fig. 1 in (a) solution and (b) thin films. | ||
In thin solid films all compounds exhibit a substantial bathochromic shift of the optical absorption and all compounds show fine structure as a consequence of vibronic transitions and coupling of transition dipoles (Fig. 2b) of nearby molecules. The magnitude of the bathochromic shift in thin films is similar for all bis-DPPs, except for BDT-L which is less shifted. The relative intensity of the longest wavelength absorption band is the highest for BDT-EH-S-BT and then reduces along the series BDT-2C6-S, BDT-S, BDT-EH-S, TT-S and DTT-S to lower values for DTT-L and BDT-L. A low relative intensity of the longest wavelength absorption band in the solid state can point to the formation of H-type aggregates.39 Especially for BDT-L, for which λmax in solution (614 nm) is larger than in the film (604 nm), the optical absorption is reminiscent of an H-type aggregate in the film.
Cyclic voltammetry was performed in dichloromethane to determine the frontier orbital energy levels of the bis-DPP molecules (Fig. 3). The corresponding cyclic voltammograms are collected in Fig. S1 (ESI†) and show that the oxidation and reduction waves are in general chemically reversible. The electrochemical band gaps determined from the differences between the potentials at the onsets of the redox waves are similar to the optical band gaps determined from the onsets of absorption (in the solution spectra) (Table 1), but consistently somewhat smaller. From the cyclic voltammetry we find that the HOMO levels of the bis-DPP molecules are at −5.515 (±0.03) eV, using −5.23 eV vs. vacuum for the ferrocene/ferrocenium redox couple. Based on these small differences in the oxidation potentials, no large differences are expected in the open-circuit voltage (Voc) of the solar cells based on these bis-DPP molecules.
 | ||
| Fig. 3 Energy levels (in eV vs. vacuum) of bis-DPP molecules measured by cyclic voltammetry. | ||
| Molecule | TT-S | DTT-S | DTT-L | BDT-S | BDT-L | BDT-EH-S | BDT-2C6-S | BDT-EH-S-BT |
|---|---|---|---|---|---|---|---|---|
| E CVg (eV) | 1.78 | 1.71 | 1.76 | 1.78 | 1.78 | 1.79 | 1.79 | 1.72 |
| λ max (nm) | 616 | 615 | 623 | 620 | 614 | 624 | 625 | 666 |
| λ onset (nm) | 689 | 684 | 691 | 662 | 657 | 669 | 669 | 711 |
| E g (eV) | 1.80 | 1.81 | 1.79 | 1.87 | 1.89 | 1.85 | 1.85 | 1.74 |
2.3. Solar cells and morphologies
Organic solar cells were fabricated by sandwiching a bulk heterojunction of the bis-DPP molecules and [60]PCBM between a transparent ITO/PEDOT:PSS front electrode and a LiF/Al back electrode on glass substrates. The active layer was deposited via spin coating from chloroform with and without co-solvents. All photoactive layers were extensively optimized for their photovoltaic performance by adjusting the solvent mixture, spin speed, and applying thermal annealing conditions. An overview of the most relevant data can be found in Tables S1–S8 (ESI†). The ESI† also contains the current density–voltage (J–V) characteristics and the external quantum efficiencies (EQEs) of the optimized devices.In Table 2 we compare the photovoltaic parameters of the optimized devices of TT-S, DTT-S and BDT-S mixed with [60]PCBM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio. The highest PCE = 4.3% is found for TT-S:[60]PCBM, similar to the value of 4.0% reported previously for this compound.61 This layer possesses the highest short-circuit current density (Jsc) and fill factor (FF) of the three derivatives. The EQE of the TT-S:[60]PCBM cells reaches a maximum of 49% (Fig. S2a, ESI†).
:1 weight ratio. The highest PCE = 4.3% is found for TT-S:[60]PCBM, similar to the value of 4.0% reported previously for this compound.61 This layer possesses the highest short-circuit current density (Jsc) and fill factor (FF) of the three derivatives. The EQE of the TT-S:[60]PCBM cells reaches a maximum of 49% (Fig. S2a, ESI†).
:1 ratioa
| Material | Annealing | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|
| a Spin coated from chloroform containing 0.2% 1-CN. | |||||
| TT-S | 110 °C 10 min | 9.46 | 0.81 | 0.56 | 4.3 |
| DTT-S | No | 6.82 | 0.76 | 0.44 | 2.3 |
| BDT-S | No | 5.95 | 0.85 | 0.53 | 2.7 |
Atomic force microscopy (AFM) height and phase images of the TT-S:[60]PCBM blend (Fig. S11, ESI†) reveal the presence of fibrous surface features and a root-mean-square surface roughness (Rq) of 6.9 nm. In transmission electron microscopy (TEM), however, the TT-S:[60]PCBM blend shows little contrast (Fig. 4a and S2b in the ESI†), suggesting intimate mixing, without large scale phase separation.
 | ||
| Fig. 4 TEM images of optimized photoactive layers of (a) TT-S:[60]PCBM, (b) DTT-S:[60]PCBM, (c) BDT-S:[60]PCBM, (d) DTT-L:[60]PCBM, and (e) BDT-L:[60]PCBM. | ||
The DTT-S:[60]PCBM solar cell (PCE = 2.3%) has a lower FF, Jsc and EQE (37%) than the optimized TT-S:[60]PCBM device. The photocurrent is strongly bias dependent and reaches a similar value as the photocurrent in the TT-S:[60]PCBM cell at a reverse bias of −1.5 V (cf. Fig. S2a and S3a in the ESI†). This demonstrates that exciton dissociation into charge-transfer states is similar in the two blends, but that charge extraction is more difficult in the DTT-S:[60]PCBM blend. This can be due to the much lower hole mobility of DTT-S (10−4 cm2 V−1 s−1) compared to TT-S (0.1 cm2 V−1 s−1), which has been reported in field-effect transistors of these materials.105 A low hole mobility reduces the rate at which photogenerated charge-transfer states dissociate into free charges and can also reduce the fill factor if the charge transport of electrons and holes is unbalanced.106 A second possible reason for the low FF and the observed field-assisted photocurrent collection is the absence of a percolating phase of DTT-S molecules, such that charge transport is hampered. Consistently, the TEM images of as-cast DTT-S:[60]PCBM blends (Fig. 4b, and S3b in the ESI†) do not show significant phase separation.
For BDT-S:[60]PCBM blends (PCE = 2.8%), the FF is enhanced compared to DTT-S:[60]PCBM, but the photocurrent and maximum EQE (31%) are lower and do not significantly increase in reverse bias (Fig. S4a, ESI†). Hence, charges are generated less efficiently than in the other two blends but are relatively easily transported. This would be consistent with a more phase-separated morphology in which pure domains facilitate charge transport but where a decreased donor–acceptor interface area reduces the exciton dissociation. The TEM images of the BDT-S:[60]PCBM blend (Fig. 4c, and S4b in the ESI†), however, do not show distinct evidence of such coarser phase separation, although there is contrast on larger length scales than in the blends of TT-S and DTT-S with [60]PCBM.
:1 ratioa
| Material | Annealing | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|
| a Spin coated from chloroform containing 0.2% 1-CN. | |||||
| DTT-L | No | 5.83 | 0.81 | 0.60 | 2.8 |
| BDT-L | No | 5.53 | 0.85 | 0.58 | 2.7 |
The fill factor of 0.60 of the DTT-L:[60]PCBM cell is comparatively high for a solution-processed small molecule solar cell and much higher than the value of 0.44 for the short chain DTT-S:[60]PCBM blends. Compared to the intimately mixed DTT-S:[60]PCBM blend, the DTT-L:[60]PCBM films display distinct contrast on a length scale of less than 50 nm in TEM (Fig. 4d and S5b in the ESI†). This coarser phase separation improves percolation of charges and enhances the fill factor.
Also the morphology of BDT-L:[60]PCBM films shows large (∼100 nm) darker [60]PCBM rich domains (Fig. 4e and S6b in the ESI†) in TEM. These images are reminiscent of the polymer–fullerene morphologies in which spinodal liquid–liquid demixing occurs before the film is dry.107 For polymer–fullerene bulk heterojunctions this predominantly happens under conditions where the polymer has good solubility and aggregation only occurs in the last stages of film drying.108 This is consistent with the present result where long side chains impart increased solubility on BDT-L compared to BDT-S. In the regions in between the dark [60]PCBM domains the blend has distinct crystalline domains as inferred from the lattice fringes in the TEM images (Fig. S6b, ESI†). The main lattice spacing estimated from the Fourier transform of the TEM image is 1.9 ± 0.1 nm (Fig. S6b, ESI†) and corresponds to the value of 2.0 nm of pure BST-L inferred from the X-ray diffraction at 2θ = 4.40° (Fig. S14, ESI†).
We see that for these two examples the longer side chains result in a coarser and more distinct phase separation with [60]PCBM as compared to the blends of the same compounds with shorter side chains. In analogy with previously studied polymer–fullerene blends we attribute this effect to an increased solubility. There are two effects that play as role. First, a higher solubility implies that spinodal liquid–liquid demixing can occur before the molecules aggregate or crystallize.108 This results in large, almost pure domains as seen in the BDT-L:[60]PCBM films. Second, the critical size at which a nucleus becomes stable against re-dissolving is larger for more soluble molecules. Hence, phase separation with narrow fibres and small crystallites can be expected for less soluble compounds.109
 | ||
| Fig. 5 TEM images of optimized photoactive layers of BDT-EH-S (a and b) and BDT-2C6-S (c and d) spin coated from chloroform containing 0.2% 1-CN before (a and c) and after (b and d) thermal annealing at 110 °C for 10 min. | ||
:1 ratio
| Materials | Annealing | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|
| a Spin coated from chloroform containing 0.2% 1-CN. b Spin coated from chloroform containing 0.3% 1,8-diiodooctane. c Spin coated from pure chloroform. | |||||
| BDT-EH-Sa | No | 6.76 | 0.92 | 0.39 | 2.4 |
| BDT-EH-Sa | 110 °C 10 min | 9.36 | 0.83 | 0.56 | 4.4 |
| BDT-2C6-Sa | No | 3.59 | 0.97 | 0.30 | 1.1 |
| BDT-2C6-Sa | 110 °C 10 min | 2.43 | 0.86 | 0.29 | 0.6 |
| BDT-2C6-Sb | No | 5.67 | 0.80 | 0.56 | 2.5 |
| BDT-EH-S-BTc | 110 °C 10 min | 9.90 | 0.77 | 0.56 | 4.3 |
After thermal annealing, the BDT-EH-S:[60]PCBM film shows small but distinguishable domain features in TEM (Fig. 5b and S8b ESI† for TEM at higher magnification) and a significantly increased PCE of 4.4%, mainly as a result of an increased photocurrent and fill factor. The maximum EQE is 52% (Fig. S8a, ESI†). For BDT-EH-S:[60]PCBM films a higher PCE of 5.79% has been reported for a layer processed from chloroform and annealed at 110 °C for 10 min.53 The photovoltaic parameters of the reported record device (Jsc = 11.97 mA cm−2, Voc = 0.84 V, FF = 0.576)53 show that the main difference with our result is a higher short-circuit current. In our hands processing from chloroform in combination with thermal annealing gave PCE = 4.3% (Table S6, ESI†).
Thermal annealing of the BDT-2C6-S:[60]PCBM films, on the other hand, results in a significant loss of the PCE, mainly due to a loss of photocurrent and a fill factor that remains low. In TEM the BDT-2C6-S:[60]PCBM active layer exhibits large worm-like domains after thermal annealing (Fig. 5d and S9b ESI† for TEM at higher magnification). The increased domain size can explain the lower photocurrent, but does not explain the very low FF, which might be due to the absence of percolating pathways for charges.
The TEM images in Fig. 5 show that in the blends of BDT-EH-S and BDT-2C6-S with [60]PCBM the degree of phase separation is similar directly after spin coating from chloroform containing 0.2% 1-CN, but that thermal annealing results in much larger domains for BDT-2C6-S:[60]PCBM than for BDT-EH-S:[60]PCBM. The difference is likely related to the different tendency of the two molecules to crystallize. X-ray diffraction (XRD) on pure (i.e. without [60]PCBM) drop-cast films of BDT-EH-S and of BDT-2C6-S shown in Fig. 6 reveals that the main diffraction peak is much more intense for BDT-2C6-S than for BDT-EH-S. This lends support to the conclusion from TEM that BDT-2C6-S crystallizes more easily than BDT-EH-S. The TEM results indicate that in as-cast films the crystallization of bis-DPP molecules is reduced in the presence of [60]PCBM and that the extent to which crystallization can be enhanced by thermal annealing depends on the molecular structure.
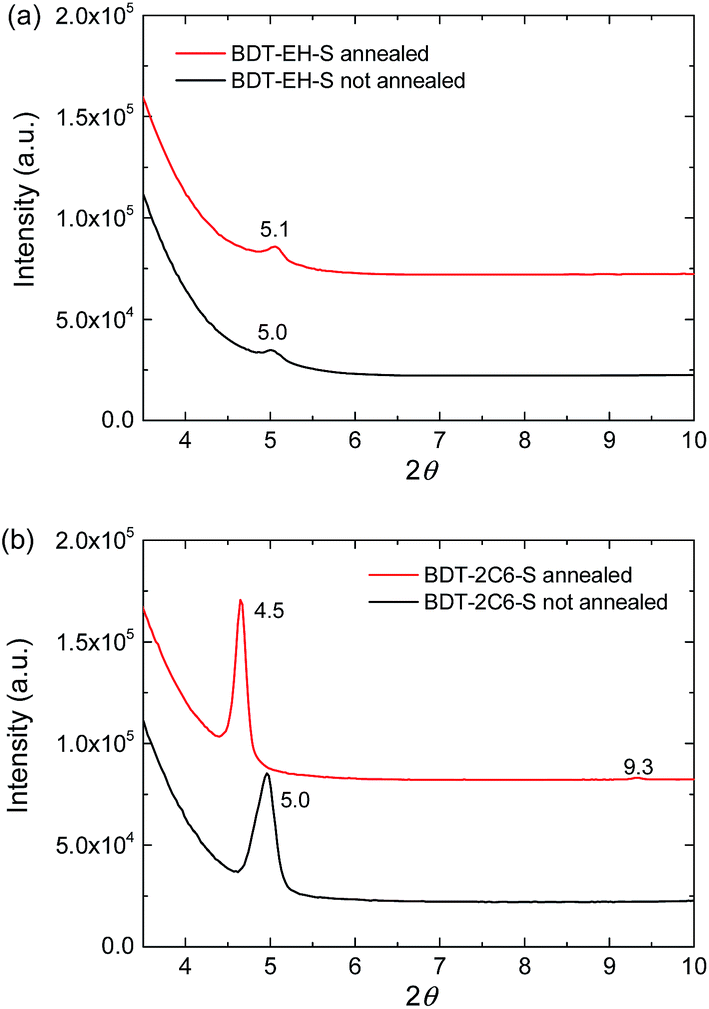 | ||
| Fig. 6 X-ray diffractograms of thin films of (a) BDT-EH-S and (b) BDT-2C6-S before and after thermal annealing. | ||
Table 4 shows that both the BDT-EH-S:[60]PCBM and BDT-2C6-S:[60]PCBM solar cells have a ∼100 mV lower open-circuit voltage after thermal annealing of the active layer. We attribute this to the aggregation of the two molecules and a concomitant shift of frontier orbital energy levels. In the XRD of the pure BDT-2C6-S films we see a narrowing and shift of the diffraction peak of BDT-2C6-S after annealing. The shift indicates a change of the lattice constants and crystal structure.
The best performance for BDT-2C6-S:[60]PCBM (PCE = 2.5% and maximum EQE = 36%) was obtained when the layer was spin coated from chloroform containing 0.3% 1,8-diiodooctane (DIO) (Table 4). The increase in Jsc and FF and concomitant decrease in Voc for this device as compared to the un-annealed layer from chloroform containing 0.2% 1-CN, suggest that the use of DIO enhances the formation of crystalline domains.
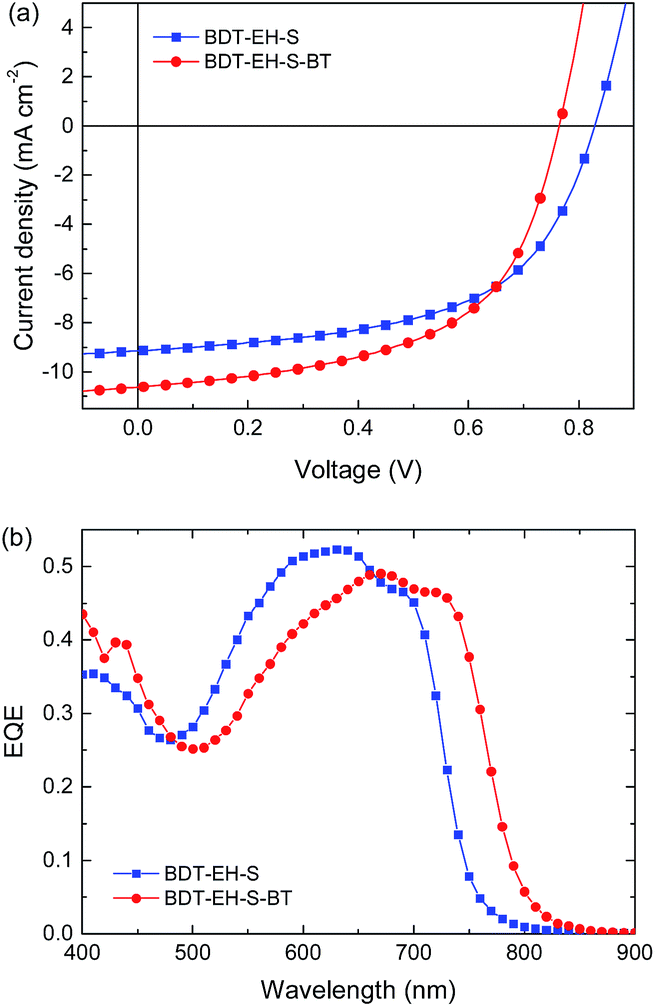 | ||
| Fig. 7 (a) J–V characteristics and (b) EQE of optimized devices of BDT-EH-S and BDT-EH-S-BT with [60]PCBM. | ||
 | ||
| Fig. 8 TEM images of optimized photoactive layers of BDT-EH-S (a) and BDT-EH-S-BT (b) with [60]PCBM after thermal annealing at 110 °C for 10 min. | ||
| Material | No co-solvent | DIO (0.2%) | o-DCB (0.2%) | 1-CN (0.2%) |
|---|---|---|---|---|
| a These PCE values were obtained from the J–V characteristics under white light illumination and can differ from the more accurate values mentioned in Tables 2–5 where the Jsc was obtained from integration of the EQE spectrum. b After thermal annealing at 110 °C for 5 or 10 min. c Using 0.5% o-DCB. d Using 0.1% DIO. e Using 0.3% DIO. | ||||
| TT-S | 2.9b | 1.8 | 2.3b | 4.8b |
| DTT-S | 2.2 | 1.8 | 1.8 | 2.5 |
| DTT-L | 1.1 | 1.2 | 1.7 | 2.6 |
| BDT-S | 1.7 | 1.5 | 1.3c | 2.7 |
| BDT-L | 0.8b | 1.2b | 1.1b | 2.3 |
| BDT-EH-S | 4.3b | 4.5b,d | 3.4c | 4.3b |
| BDT-2C6-S | 1.5b | 2.5e | 1.1 | 1.2 |
| BDT-EH-S-BT | 4.6b | 3.7 | 4.0b | 4.3b |
| Material | Solvent | As cast | Annealedb | +/− |
|---|---|---|---|---|
| a These PCE values were obtained from the J–V characteristics under white light illumination and can differ from the more accurate values mentioned in Tables 1–4, where the Jsc was obtained from integration of the EQE spectrum. b At 110 °C for 5 or 10 min. | ||||
| TT-S | CHCl3/1-CN (0.2%) | 3.9 | 4.8 | + |
| DTT-S | CHCl3/1-CN (0.2%) | 2.5 | 1.1 | − |
| DTT-L | CHCl3/1-CN (0.2%) | 2.6 | 1.6 | − |
| BDT-S | CHCl3/1-CN (0.2%) | 2.7 | 1.8 | − |
| BDT-L | CHCl3/1-CN (0.2%) | 2.3 | 1.9 | − |
| BDT-EH-S | CHCl3/1-CN (0.2%) | 2.5 | 4.3 | + |
| BDT-2C6-S | CHCl3/DIO (0.2%) | 1.9 | 0.5 | − |
| BDT-EH-S-BT | CHCl3 | 4.2 | 4.6 | + |
The role of the co-solvent in solution-processed small-molecule solar cells remains a point of discussion. A very strong dependence of the PCE on subtle changes in the amount of co-solvent has been noted before for small-molecule bulk-heterojunctions solar cells,110 but also less pronounced effects have been reported.111 For DPP-based polymers it was recently shown that the solubility of the DPP-polymer determines the width of the polymer fibrils formed.109 Co-solvents that provide a lower solubility for the DPP-polymer, gave smaller domain sizes and higher PCEs. In general the effect of the co-solvent for the bis-DPP:[60]PCBM mixtures is less outspoken than for the polymers and there is no obvious trend in the data. There is an important difference, between co-solvents used for small-molecule and for polymer solar cells. The amount of co-solvent that gives optimized performance is smaller for bis-DPP small molecules (∼0.2 vol%) than for DPP polymer solar cells (>3 vol%). Because the co-solvents have a lower evaporation rate than the main solvent, this implies that for polymer solar cells the last phase of film drying where the main solvent has evaporated, the deposition is almost exclusively from the co-solvent. In contrast, for small-molecule bis-DPPs, where the amount of co-solvent is a factor of ten less, the primary solvent plays a significant role during the entire deposition. In fact, for the bis-DPP molecules the optimal amount of co-solvent of about 0.2 vol% (corresponding to 2.4 to 3.6 mg ml−1, depending on the co-solvent) is less than the concentration of the semiconductors (typical total concentration used 10–20 mg ml−1).
3. Conclusions
The effects of structural variations in the core, the side chains, and the end groups on the photovoltaic performance has been studied in a series of bis-DPP small molecules for organic solar cells in an attempt to understand structure – processing – morphology – performance relationships. Molecules BDT-EH-S and BDT-EH-S-BT reach PCEs of ∼4.3% in blends with [60]PCBM after thermal annealing. Each of these blends has a morphology that features small, but distinct domains. For TT-S the PCE also reaches 4.3% in combination with [60]PCBM. In this case TEM gives no clear evidence of phase separation, but AFM shows distinct surface features.Molecules with longer side chains (DTT-L and BDT-L) give coarser phase separation because they can give rise to spinodal demixing of the solution and because their critical nucleus size is larger. To achieve small domain sizes that provide efficient exciton dissociation and good efficiency, the solubility of the compounds is a critical parameter that should not be too high.
For the bis-DPP molecules the use of co-solvent results in an improved solar cell performance after spin coating. While co-solvents such as DIO and o-DCB can be used, 1-CN provided in general the best performance. For the bis-DPPs molecules the amount of co-solvent required for best performance is usually 0.2 vol%, and hence much less than typical values (3 vol% to 10 vol%) needed for reaching optimized performance for DPP polymers.107–109 For DPP polymers we have previously established that the co-solvent induces polymer aggregation in the last stages of the drying where the total solvent content has decreased to about 80% and the remaining solvent fraction is mainly comprised of the high-boiling co-solvent.108 At the low concentrations of co-solvent used for the bis-DPP molecules, the mechanism is likely to be different, because the amount of co-solvent in the casting solution is initially of the same order, or even less, as that of the semiconductors. Thus the film will be virtually dry before the co-solvent has become the main solvent component.
Thermal annealing improves the efficiency of the cells of TT-s, BDT-EH-S and BDT-EH-S-BT where casting results in intimately mixed films. By blending with [60]PCBM the crystallization of these bis-DPP is retarded and it can be accomplished by thermal annealing. However, thermal annealing can also decrease the device performance. This occurs when the initial phase separation is already coarse (DTT-L and BDT-L), but also can also happen in cases (DTT-S, BDT-S, and BDT-2C6-S) where the initial morphology is finely mixed. In the latter cases we find that the thermal annealing results in a significantly reduced fill factor, which signifies that charge collection is hampered, despite a more crystalline morphology.
With these results, some structural factors for designing successful bis-DPP molecules for solution-processed organic solar cells emerge. Solubility and tendency to crystallize are important to achieve the small domain size (10–20 nm) which is necessary for a good performance. Yet, strong effects are seen with rather subtle structural changes and, hence, evolutionary rather than rational design of successful molecules will remain the most likely option to progress in this field for some time.
Acknowledgements
This research forms part of the research programme of the Dutch Polymer Institute (DPI), projects #734 and #762. The research leading to these results has further received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement No. 339031. The research is part of the Solliance OPV program and has received funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035).Notes and references
- S.-H. Liao, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- S. Zhang, L. Ye, W. Zhao, B. Yang, Q. Wang and J. Hou, Sci. China: Chem., 2015, 58, 248–256 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed.
- L. Lu, T. Zheng, T. Xu, D. Zhao and L. Yu, Chem. Mater., 2015, 27, 537–543 CrossRef CAS.
- H. Zong, C.-Z. Li, J. Carpenter, H. Ade and A. K.-Y. Jen, J. Am. Chem. Soc., 2015, 137, 7616–7619 CrossRef PubMed.
- T. Vangerven, P. Verstappen, J. Drijkoningen, W. Dierckx, S. Himmelberger, A. Salleo, D. Vanderzande, W. Maes and J. V. Manca, Chem. Mater., 2015, 27, 3726–3732 CrossRef CAS.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed.
- S. Roquet, A. Cravino, P. Leriche, O. Aleveque, P. Frere and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS PubMed.
- N. M. Kronenberg, M. Deppisch, F. Würthner, H. W. A. Lademann, K. Deing and K. Meerholz, Chem. Commun., 2008, 6489–6491 RSC.
- U. Mayerhöffer, K. Deing, K. Gruß, H. Braunschweig, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2009, 48, 8776–8779 CrossRef PubMed.
- A. K. K. Kyaw, D. H. Wang, D. Wynands, J. Zhang, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Nano Lett., 2013, 13, 3796–3801 CrossRef CAS PubMed.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341–8344 CrossRef CAS PubMed.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- Z. Yi, S. Wang and Y. Liu, Adv. Mater., 2015, 27, 3589–3606 CrossRef CAS PubMed.
- B. Tamayo, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545–11551 CrossRef.
- B. Walker, A. B. Tamayo, X.-D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T.-Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS.
- A. B. Tamayo, X.-D. Dang, B. Walker, J. Seo, T. Kent and T.-Q. Nguyen, Appl. Phys. Lett., 2009, 94, 103301 CrossRef.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- B.-S. Jeong, H. Choi, N. Cho, H. M. Ko, W. Lim, K. Song, J. K. Lee and J. Ko, Sol. Energy Mater. Sol. Cells, 2011, 95, 1731–1740 CrossRef CAS.
- Y. Zhang, X.-D. Dang, C. Kim and T.-Q. Nguyen, Adv. Energy Mater., 2011, 1, 610–617 CrossRef CAS.
- J. Mei, K. R. Graham, R. Stadler, S. P. Tiwari, H. Cheun, J. Shim, M. Yoshi, C. Nuckolls, B. Kippelen, R. K. Castellano and J. R. Reynolds, Chem. Mater., 2011, 23, 2285–2288 CrossRef CAS.
- W. Kylberg, P. Sonar, J. Heier, J.-N. Tisserant, C. Müller, F. Nüesch, Z.-L. Chen, A. Dodabalapur, S. Yoon and R. Hany, Energy Environ. Sci., 2011, 4, 3617–3624 Search PubMed.
- E. Ripaud, D. Demeter, T. Rousseau, E. Boucard-Cétol, M. Allain, R. Po, P. Leriche and J. Roncali, Dyes Pigm., 2012, 95, 126–133 CrossRef CAS.
- Y. Lin, P. Cheng, Y. Li and X. Zhan, Chem. Commun., 2012, 48, 4773–4775 RSC.
- Y. Liu, X. Du, Z. Xiao, J. Cao, S. Tan, Q. Zuo and L. Ding, Synth. Met., 2012, 162, 1665–1671 CrossRef CAS.
- J. H. Liu, B. Walker, A. Tamayo, Y. Zhang and T. Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS.
- H. Wang, F. Liu, L. Bu, J. Gao, C. Wang, W. Wei and T. P. Russell, Adv. Mater., 2013, 25, 6519–6525 CrossRef CAS PubMed.
- V. S. Gevaerts, E. M. Herzig, M. Kirkus, K. H. Hendriks, M. M. Wienk, J. Perlich, P. Müller-Buschbaum and R. A. J. Janssen, Chem. Mater., 2014, 26, 916–926 CrossRef CAS.
- Q.-R. Yin, J.-S. Miao, Z. Wu, Z.-F. Chang, J.-L. Wang, H.-B. Wu and Y. Cao, J. Mater. Chem. A, 2015, 3, 11575–11586 RSC.
- J.-L. Wang, Z. Wu, J.-S. Miao, K.-K. Liu, Z.-F. Chang, R.-B. Zhang, H.-B. Wu and Y. Cao, Chem. Mater., 2015, 27, 4338–4348 CrossRef CAS.
- M. E. Farahat, D. Patra, C.-H. Lee and C.-W. Chu, ACS Appl. Mater. Interfaces, 2015, 7, 22542–22550 Search PubMed.
- S. Loser, C. J. Bruns, H. Miyauchi, R. C. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 21, 8142–8145 CrossRef PubMed.
- S. Loser, H. Miyauchi, J. W. Hennek, J. Smith, C. Huang, A. Facchetti and T. J. Marks, Chem. Commun., 2012, 48, 8511–8513 RSC.
- P.-L. T. Boudreault, J. W. Hennek, S. Loser, R. P. Ortiz, B. J. Eckstein, A. Facchetti and T. J. Marks, Chem. Mater., 2012, 24, 2929–2942 CrossRef CAS.
- D. Sahu, C.-H. Tsai, H.-Y. Wei, K.-C. Ho, F.-C. Chang and C.-W. Chu, J. Mater. Chem., 2012, 22, 7945–7953 RSC.
- W. W. H. Wong, J. Subbiah, S. R. Puniredd, B. Purushothaman, W. Pisula, N. Kirby, K. Müllen, D. J. Jones and A. B. Holmes, J. Mater. Chem., 2012, 22, 21131–21137 RSC.
- J. W. Lee, Y. S. Choi and W. H. Jo, Org. Electron., 2012, 13, 3060–3066 CrossRef CAS.
- J. K. Park, C. Kim, B. Walker, T.-Q. Nguyen and J. H. Seo, RSC Adv., 2012, 2, 2232–2234 RSC.
- J.-Y. Pan, L.-J. Zuo, X.-L. Hu, W.-F. Fu, M.-R. Chen, L. Fu, X. Gu, H.-Q. Shi, M.-M. Shi, H.-Y. Li and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 972–980 Search PubMed.
- J. Huang, C. Zhan, X. Zhang, Y. Zhao, Z. Lu, H. Jia, B. Jiang, J. Ye, S. Zhang, A. Tang, Y. Liu, Q. Pei and J. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2033–2039 Search PubMed.
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724–728 CrossRef CAS.
- Y. Lin, L. Ma, Y. Li, Y. Liu, D. Zhu and X. Zhan, Adv. Energy Mater., 2013, 3, 1166–1170 CrossRef CAS.
- C. M. Proctor, C. Kim, D. Neher and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 3584–3594 CrossRef CAS.
- B. Walker, J. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo, G. C. Bazan and T.-Q. Nguyen, Energy Environ. Sci., 2013, 6, 952–962 Search PubMed.
- Y.-A. Duan, Y. Geng, H.-B. Li, J.-L. Jin, Y. Wu and Z.-M. Su, J. Comput. Chem., 2013, 34, 1611–1619 CrossRef CAS PubMed.
- L. Li, Y. Huang, J. Peng, Y. Cao and X. Peng, J. Mater. Chem. A, 2013, 1, 2144–2150 RSC.
- A. Tang, L. Li, Z. Lu, J. Huang, H. Jia, C. Zhan, Z. Tan, Y. Li and J. Yao, J. Mater. Chem. A, 2013, 1, 5747–5757 RSC.
- W. Li, M. Kelchtermans, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2013, 1, 15150–15157 RSC.
- L. Zhang, S. Zeng, L. Yin, C. Ji, K. Li, Y. Li and Y. Wang, New J. Chem., 2013, 37, 632–639 RSC.
- Y. S. Choi and W. H. Jo, Org. Electron., 2013, 14, 1621–1628 CrossRef CAS.
- A. Guerrero, S. Loser, G. Garcia-Belmonte, C. J. Bruns, J. Smith, H. Miyauchi, S. I. Stupp, J. Bisquert and T. J. Marks, Phys. Chem. Chem. Phys., 2013, 15, 16456–16462 RSC.
- J. Huang, X. Wang, X. Zhang, Z. Niu, Z. Lu, B. Jiang, Y. Sun, C. Zhan and J. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3853–3862 Search PubMed.
- Q.-C. Yu, W.-F. Fu, J.-H. Wan, X.-F. Wu, M.-M. Shi and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5798–5809 Search PubMed.
- S.-Y. Liu, W.-Q. Liu, J.-Q. Xu, C.-C. Fan, W.-F. Fu, J. Ling, J.-Y. Wu, M.-M. Shi, A. K.-Y. Jen and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6765–6775 Search PubMed.
- M. Jung, Y. Yoon, J. H. Park, W. Cha, A. Kim, J. Kang, S. Gautam, D. Seo, J. H. Cho, H. Kim, J. Y. Choi, K. H. Chae, K. Kwak, H. J. Son, M. J. Ko, H. Kim, D.-K. Lee, J. Y. Kim, D. H. Choi and B. S. Kim, ACS Nano, 2014, 8, 5988–6003 CrossRef CAS PubMed.
- W. Shin, T. Yasuda, Y. Hidaka, G. Watanabe, R. Arai, K. Nasu, T. Yamaguchi, W. Murakami, K. Makita and C. Adachi, Adv. Energy Mater., 2014, 4, 1400879 CrossRef.
- Z. Niu, X. Wang, J. Huang, A. Tang, Y. Sun and C. Zhan, Asian J. Org. Chem., 2014, 3, 948–952 CrossRef CAS.
- Q.-C. Yu, W.-F. Fu, H.-Y. Wang, X.-F. Wu, J.-H. Wan, M.-M. Shi and H.-Z. Chen, Asian J. Org. Chem., 2014, 3, 984–993 CrossRef CAS.
- T. Harschneck, N. Zhou, E. F. Manley, S. J. Lou, X. Yu, M. R. Butler, A. Timalsina, R. Turrisi, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Chem. Commun., 2014, 50, 4099–4101 RSC.
- Z. Du, W. Chen, S. Wen, S. Qiao, Q. Liu, D. Ouyang, N. Wang, X. Bao and R. Yang, ChemSusChem, 2014, 7, 3319–3327 CrossRef CAS PubMed.
- H. Qin, L. Li, F. Guo, S. Su, J. Peng, Y. Cao and X. Peng, Energy Environ. Sci., 2014, 7, 1397–1401 Search PubMed.
- H. Bai, P. Cheng, Y. Wang, L. Ma, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2014, 2, 778–784 RSC.
- W. Liu, S. Liu, N. K. Zawack, T. R. Andersen, P. Cheng, L. Fu, M. Chen, W. Fu, E. Bundgaard, M. Jørgensen, X. Zhan, F. C. Krebs and H. Chen, J. Mater. Chem. A, 2014, 2, 19809–19814 RSC.
- Y. Chen, A. Tang, X. Zhang, Z. Lu, J. Huang, C. Zhan and J. Yao, J. Mater. Chem. A, 2014, 2, 1869–1876 RSC.
- C. Yu, Z. Liu, Y. Yang, J. Yao, Z. Cai, H. i. Luo, G. Zhang and D. Zhang, J. Mater. Chem. C, 2014, 2, 10101–10109 RSC.
- S. Y. Liu, W. F. Fu, J. Q. Xu, C. C. Fan, H. Jiang, M. Shi, H. Y. Li, J. W. Chen, Y. Cao and H. Z. Chen, Nanotechnology, 2014, 25, 014006 CrossRef PubMed.
- Y. Zhang, H. Tan, M. Xiao, X. Bao, Q. Tao, Y. Wang, Y. Liu, R. Yang and W. Zhu, Org. Electron., 2014, 15, 1173–1183 CrossRef CAS.
- H.-F. Feng, W.-F. Fu, L. Li, Q.-C. Yu, H. Lu, J.-H. Wan, M.-M. Shi, H. Z. Chen, Z. Tan and Y. Li, Org. Electron., 2014, 15, 2575–2586 CrossRef CAS.
- A. M. Raynor, A. Gupta, H. Patil, A. Bilic and S. V. Bhosale, RSC Adv., 2014, 4, 57635–57638 RSC.
- H. Patil, A. Gupta, A. Bilic, S. V. Bhosale and S. V. Bhosale, Tetrahedron Lett., 2014, 55, 4430–4432 CrossRef CAS.
- X. Duan, M. Xiao, J. Chen, X. Wang, W. Peng, L. Duan, H. Tan, G. Lei, R. Yang and W. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 18292–18299 Search PubMed.
- J.-H. Kim, J. B. Park, H. Yang, I. H. Jung, S. C. Yoon, D. Kim and D.-H. Hwang, ACS Appl. Mater. Interfaces, 2015, 7, 23866–23875 Search PubMed.
- A. Tang, C. Zhan and J. Yao, Adv. Energy Mater., 2015, 5, 1500059 CrossRef.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- Z. Tang, B. Liu, A. Melianas, J. Bergqvist, W. Tress, Q. Bao, D. Qian, O. Inganäs and F. Zhang, Adv. Mater., 2015, 27, 1900–1907 CrossRef CAS PubMed.
- J. W. Jung, T. P. Russell and W. H. Jo, Chem. Mater., 2015, 27, 4865–4870 CrossRef CAS.
- J. W. Jung and W. H. Jo, Chem. Mater., 2015, 27, 6038–6043 CrossRef CAS.
- M. Jung, D. Seo, K. Kwak, A. Kim, W. Cha, H. Kim, Y. Yoon, M. J. Ko, D.-K. Lee, J. Y. Kim, H. J. Son and B. S. Kim, Dyes Pigm., 2015, 115, 23–34 CrossRef CAS.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. Russell, R. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS PubMed.
- B. Chen, Y. Yang, P. Cheng, X. Chen, X. Zhan and J. Qin, J. Mater. Chem. A, 2015, 3, 6894–6900 RSC.
- D. Qian, B. Liu, S. Wang, S. Himmelberger, M. Linares, M. Vagin, C. Müller, Z. Ma, S. Fabiano, M. Berggren, A. Salleo, O. Inganäs, Y. Zou and F. Zhang, J. Mater. Chem. A, 2015, 3, 24349–24357 RSC.
- N. Zhou, S. Vegiraju, X. Yu, E. F. Manley, M. R. Butler, M. J. Leonardi, P. Guo, W. Zhao, Y. Hu, K. Prabakaran, R. P. H. Chang, M. A. Ratner, L. X. Chen, A. Facchetti, M. C. Chen and T. J. Marks, J. Mater. Chem. C, 2015, 3, 8932–8941 RSC.
- Y. Zhang, M. Xiao, N. Su, J. Zhong, H. Tan, Y. Wang, Y. Liu, Y. Pei, R. Yang and W. Zhu, Org. Electron., 2015, 17, 198–207 CrossRef CAS.
- W. Liu, H. Shi, W. Fu, L. Zuo, L. Wang and H. Chen, Org. Electron., 2015, 25, 219–224 CrossRef CAS.
- S. Wang, J. Yang, Z. Zhang, Y. Hu, X. Cao, H. Li, Y. Tao, Y. Li and W. Huang, RSC Adv., 2015, 5, 68192–68199 RSC.
- T. W. Lee, D. H. Lee, T. R. Hong, J. Shin, M. J. Cho and D. H. Choi, Synth. Met., 2015, 206, 24–32 CrossRef CAS.
- J. Chen, M. Xiao, F. Meng, L. Duan, H. Tan, Y. Wang, Y. Liu, R. Yang and W. Zhu, Synth. Met., 2015, 199, 400–407 CrossRef CAS.
- D. Yu, Y. Liu, M. Xiao, Q. Fan, W. Su, X. Li, H. Tan, Y. Wang, R. Yang and W. Zhu, Dyes Pigm., 2016, 125, 151–158 CrossRef CAS.
- X. Liu, C. Huang, W. Shen, R. He and M. Li, J. Mol. Model., 2016, 22, 15 CrossRef PubMed.
- T. Liang, L. Xiao, C. Liu, K. Gao, H. Qin, Y. Cao and X. Peng, Org. Electron., 2016, 29, 127–134 CrossRef CAS.
- J. Chen, M. Xiao, L. Duan, Q. Wang, H. Tan, N. Su, Y. Liu, R. Yang and W. Zhu, Phys. Chem. Chem. Phys., 2016, 18, 1507–1515 RSC.
- Y. Lin, J. Wang, T. Li, Y. Wu, C. Wang, L. Han, Y. Yao, W. Ma and X. Zhan, J. Mater. Chem. A, 2016, 4, 1486–1494 RSC.
- J. Wang, K. Shi, Y. Suo, Y. Lin, G. Yu and X. Zhan, J. Mater. Chem. C, 2016, 4, 3781–3791 RSC.
- C. Lu and W. C. Chen, Chem.–Asian J., 2013, 8, 2813–2821 CrossRef CAS PubMed.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS PubMed.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS PubMed.
- J. J. van Franeker, G. H. L. Heintges, C. Schaefer, G. Portale, W. Li, M. M. Wienk, P. van der Schoot and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 11783–117945 CrossRef CAS PubMed.
- C. J. Takacs, Y. Sun, G. C. Welch, L. A. Perez, X. Liu, W. Wen, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 16597–16606 CrossRef CAS PubMed.
- H. Wang, F. Liu, L. Bu, J. Gao, C. Wang, W. Wei and T. P. Russell, Adv. Mater., 2013, 25, 6519–6525 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphs and tables. See DOI: 10.1039/c6ta01533f |
| This journal is © The Royal Society of Chemistry 2016 |