
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of flux synthesis on SrNbO2N particles for photoelectrochemical water splitting†
Masanori
Kodera
,
Haruki
Urabe
,
Masao
Katayama
,
Takashi
Hisatomi
,
Tsutomu
Minegishi
 and
Kazunari
Domen
*
and
Kazunari
Domen
*
Department of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
First published on 7th April 2016
Abstract
An investigation was carried out on the effects of using a flux during the fabrication of SrNbO2N particles on the photoelectrochemical (PEC) properties of CoPi/SrNbO2N/Nb/Ti photoanodes prepared by a particle transfer method. The type of flux and the synthesis conditions were found to affect the morphology and crystallinity of oxide precursors and subsequently nitrided SrNbO2N. By a suitable choice of flux synthesis conditions, the PEC performance of the photoanodes could be improved. Oxide precursors treated with a NaI flux were solid solutions of NaNbO3 and Sr4Nb2O9, and had a perovskite-type structure. When the precursors were first pre-calcined, larger secondary SrNbO2N particles with a higher degree of crystallinity were obtained. As a result, a photocurrent density of 1.5 mA cm−2 at 1.23 VRHE was achieved under simulated sunlight (AM 1.5G) in an alkaline solution (pH 13).
Introduction
Recently, there is increasing awareness that fossil fuels are in danger of becoming depleted and are responsible for global warming.1 Therefore, the development of new alternative energy sources has become a strong priority. To this end, hydrogen has received a considerable amount of attention as a renewable green fuel, and photoelectrochemical (PEC) water splitting under sunlight is a promising method for clean hydrogen production.2,3 Among several possible cell configurations, p/n PEC cells consisting of a photoanode and a photocathode are considered to be potential candidates for water splitting without the need for an external bias. In order for this technology to be practical, a solar-to-hydrogen (STH) conversion efficiency of 10% is considered to be a requirement. To achieve this, and assuming that a quantum efficiency (QE) of 60–70% can be obtained in the desired wavelength range, photoelectrodes that can absorb light at wavelengths greater than 600 nm must be used. Thus, the development of photoelectrodes with narrow bandgaps (<2.0 eV) is required.There have been reports of (oxy)nitrides such as Ta3N5 and LaTiO2N, with absorption edges at about 600 nm, both in the form of powder suspensions and as photoelectrodes.4–7 In particular, perovskite-type oxynitrides with the general formula AB(O,N)3 have been intensively studied.8,9 In the last several years, reports focusing on Ta-based oxynitrides such as BaTaO2N have appeared.10,11 However, there have been few studies on Nb-based perovskite-type oxynitrides, especially for use as photoelectrodes.12,13 The latter are an attractive material group because their absorption edge is longer than that of other perovskite-type oxynitrides. Furthermore, Nb is more abundant and less expensive than Ta.
SrNbO2N is a perovskite-type oxynitride with a bandgap of 1.8 eV, and in a powder suspension it can produce both oxygen and hydrogen in the presence of suitable sacrificial reagents.14,15 Some of the present authors previously reported that SrNbO2N exhibited n-type semiconducting characteristics and acted as a photoanode in a PEC water splitting cell.16 Its PEC performance, however, was very low as expected from its wide light absorption range (λ < 700 nm), and it is therefore necessary to improve it. In the previous study, SrNbO2N particles were synthesized using a polymerized complex technique, and photoanodes were prepared using a particle transfer method.17 The resulting particles were aggregates of small primary particles with sizes of a few hundred nanometres. The limited photocurrent density of the SrNbO2N photoelectrodes could be attributed to the low crystallinity of these particles and the electrical resistance at the grain boundaries, which promoted the recombination of the photo-excited carriers. Stability under illumination was another problem for this material. When CoOx was used as a cocatalyst for water oxidation, the photocurrent rapidly decreased and became almost zero after 10 min at a potential of 1.0 VRHE.
The flux method has been applied to obtain highly crystalline materials. It is widely applied to oxide photocatalysts18 while there are only few studies on the flux method to oxynitrides and/or their precursors. For example, La2Ti2O7 synthesized using an NaCl–KCl flux had high crystallinity, and LaTiO2N obtained by nitridation of this material exhibited higher photocatalytic oxygen evolution activity from an Ag+ containing solution than LaTiO2N produced using other methods.6 BaTaO2N formed by nitriding an oxide precursor in the presence of a KCl flux was shown to have a cubic morphology and excellent crystallinity, resulting in a higher photocatalytic activity than a similar compound produced without a flux treatment.19 Thus, the flux method is expected to be effective for improving photocatalytic activity by enhancing the crystallinity of particles.
Here, we report the synthesis of SrNbO2N powder using different types of flux and different flux treatment conditions, and an evaluation of the PEC performance of photoanodes produced using these samples. An investigation was carried out on the effects of using a flux treatment at different stages in the preparation process, the effects of the type of flux, and the effects of pre-calcination on the oxide precursors.
Experimental
Synthesis procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb:NaCl = 1:1:5. The mixture was heated in an alumina crucible for 5 h at 1423 K in air, then cooled to 1073 K at a rate of 1 K min−1, and finally allowed to cool to room temperature. The obtained oxide precursor was then heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1.
:Nb:NaCl = 1:1:5. The mixture was heated in an alumina crucible for 5 h at 1423 K in air, then cooled to 1073 K at a rate of 1 K min−1, and finally allowed to cool to room temperature. The obtained oxide precursor was then heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1.
As a flux treatment during nitridation, SrNbO2N was prepared by the following procedure. A mixture of SrCO3, Nb2O5 and NaCl in a molar ratio of Sr:Nb:NaCl = 1:1:5 in an alumina boat was heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1, and then cooled to room temperature without interrupting the NH3 flow. The resulting oxynitride was washed thoroughly with water, filtered, and dried at room temperature.
The third method was a post-nitridation flux treatment of SrNbO2N. About 0.3 g of quartz wool was loaded with a mixture of SrCO3 and Nb2O5 in a molar ratio of Sr:Nb = 1:1, and heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1 to obtain SrNbO2N. The sample was mixed with a NaCl flux in a molar ratio of SrNbO2N:NaCl = 1:5, and heated for 3 h at 1173 K under an NH3 flow of 250 mL min−1 in an alumina boat. The final oxynitride was washed thoroughly with water, filtered, and dried.
As a reference, a SrNbO2N sample produced without any flux treatment was also prepared. About 0.3 g of quartz wool was loaded with a mixture of SrCO3 and Nb2O5 in a molar ratio of Sr:Nb = 1:1, and heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1. In the remainder of this paper, this sample is referred to as SNON (no flux). Similarly, oxynitride samples produced using a flux treatment during oxide synthesis, during nitridation, and the following nitridation are referred to as SNON (oxide), SNON (nitridation), and SNON (post), respectively.
:Nb:AX = 1:1:5. The mixture was heated in an alumina crucible for 5 h at 1423 K in air, cooled to 1073 K at a rate of 1 K min−1, and then allowed to cool to room temperature. As a reference, a sample heated without a flux was also prepared. In the nitridation process, the obtained oxide precursors were then heated for 15 h at 1173 K under an NH3 flow of 250 mL min−1.
The oxide precursors produced without and with a flux, and the resulting nitrided samples are referred to as SNO (no flux), SNO (AX), SNON (no flux) and SNON (AX), respectively.
:Nb molar ratio of 1:1 was first heated in an alumina crucible for 5 h at 1423 K in air, cooled to 1073 K at a rate of 1 K min−1, and then allowed to cool to room temperature. Subsequently, the product was treated by a flux (NaCl, RbCl, or NaI) using the same procedure as mentioned in (2). In the nitridation process, the oxide was heated for 15–20 h at 1173 or 1223 K under an NH3 flow of 250 mL min−1.
The pre-calcined oxide precursors and nitrided samples are referred to as SNO (AX, pre-calcined), SNON (AX, pre-calcined).
Characterization
The samples were characterized using X-ray diffraction (XRD; Rigaku, RINT-Ultima III), UV-Vis-NIR diffuse-reflectance spectroscopy (DRS; JASCO, V-670DS), field-emission scanning electron microscopy (FE-SEM; Hitachi, S-4700), and field-emission transmission electron microscopy (FE-TEM; JEOL, JEM-2800). Selected area electron diffraction (SAED) patterns were acquired in the same FE-TEM system. Elemental analysis was performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES; Shimadzu, ICPS-8100), and energy dispersive X-ray spectroscopy (EDX; Horiba, EMAX-7000). To estimate the faradaic efficiency of the SrNbO2N photoelectrodes under light irradiation, the evolved gases were analysed using micro-gas chromatography (GC; GC3000A, Inficon). Structural parameters were refined by Rietveld analysis using the RIETAN-FP code.20Preparation of SrNbO2N photoelectrodes
Photoelectrodes were prepared using the particle transfer method.21 SrNbO2N powder (30 mg) was dispersed in isopropanol (1 mL). After ultrasonication for more than 5 min, the suspension was deposited dropwise onto a glass plate. Nb and Ti bilayer films were deposited on the samples using a radio-frequency (RF) magnetron sputtering system (ULVAC, MNS-2000-RFG3). A Nb contact layer was first deposited at an RF power of 100 W for 5 min under an Ar pressure of 0.1 Pa. Subsequently, a Ti conductor layer was deposited at an RF power of 200 W for 3 h. The metal layers holding the photocatalyst powder were peeled off and transferred onto another glass plate using carbon tape. Residual particles physically attached to the main particle layer were removed by ultrasonication. Finally, an electrical wire was connected using indium solder, which was then covered with epoxy resin.Surface modification of photoelectrodes using a CoPi catalyst
Before the PEC measurements, the SrNbO2N photoelectrodes were loaded with a CoPi cocatalyst by electrodeposition.22 Co(NO3)2 (1 mM) was dissolved in an aqueous solution buffered with 0.1 M potassium phosphate at pH 6.9. The electrode samples were immersed in the electrolyte solution and the electrode potential was held at 1.7 V vs. the reversible hydrogen electrode (RHE) for 200 s under darkness to anodically deposit CoPi on the surface of the electrodes.Photoelectrochemical measurements
PEC measurements were carried out using a three-electrode configuration under intermittent illumination by AM 1.5G simulated sunlight (San'ei Denki, XES-40S2-CE). A Pt wire and an Ag/AgCl electrode were used as the counter and reference electrodes, respectively. The potential was converted into RHE units using the Nernst's equation (ERHE = EAg/AgCl + 0.0591 × pH +0.199). The potential was swept from positive to negative at 10 mV s−1. The electrolyte was a 0.2 M aqueous sodium phosphate solution with its pH adjusted to 13 by NaOH addition.Results and discussion
Flux treatment at different stages
The flux method was used at three different stages: during the synthesis of oxide precursors, nitridation, or post-nitridation treatment of oxynitrides. Fig. 1A shows XRD patterns of nitrided samples produced using an NaCl flux at different stages in the preparation process, together with that of the reference sample for which no flux was used. For all samples, the dominant phase was SrNbO2N. Compared to SNON (no flux), the intensity of the diffraction peaks for all of the flux treated samples increased remarkably, indicating that highly crystalline SrNbO2N was obtained. The full width at half maximum (FWHM) of the (110) peak for SNON (oxide), SNON (nitridation), and SNON (post) was 0.24, 0.11, and 0.16, respectively, indicating that the crystallinity was improved in the order SNON (oxide), SNON (post), and SNON (nitridation). | ||
| Fig. 1 (A) XRD patterns, (B) SEM images and (C) current–potential curves of SrNbO2N particles prepared (a) without a flux, and using an NaCl flux (b) during oxide preparation, (c) during nitridation, and (d) following nitridation. The black triangles in the XRD patterns indicate NbOxNy. The white bars in the SEM images represent 1 μm. The PEC measurements were performed under AM 1.5G irradiation in 0.2 M Na3PO4 with the pH adjusted to 13 by NaOH addition. | ||
The XRD patterns also indicated the presence of an impurity phase of NbOxNy (such as NbO0.1N0.9, PDF 00-025-1360), except for the case of SNON (oxide). In Fig. S1,† the DRS spectrum of SNON (oxide) is seen to exhibit relatively high background absorption at wavelengths longer than the absorption edge. This can presumably be attributed to anion vacancies and/or reduced Nb species in SrNbO2N that were produced during nitridation.17 These results suggest that the presence of nonstoichiometric species cannot be avoided using these flux treatments.
Fig. 1B shows the SEM images of the four different samples. It can be seen that SNON (no flux) and SNON (oxide) both have a porous morphology, similar to that previously reported for BaTaO2N.10 During nitridation of the oxide precursors, it is thought that many pores are formed as a result of replacing O with N atoms. On the other hand, in the cases of SNON (nitridation) and SNON (post), the surfaces of particles are smooth. The flux treatment during or after nitridation thus promoted crystal growth, resulting in particles with clear crystal habits.
Fig. 1C shows the PEC properties of SrNbO2N photoelectrodes prepared from the same samples. It can be seen that the flux treatment caused a clear enhancement of the anodic photocurrent, with SNON (oxide) exhibiting the largest photocurrent density. Therefore, a detailed investigation was carried out on the effects of the type of flux used for the oxide precursors on the PEC properties of SrNbO2N.
Effects of the flux type used for oxide precursors
Fig. 2A shows the XRD patterns of oxide precursors prepared with different types of fluxes. It is clear that the type of flux used had an effect on both the crystal structure and the phases that were present. The precursor calcined without a flux was a mixture of Sr5Nb4O15 and Sr2Nb2O7. In contrast, Cl- and Br-based fluxes produced Sr2Nb2O7 as the dominant phase. In addition, the use of KCl and RbCl led to the formation of impurity phases such as KSr2Nb3O10 and RbSr2Nb3O10, respectively, and a small amount of K or Rb could be detected by SEM-EDX (Table S1†). On the other hand, SNO (NaCl), SNO (NaBr) and SNO (SrCl2) did not contain such impurity phases, although a small amount of Sr5Nb4O15 was present. In the case of SNO (KI) and SNO (CsI), the dominant phase was Sr5Nb4O15, but K or Cs containing impurity phases, such as CsSr2Nb3O10, were identified.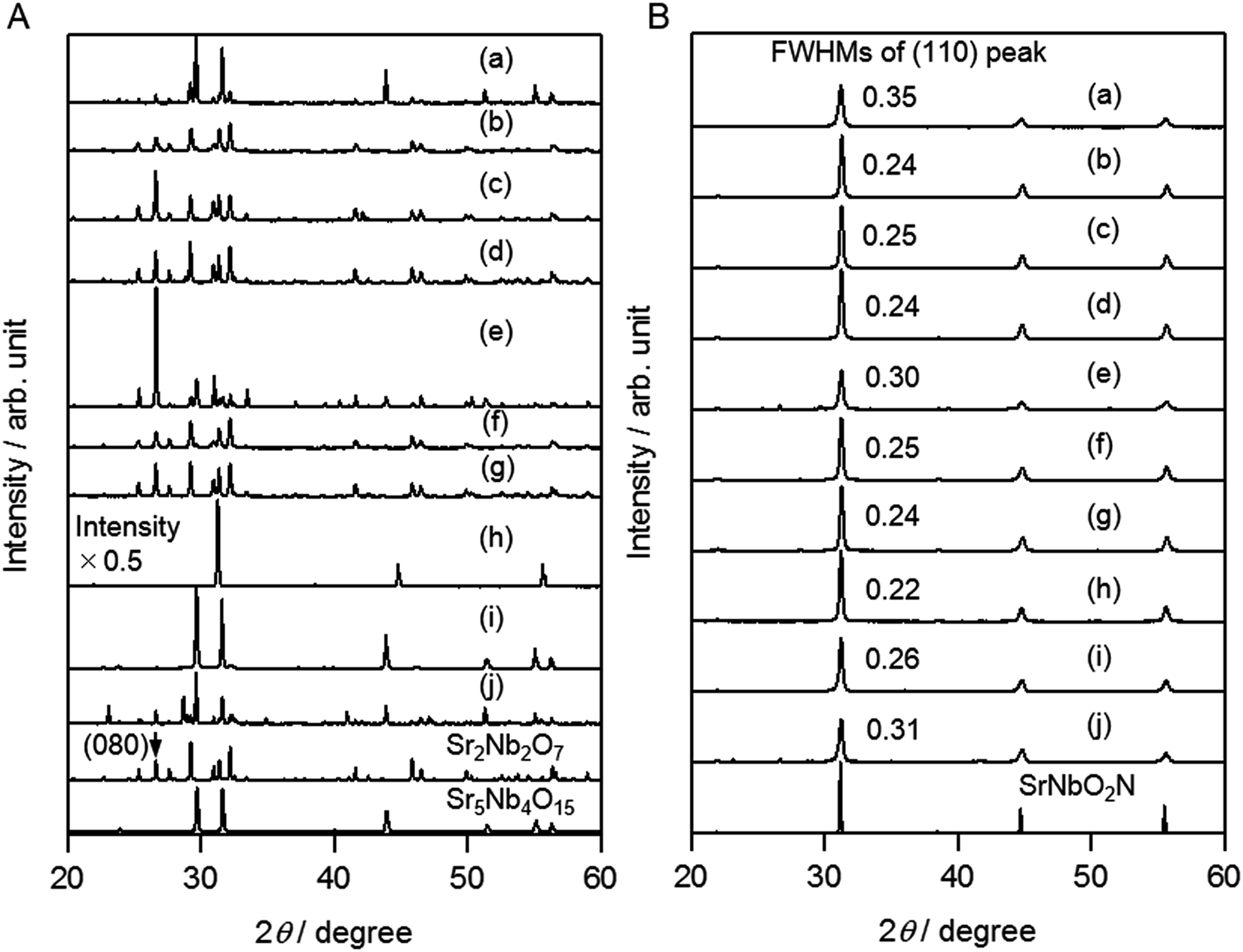 | ||
| Fig. 2 XRD patterns of (A) oxide precursors and (B) SrNbO2N particles produced (a) without flux, and with (b) NaCl, (c) KCl, (d) RbCl, (e) SrCl2, (f) NaBr, (g) KBr, (h) NaI, (i) KI, and (j) CsI fluxes. As references, patterns of Sr2Nb2O7 (PDF 00-052-0321), Sr5Nb4O15 (PDF 00-048-0421), and SrNbO2N (PDF 00-039-0675) are also shown. | ||
For SNO (NaI), the product was much different from the oxide precursors treated with the other kinds of flux. First, the XRD pattern in Fig. 2A indicates that it has a perovskite-type structure. In addition, in the DRS spectrum in Fig. S2k,† no optical absorption occurs in the visible region. ICP-AES and TEM-EDX measurements were performed to determine the composition of this sample, and the results are shown in Table S2.† It can be seen that a certain amount of Na is present. The nominal composition was determined to be Na0.36Sr0.87Nb0.78O3.
It has been reported that SrNbO3 is a perovskite-type oxide which has a red colour because the valence state of Nb is Nb4+.23,24 Na(1−x)SrxNbO3 also has a perovskite structure, and its colour is from light to deep blue due to the presence of Nb4+ species.25 Based on the DRS results in Fig. S2,† the SNO (NaI) produced in the present study was neither SrNbO3 nor Na(1−x)SrxNbO3.
The most likely possibility is that SNO (NaI) is a solid solution of NaNbO3 and Sr4Nb2O9. NaNbO3 is a perovskite-type oxide (PDF 01-075-2102), and Sr4Nb2O9, which can be denoted as (Sr(Sr1/3Nb2/3)O3)3, is also a perovskite-related oxide (PDF 01-073-9070). Moreover, neither of these compounds exhibit optical absorption in the visible region. Based on the nominal composition determined above for SNO (NaI), the solid solution can be expressed as (NaNbO3)0.35–(Sr4/3Nb2/3O3)0.65. The results of Rietveld refinement based on the solid solution model (see Table S3†) are shown in Fig. 3. Simulated diffraction peaks were well fitted to the experimental ones of SNO (NaI), which offers further support for the presence of a solid solution. Onishi et al. also reported a similar solid solution system of NaTaO3 and Sr4Ta2O9.26 The incorporation of Na is thought to occur as follows. During heat treatment at a temperature higher than the melting point of the flux, evaporation will occur to produce AX vapour. In the case of NaI, however, not only NaI but also I2 vapour is expected to be produced. In fact, a dark purple gas was observed during the flux treatment. As a result, the excess Na+ is incorporated into the oxide, accompanied by absorption of oxygen from air. SNO (KI) and SNO (CsI) also contained certain amounts of K and Cs, respectively, supporting the above explanation.
 | ||
| Fig. 3 XRD pattern of SNO (NaI) and the results of Rietveld refinement. The crosses and solid curve represent the observed and calculated patterns, respectively, and the difference between the two is shown at the bottom. The vertical marks indicate the Bragg diffraction positions for (NaNbO3)0.35–(Sr4/3Nb2/3O3)0.65 solid solution. Reliability factors; RWP = 8.283%, RB = 1.450%. | ||
The SEM images of oxide precursors prepared with different types of flux are shown in Fig. 4. There is a clear dependence of the morphology on the type of flux used. Without a flux, the particles are generally irregular in shape. In SNO (NaCl), SNO (NaBr), and SNO (RbCl), the particles are column-like, while in SNO (KCl) and SNO (KBr), they are plate-like. In the XRD patterns in Fig. 2A, the relative peak intensities are different for SNO (NaCl) and SNO (KCl). The intensity of the Sr2Nb2O7 (080) diffraction peak in SNO (KCl) is much higher than that in SNO (NaCl), which is consistent with the plate-like morphology of SNO (KCl) in the SEM images. In SNO (SrCl2), SNO (NaI) and SNO (CsI), relatively large particles were present. Fig. S4† shows TEM images and a SAED pattern of a single secondary SNO (NaI) particle. Clear lattice fringes and diffraction spots are observed, indicating that the particle is most likely a single crystal, which is also consistent with the presence of a solid solution.
 | ||
| Fig. 4 SEM images of oxide precursors produced (a) without flux, and with (b) NaCl, (c) KCl, (d) RbCl, (e) SrCl2, (f) NaBr, (g) KBr, (h) NaI, (i) KI, (j) CsI, and (k) SrCl2 fluxes. The scale bars in (a)–(j) are 2 μm, and that in (k) is 20 μm. | ||
As indicated by the XRD results in Fig. 2B, after nitridation of SNO (AX), SrNbO2N was the main phase in all cases. Compared to the SNON (no flux) sample, the FWHM of the (110) diffraction peak was smaller for all of the SNON (AX) samples, indicating higher crystallinity. For SNON (SrCl2) and SNON (CsI), diffraction peaks associated with the oxide precursors were also observed, suggesting that nitridation was not complete, probably due to large particle size. On the other hand, for SNON (NaI), only diffraction peaks assigned to SrNbO2N were observed. It should be noted that the ICP-AES analysis revealed that almost all of the Na species in SNO (NaI) evaporated during nitridation (Table S2b†). In addition, as seen in Fig. S2,† a clear absorption edge associated with SrNbO2N was present. This suggests that SNON (NaI) was completely nitrided, regardless of the relatively large particle size. Thus, nitridation of a perovskite-type oxide precursor to produce a perovskite-type oxynitride without a structural transformation proceeded under relatively mild conditions even when the precursor oxide had large, well crystallized particles. The lattice constants of SNON (no flux) and SNON (AX) refined by Rietveld analysis are shown in Table S4.† No clear trends depending on the type of flux were observed.
The SEM images of SNON (no flux) and SNON (AX) in Fig. S3† show that the morphology of the original oxide precursor particles was maintained in the form of secondary particles after nitridation. The primary particles had sizes on the order of 100 nm regardless of the type of flux.
Fig. 5 shows the PEC properties of the nitrided samples. The photocurrent density of all SNON (AX) samples is seen to be higher than that of SNON (no flux). This is probably due to the higher crystallinity of the primary particles, which results in less recombination of photoexcited carriers in the bulk. Despite the smallest FWHM of SNON (NaI), it did not exhibit the highest photocurrent. One possibility is that other properties such as the morphology or the existence of impurity also affect their PEC performances. At the present stage, these effects are not fully understood. After nitridation, however, most of the flux components evaporated and as a result, less than 1 mol% cation was observed as shown in Table S2.† Thus the effects of doping of cations seem limited. The highest photocurrent density of 1.0 mA cm−2 at 1.23 VRHE was obtained for SNON (RbCl), which is more than 1.5 times higher than the previously reported value of 0.6 mA cm−2.17
 | ||
| Fig. 5 Current density–potential curves of SrNbO2N obtained from oxide precursors prepared (a) without flux, and with (b) NaCl, (c) KCl, (d) RbCl, (e) SrCl2, (f) NaBr, (g) KBr, (h) NaI, (i) KI, and (j) CsI fluxes. The PEC measurements were performed under AM 1.5G irradiation in 0.2 M Na3PO4 (pH adjusted to 13 by NaOH addition). | ||
Effects of pre-calcination of the oxide precursor
As discussed above, the flux synthesis of the oxide precursors was found to be effective in improving the PEC properties of the final oxynitrides. In an attempt at further improvement, pre-calcination of the oxide precursors was carried out. The XRD patterns of SNO (AX) and SNO (AX, pre-calcined) are shown in Fig. S5A.† In the case of the NaCl and RbCl fluxes, pre-calcination caused an increase in the intensity of the diffraction peaks for the oxide precursors, indicating an improvement in crystallinity. On the other hand, there was no substantial difference in the case of NaI. The SEM images of the oxide precursors with and without pre-calcination are shown in Fig. S6.† It can be seen that in all cases, pre-calcination resulted in larger particles.Fig. S5A† also shows XRD patterns after nitridation of the above oxide precursors. After nitridation for 15 h at 1173 K, oxide diffraction peaks still remained for SNON (NaCl, pre-calcined) and SNON (RbCl, pre-calcined), although the SrNbO2N phase was dominant. After nitridation for 20 h at 1223 K, both oxide precursors were completely nitrided to SrNbO2N. More severe nitridation conditions were required because of the improvement in the crystallinity of the oxide precursors by pre-calcination. On the other hand, for SNON (NaI, pre-calcined), nitridation for 15 h at 1173 K was enough to obtain single-phase SrNbO2N even though the particle size became larger by pre-calcination. In addition, nitridation for 20 h at 1223 K seemed to be too severe, resulting in a reduction in the peak intensities.
The PEC properties of this series are shown in Fig. 6 and S5.† Regardless of the type of flux, a combination of pre-calcination and appropriate nitridation conditions improved the photocurrent density. In particular, SNON (NaI, pre-calcined) exhibited a value of 1.5 mA cm−2 at 1.23 VRHE, which is the highest ever reported for photoanodes fabricated from Nb-based perovskite-type oxynitrides.
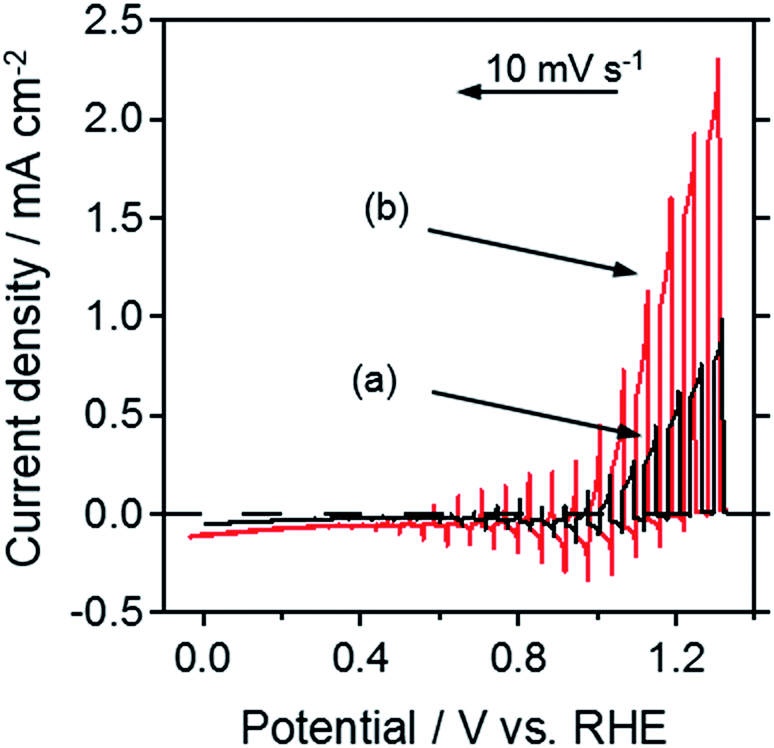 | ||
| Fig. 6 Current–potential curves of SrNbO2N prepared from oxide precursors using a NaI flux (a) without pre-calcination, and (b) with pre-calcination, under AM 1.5G irradiation in 0.2 M Na3PO4 (pH adjusted to 13 by NaOH addition). | ||
The cross-sectional and top-viewed SEM images of SNON (NaI, pre-calcined)/Nb/Ti photoelectrodes are shown in Fig. S7.† Almost a monolayer of secondary particles with sizes of several micrometres is seen to be present on the metal layers. As has been previously proposed, the larger particle size that is possible using the particle transfer method offers advantages in terms of achieving sufficient light absorption.17 Therefore, the improvement in the PEC properties was considered to be attributed not only to the high crystallinity of the particles, but also to large secondary particles with sizes on the order of micrometres. Moreover, it is likely that the relatively mild nitridation conditions also played a role, partly because the formation of nonstoichiometric species such as Nb4+ was suppressed.
Finally, to confirm that the observed anodic photocurrent originated from a water splitting reaction, an analysis of the gas evolved during the reaction was carried out. Fig. 7 shows the time course of the photocurrent density for SNON (NaI, pre-calcined), and of the evolved amount of oxygen and hydrogen. A photocurrent was clearly detected for over 2 h at a potential of 1.2 VRHE. Compared to previous results, the stability of electrodes much improved as well as photocurrent density. It is likely that improved charge transfer decreased the possibility of the occurrence of an undesired reaction such as self-oxidation. As shown in Fig. S8,† surface modification with an appropriate amount of CoPi was effective to improve stability. Moreover, the gases evolved in an almost stoichiometric ratio even though the faradaic efficiency for water oxidation was estimated to be about 80%. This is probably caused by the problem of stirring and self-oxidation. At the initial stage, it takes a certain time until evolved gases become uniform, resulting in the lower faradaic efficiency. At the later stage, self-oxidation seems to play the major role in the reduced faradaic efficiency.
 | ||
| Fig. 7 Time course of (A) photocurrent density and (B) hydrogen and oxygen evolution for SrNbO2N prepared from pre-calcined oxide precursors using a NaI flux. A voltage of 1.2 VRHE was applied under AM 1.5G irradiation in 0.2 M Na3PO4 (pH adjusted to 13 by NaOH addition). The geometric surface area of the electrode was 1.78 cm2. The dashed lines in (B) indicate estimated amounts of gases from the current–time plot assuming a faradaic efficiency of 100%. | ||
Methods for achieving further improvements in the photocurrent, stability and faradaic efficiency using Nb-based photoanodes are now under investigation.
Conclusions
The effects of flux treatments during the synthesis of SrNbO2N particles on their PEC properties were investigated. The flux synthesis of oxide precursors improved the photocurrent density, and pre-calcination before the flux treatment further improved the PEC performance. NaI flux-treated SrNbO2N with pre-calcination exhibited a photocurrent density of 1.5 mA cm−2 at 1.23 VRHE under AM 1.5G irradiation.Acknowledgements
This work was supported in part by Grants-in-Aid for Specially Promoted Research (No. 23000009) and “Nanotechnology Platform” from the Ministry of Education, Culture Sports Science and Technology (MEXT), Japan, and collaboration with Companhia Brasileira de Metalurgia e Mineração (CBMM). The authors would like to acknowledge Dr Taro Yamada at the University of Tokyo for performing the ICP measurements and also acknowledge Dr Yosuke Goto at the University of Tokyo for performing the Rietveld analysis.References
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- L. C. Seitz, Z. Chen, A. J. Forman, B. A. Pinaud, J. D. Benck and T. F. Jaramillo, ChemSusChem, 2014, 7, 1372–1385 CrossRef CAS PubMed.
- E. Nurlaela, S. Ould-Chikh, I. Llorens, J.-L. Hazemann and K. Takanabe, Chem. Mater., 2015, 27, 5685–5694 CrossRef CAS.
- J. Seo, T. Takata, M. Nakabayashi, T. Hisatomi, N. Shibata, T. Minegishi and K. Domen, J. Am. Chem. Soc., 2015, 137, 12780–12783 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- M. Matsukawa, R. Ishikawa, T. Hisatomi, Y. Moriya, N. Shibata, J. Kubota, Y. Ikuhara and K. Domen, Nano Lett., 2014, 14, 1038–1041 CrossRef CAS PubMed.
- J. Xu, C. Pan, T. Takata and K. Domen, Chem. Commun., 2015, 51, 7191–7194 RSC.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2858 CrossRef.
- K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 2227–2230 CrossRef CAS PubMed.
- M. Higashi, Y. Yamanaka, O. Tomita and R. Abe, APL Mater., 2015, 3, 104418 CrossRef.
- T. Hisatomi, C. Katayama, K. Teramura, T. Takata, Y. Moriya, T. Minegishi, M. Katayama, H. Nishiyama, T. Yamada and K. Domen, ChemSusChem, 2014, 7, 2016–2021 CrossRef CAS PubMed.
- T. Hisatomi, C. Katayama, Y. Moriya, T. Minegishi, M. Katayama, H. Nishiyama, T. Yamada and K. Domen, Energy Environ. Sci., 2013, 6, 3595–3599 CAS.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 74–78 CrossRef CAS PubMed.
- J. Wang, X. Wang, B. Liu, X. Li and M. Cao, Mater. Lett., 2015, 152, 131–134 CrossRef CAS.
- K. Maeda, M. Higashi, B. Siritanaratkul, R. Abe and K. Domen, J. Am. Chem. Soc., 2011, 133, 12334–12337 CrossRef CAS PubMed.
- H. Urabe, T. Hisatomi, T. Minegishi, J. Kubota and K. Domen, Faraday Discuss., 2014, 176, 213–223 RSC.
- J. Boltersdorf, N. King and P. A. Maggard, CrystEngComm, 2015, 17, 2225–2241 RSC.
- M. Hojamberdiev, K. Yubuta, J. J. M. Vequizo, A. Yamakata, S. Oishi, K. Domen and K. Teshima, Cryst. Growth Des., 2015, 15, 4663–4671 CAS.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CrossRef CAS.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120–1124 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- X. Xu, C. Randorn, P. Efstathiou and J. T. Irvine, Nat. Mater., 2012, 11, 595–598 CrossRef CAS PubMed.
- N. Peng, J. T. Irvine and A. G. Fitzgerald, J. Mater. Chem., 1998, 8, 1033–1038 RSC.
- B. Ellis, J.-P. Doumerc, M. Pouchard and P. Hagenmuller, Mater. Res. Bull., 1984, 19, 1237–1243 CrossRef CAS.
- L. An and H. Onishi, ACS Catal., 2015, 5, 3196–3206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional diffuse reflectance spectra, XRD patterns, TEM images, SEM-EDX measurements and current–potential curves are included. See DOI: 10.1039/c6ta00971a |
| This journal is © The Royal Society of Chemistry 2016 |