
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic liquid-derived Fe–N/C catalysts for highly efficient oxygen reduction reaction without any supports, templates, or multi-step pyrolysis†
Fei
He
a,
Xinghua
Chen
a,
Yanfei
Shen
b,
Ying
Li
a,
Anran
Liu
a,
Songqin
Liu
a,
Toshiyuki
Mori
c and
Yuanjian
Zhang
*a
aJiangsu Province Hi-Tech Key Laboratory for Bio-Medical Research, Jiangsu Optoelectronic Functional Materials and Engineering Laboratory, School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China. E-mail: Yuanjian.Zhang@seu.edu.cn
bMedical School, Southeast University, Nanjing 210009, China
cGlobal Research Center for Environment and Energy Based on Nanomaterials Science (GREEN), National Institute for Materials Sciences (NIMS), 1-1 Namiki, Ibaraki, 305-0044, Japan
First published on 4th April 2016
Abstract
The development of efficient and low-cost non-precious-metal electrocatalysts such as Fe–N/C for the oxygen reduction reaction (ORR) is crucial for fuel cells and metal–air batteries. Here, we report a class of highly efficient Fe–N/C electrocatalysts derived from versatile imidazolium-based ionic liquids (ILs) with a halogen-coordinated iron anion. Without any supports, templates, or multi-step pyrolysis, the as-prepared Fe–N/C catalysts exhibited superior activity in the ORR to the state-of-the-art Pt/C in alkaline electrolytes by 44 mV in half-wave potential. More interestingly, owing to the versatile configuration of the imidazolium-based IL precursors, a diverse range of catalyst structures was successfully modulated. Based on this, it was clearly revealed that the electrical conductivity, type/amount of N dopants, and “effective porosity” (not the conventional total surface area) jointly determined the electrocatalytic activity. A pivotal radar chart is further proposed to successfully predict activity in the ORR merely from the structures of Fe–N/C catalysts. The proposed ILs platform would provide a valuable toolbox for the molecular design of precursors to tune the structures of Fe–N/C electrocatalysts towards excellent activity in the ORR in a highly flexible manner, and facilitate the long-term challenging study of relationships between processing, structure and activity.
Introduction
The replacement of noble metals in key technologies such as proton exchange membrane fuel cells (PEMFCs) and metal–air batteries is of global importance for sustainable energy.1–5 An area of particular promise for non-precious-metal catalysts is doped carbon.6–8 Inspired by macrocyclic cobalt phthalocyanines, which have well-defined nitrogen-coordinated metal ion centers,9 M–N/C catalysts (M represents, e.g., Fe, Co, and Cu) have been commonly developed by the high-temperature pyrolysis of mixtures of transition metals and C/N-containing molecules.10–15 In general, the activities in the ORR of M–N/C catalysts obtained by conventional preparation strategies, including the physical mixture of individual C/N/M-containing precursors,16,17 coordination of N-containing polymers with metal ions18 and wet impregnation,19 are significantly influenced by several factors such as the molecular structure of precursors,20,21 the extent of the dispersion/coordination of metal ions in precursors,22,23 and the diversity of interfaces among precursors, templates and supports.24,25 In this context, the validity of investigating the structure–function relationships of M–N/C catalysts is rendered questionable by the complexities of these factors. Therefore, it is highly desirable to minimize the number of variable conditions in a system in a relatively acceptable way. In this regard, ionic liquids (ILs), which have otherwise been recognized simply as green solvents, may have the potential to address this challenge via the free combination of task-specific anions and cations.26,27 Very recently, several pioneering works revealed that M–N/C catalysts derived from some ILs exhibit outstanding electrocatalytic activity in the ORR, but additional carbon supports,28,29 porous hard/soft templates30,31 or sophisticated multiple pyrolysis steps29 were required, which still makes the research process complicated.Here, we report that imidazolium-based ILs with a halogen-coordinated iron anion can be developed as a class of single precursors for the one-step pyrolysis of Fe–N/C catalysts with excellent electrocatalytic activity in the ORR, which even surpassed that of commercially available Pt/C in alkaline electrolytes. It was observed that with respect to previous precursors the imidazolium-based ILs were affected by fewer of the aforementioned influences in preparing Fe–N/C catalysts because of their uniform homogeneous distribution of Fe ions on the molecular level and no requirement for any supports, templates or multiple pyrolysis steps. Interestingly, the versatile configurations of the imidazolium-based IL precursors could modulate a diverse range of features of catalysts such as porosity, surface area, graphitization, electrical conductivity, and doping by N atoms in the as-prepared Fe–N/C catalysts. As was successfully demonstrated, this in turn largely facilitated the discovery of structure–activity relationships and made the prediction of the activity in the ORR of Fe–N/C catalysts merely from their structural features possible. The proposed ILs platform would provide the scope to modulate the molecular design of precursors for synthesizing a range of M–N/C catalysts with different structures towards excellent activity in the ORR in a highly flexible manner and facilitate the long-term challenging study of relationships between processing, structure and activity.
Experimental section
Chemicals
All chemicals and solvents were of analytical grade and were purchased from Aladdin Chemistry Co., Ltd. or Sinopharm Chemical Reagent Co., Ltd. and used without further purification. Ultrapure water (18.2 MΩ cm) was obtained from a Thermal Smart2 water purification system (USA).Preparation of ionic liquid precursors
Detailed synthetic procedures of MIm + Fe and each IL (IL-1, IL-2, IL-3, IL-3′, and IL-4) are listed as follows. The general approach used for the preparation of ionic liquid precursors is shown in Scheme 1.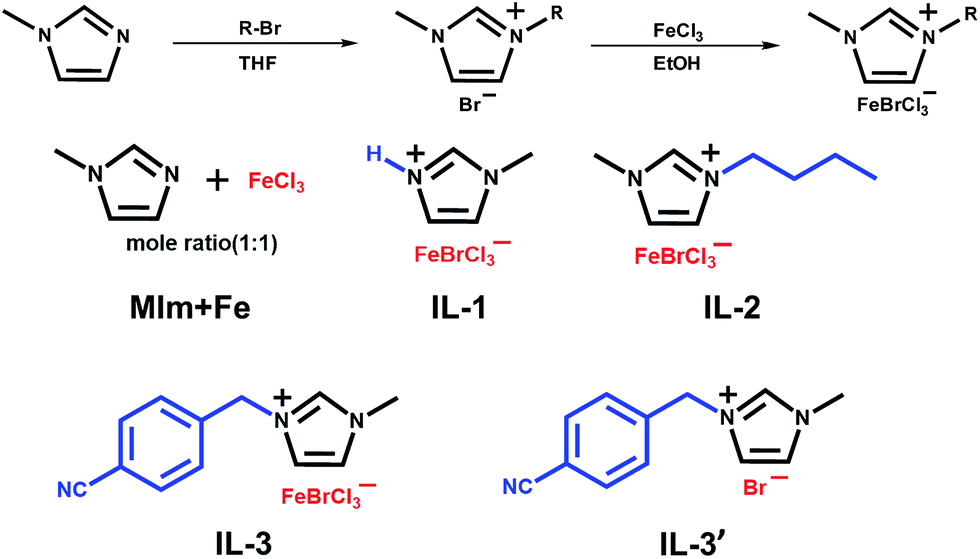 | ||
| Scheme 1 General synthetic steps and chemical structures of imidazolium-based ionic liquid precursors (IL-1, IL-2, IL-3 and IL-3′) and a physical mixture of N-methylimidazole and ferric chloride (MIm + Fe). | ||
Synthesis of MIm + Fe
N-Methylimidazole (8.21 g, 100 mmol), anhydrous FeCl3 (16.2 g, 100 mmol) and dry EtOH (100 mL) were mixed in a round-bottom flask and stirred at RT under a N2 atmosphere for 8 h. After that, EtOH was removed via rotary evaporation. The product was dried for 10 h at 80 °C under vacuum to yield MIm + Fe.Synthesis of IL-1
N-Methylimidazole hydrobromide was prepared by an acid–base neutralization reaction. N-Methylimidazole (8.21 g, 100 mmol) and dry THF (50 mL) were mixed in a round-bottom flask with magnetic stirring. After cooling to 0 °C in an ice-water bath, an excess of hydrogen bromide, which was prepared from phosphorus tribromide and water, was added to the reaction mixture for 2 h. Then, THF was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The top layer was decanted off and this procedure was repeated three times. The collected oil was dried for 6 h at 80 °C under vacuum to yield N-methylimidazole hydrobromide (15.8 g, yield: 97%). 1H NMR (DMSO-d6, δ, ppm, TMS): 3.88 (3H, CH3N–), 7.69, 7.74 and 9.18 (3H, imidazole protons), 14.21 (1H, –N+H–). Then, N-methylimidazole hydrobromide (15.8 g, 97 mmol), anhydrous FeCl3 (15.7 g, 97 mmol) and dry EtOH (100 mL) were mixed in a round-bottom flask and stirred at RT under a N2 atmosphere for 8 h. After that, EtOH was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether three times. The collected dark brown oil was dried for 10 h at 80 °C under vacuum to yield IL-1 (29.9 g, yield: 95%). Raman (cm−1): 221, 242 and 266 (Fe–Br), 328 (Fe–Cl).Synthesis of IL-2
1-Butyl-3-methylimidazolium bromide was prepared by a quaternization reaction.32,33N-Methylimidazole (8.21 g, 100 mmol), 1-bromobutane (13.7 g, 100 mmol), and dry THF (50 mL) were mixed in a round-bottom flask and stirred with refluxing at 80 °C under a N2 atmosphere for 24 h. After cooling to RT, THF was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The top layer was decanted off and this procedure was repeated three times. The collected oil was dried for 6 h at 80 °C under vacuum to yield 1-butyl-3-methylimidazolium bromide (20.8 g, yield: 95%). 1H NMR (DMSO-d6, δ, ppm, TMS): 0.89 (3H, CH3CH2CH2–), 1.25 (2H, CH3CH2CH2–), 1.76 (2H, CH3CH2CH2–), 3.87 (3H, CH3N–), 4.19 (2H, –CH2CH2N–), 7.76, 7.83 and 9.29 (3H, imidazole protons). Then, 1-butyl-3-methylimidazolium bromide (20.8 g, 95 mmol), anhydrous FeCl3 (15.4 g, 95 mmol) and dry EtOH (100 mL) were mixed in a round-bottom flask and stirred at RT under a N2 atmosphere for 8 h. After that, EtOH was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The collected dark brown oil was dried for 10 h at 80 °C under vacuum to yield IL-2 (34.4 g, yield: 95%). Raman (cm−1): 221, 242 and 266 (Fe–Br), 328 (Fe–Cl).Synthesis of IL-3
1-(4-Cyanobenzyl)-3-methylimidazolium bromide (IL-3′) was prepared by a quaternization reaction.32,33N-Methylimidazole (8.21 g, 100 mmol), 4-cyanobenzyl bromide (19.6 g, 100 mmol), and dry THF (50 mL) were mixed in a round-bottom flask and stirred with refluxing at 80 °C under a N2 atmosphere for 24 h. After cooling to RT, THF was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The top layer was decanted off and this procedure was repeated three times. The collected solid was dried for 6 h at 80 °C under vacuum to yield 1-(4-cyanobenzyl)-3-methylimidazolium bromide (26.4 g, yield: 95%). 1H NMR (DMSO-d6, δ, ppm, TMS): 3.87 (3H, CH3N–), 5.58 (2H, CNC6H4CH2N–), 7.60, 7.62, 7.92 and 7.94 (4H, benzene ring protons), 7.77, 7.84 and 9.32 (3H, imidazole protons). Then, 1-(4-cyanobenzyl)-3-methylimidazolium bromide (26.4 g, 95 mmol), anhydrous FeCl3 (15.4 g, 95 mmol) and dry EtOH (100 mL) were mixed in a round-bottom flask and stirred at RT under a N2 atmosphere for 8 h. After that, EtOH was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The collected solid was dried for 10 h at 80 °C under vacuum to yield IL-3 (39.7 g, yield: 95%). Raman (cm−1): 221, 242 and 266 (Fe–Br), 328 (Fe–Cl).Synthesis of IL-4
1-Benzyl-3-methylimidazolium bromide was prepared by a quaternization reaction.32,33N-Methylimidazole (8.21 g, 100 mmol), benzyl bromide (17.1 g, 100 mmol), and dry THF (50 mL) were mixed in a round-bottom flask and stirred with refluxing at 80 °C under a N2 atmosphere for 24 h. After cooling to RT, THF was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The top layer was decanted off and this procedure was repeated three times. The collected oil was dried for 6 h at 80 °C under vacuum to yield 1-benzyl-3-methylimidazolium bromide (24.0 g, yield: 95%). 1H NMR (DMSO-d6, δ, ppm, TMS): 3.86 (3H, CH3N–), 5.44 (2H, C6H5CH2N–), 7.35–7.45 (5H, benzene ring protons), 7.72, 7.81 and 9.31 (3H, imidazole protons). Then, 1-benzyl-3-methylimidazolium bromide (24.0 g, 95 mmol), anhydrous FeCl3 (15.4 g, 95 mmol) and dry EtOH (100 mL) were mixed in a round-bottom flask and stirred at RT under a N2 atmosphere for 8 h. After that, EtOH was removed via rotary evaporation. The crude product was rinsed with cold diethyl ether. The collected dark brown oil was dried for 10 h at 80 °C under vacuum to yield IL-4 (36.6 g, yield: 93%). Raman (cm−1): 221, 242 and 266 (Fe–Br), 328 (Fe–Cl).Preparation of catalysts
Doped carbon catalysts were prepared by heating the respective precursors in a furnace at 600 °C, 700 °C, 750 °C and 800 °C for 1 h in a nitrogen atmosphere at a ramp rate of 10 °C min−1. Thereafter, the obtained samples were ground to a powder, transferred to a round-bottom flask, ultrasonically dispersed in 25 mL of 37 wt% HCl, and stirred at RT overnight to remove excess iron species. Then, the doped carbonaceous part was isolated by centrifugation, washed with water and dried in vacuum at 80 °C overnight. The respective linear sweep voltammetry (LSV) curves showed that the catalyst calcined at 750 °C exhibited the best activity in the ORR (Fig. S9, see ESI†). Acid treatment resulted in an increase in activity in the ORR accompanied by a decrease in Fe content such as in IL-3-C (Fig. S11 and Table S1, see ESI†). After acid treatment, the total Fe content in all catalysts was estimated to range from 0.23 wt% to 0.68 wt% by XPS (Table S1, see ESI†), which was sufficient for an effective catalytic ORR.25 Therefore, the influence of the concentration of Fe species on the ORR was not comprehensively studied in this work.Electrochemical measurements
The evaluation of the activity in the ORR of various electrocatalysts and commercially available Pt/C (HiSPEC™ 3000, 20 wt% Pt on carbon black, Johnson Matthey) was performed in a standard three-electrode glass cell on a CHI700E workstation (CH Instruments, USA). A rotating disk glassy carbon electrode modified by an electrocatalyst was used as the working electrode. Pt wire and Ag/AgCl (saturated KCl) were used as the counter and reference electrodes, respectively. The procedure of modification of the working electrode was as follows: 10 μL of a suspension of an electrocatalyst in ethanol (2 mg mL−1) was cast on the pre-polished surface of a rotating glassy carbon electrode (3 mm diameter) or a rotating ring-disk electrode (4 mm diameter), resulting in a catalyst loading of 283 μg cm−2. The electrode was allowed to dry at RT and heated in air at 60 °C for 15 min. After that, 5 μL Nafion (0.05 wt%) was further cast on the surface of the electrode. Then, the electrode was again dried at RT and heated in air at 60 °C for 15 min.For the ORR test, 0.1 M KOH (pH = 13), 0.1 M Na2HPO4/NaH2PO4 solution (PBS, pH = 7.4), and 0.5 M H2SO4 for alkaline, neutral and acidic conditions, respectively, were purged with high-purity O2 gas for 30 minutes and thereafter the electrolyte was protected by a flow of O2. Activity in the ORR was measured using linear sweep voltammetry (LSV) at the rotating electrode (RRDE-3A, BAS, Japan). LSV was performed at a scan rate of 10 mV s−1. The onset potential (Eonset) was defined as the potential at which the current density reached 5% of the diffusion-limiting current density in the cathodic scan. The half-wave potential (E1/2) was defined as the potential at which the current density reached 50% of the diffusion-limiting current density in the cathodic scan.5
The kinetic limiting current density (JK) for the ORR can be derived from the experimental data using the Koutecky–Levich equation for rotating disk electrodes:34
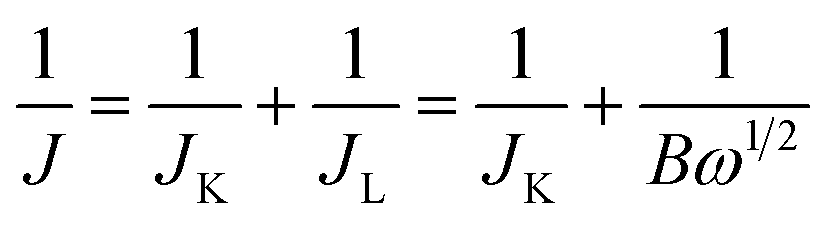 | (1) |
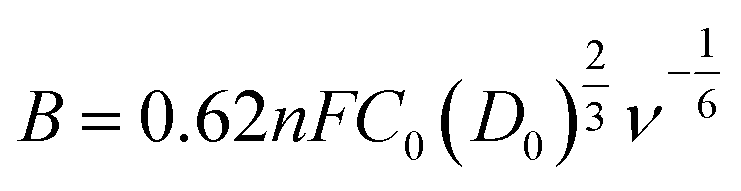 | (2) |
| JK = nFkC0 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the concentration of O2, D0 is the diffusion coefficient of O2, ν is the kinematic viscosity of the electrolyte, and k is the rate constant for electron transfer. Because the electrolyte was O2-saturated 0.1 M KOH in this report, the values of C0, D0 and ν were 1.2 × 10−3 M, 1.9 × 10−5 cm2 s−1, and 0.01 cm2 s−1, respectively.
485 C mol−1), C0 is the concentration of O2, D0 is the diffusion coefficient of O2, ν is the kinematic viscosity of the electrolyte, and k is the rate constant for electron transfer. Because the electrolyte was O2-saturated 0.1 M KOH in this report, the values of C0, D0 and ν were 1.2 × 10−3 M, 1.9 × 10−5 cm2 s−1, and 0.01 cm2 s−1, respectively.
To determine the yield of H2O2, the ring potential was set to 0.5 V (Ag/AgCl) to oxidize the H2O2 transferred from the GC disk electrode. The yield of H2O2 and the electron transfer number (n) were calculated by the following equations:35
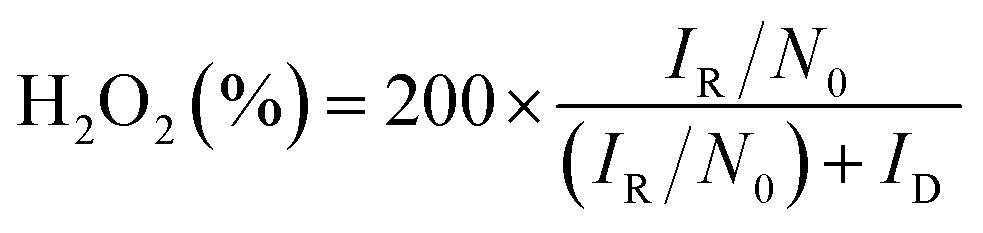 | (4) |
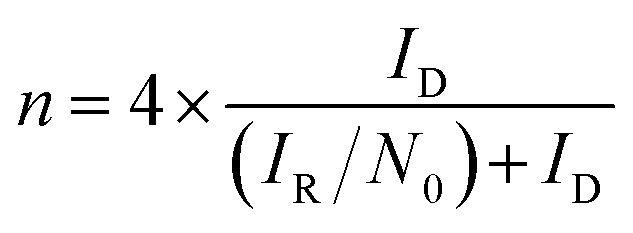 | (5) |
Characterization
NMR spectra were recorded on a Bruker Avance III 400 MHz NMR spectrometer (Germany). Thermogravimetric analysis (TGA) was conducted on a PerkinElmer Pyris 1 analyzer. Scanning electron microscopy (SEM) images were recorded on a Hitachi S-4800 electron microscope (Japan). Transmission electron microscopy (TEM) was carried out on a FEI Tecnai G2 microscope (USA) at an accelerating voltage of 200 kV. Nitrogen adsorption–desorption isotherms were measured at 77 K with a Quantachrome Autosorb iQ one-station adsorption instrument (USA). Samples were degassed at 300 °C for 8 h at reduced pressure before measurement. The surface area was calculated by the Brunauer–Emmett–Teller (BET) method. The surface area of micropores and mesopores was calculated by the V–t method. The pore size distributions (PSD) were calculated based on the model of slit/cylindrical pores on carbon using the adsorption branch of the isotherms by the QSDFT method. Raman spectra were recorded on a Thermo DXR 532 Raman spectrometer (USA) with a laser excitation wavelength of 532 nm. X-ray diffraction (XRD) was carried out on a Rigaku SmartLab 3 (Japan) X-ray diffractometer equipped with graphite-monochromated Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS) experiments were performed with a Thermo Theta Probe (USA) spectrometer using monochromated Al Kα X-rays at hν = 1486.6 eV. Peak positions were internally referenced to the C 1s peak at 284.6 eV. Conductivity measurements were performed by confining a sample between two iron discs and measuring the electrical resistance of the sample by chronocoulometry on a Gamry Reference 600 potentiostat (USA).Results and discussion
In the first set of experiments, the H-substituted imidazolium-based IL bearing a halogen-coordinated iron anion (IL-1, Scheme 1) was directly pyrolyzed at 750 °C under a N2 atmosphere without any supports, templates or multiple pyrolysis steps (see details in the Experimental section), and the as-obtained product was denoted as IL-1-C. The cyclic voltammetry (CV) curve of IL-1-C showed a well-defined cathodic peak in O2-saturated 0.1 M KOH with a peak potential that was 49 mV higher than that of commercially available Pt/C (20 wt%), which suggests that IL-1-C has excellent electrocatalytic activity in the ORR (Fig. S1, see ESI†). To exclude diffusion-controlled current, hydrodynamic linear sweep voltammetry (LSV) using a rotating disk electrode (RDE) was further performed. It was observed that the onset potential (Eonset = −11 mV) and half-wave potential (E1/2 = −99 mV) of IL-1-C were 56 mV and 44 mV higher, respectively, than those of Pt/C (Fig. 1a), which was in agreement with the Tafel plots in the low overpotential region (Fig. 1b). By varying the speed of rotation (Fig. S2, see ESI†) and using the Koutecky–Levich (K–L) equation, the kinetic limiting current density (JK) was calculated and found to be comparable to that of Pt/C (Fig. S3, see ESI†). Rotating ring-disk electrode (RRDE) measurements showed that the yields of H2O2 during the ORR in the kinetic limiting region for IL-1-C were below 5.5%. Based on the results of the RRDE measurements and eqn (5) (see details in the Experimental section), the calculated electron transfer number (n) for the ORR was 3.89–4.00, which was similar to that of Pt/C (Fig. 1c). Moreover, as indicated by the fact that the Tafel slope was almost the same (∼60 mV dec−1), the transfer of the first electron in the ORR catalyzed by IL-1-C was probably the rate-determining step, as with Pt/C.36 The as-prepared IL-1-C also exhibited significant catalytic activity in the ORR in neutral and acidic solutions, but this was poorer in comparison with the activity in alkaline solution, which was presumably because of the different surface charge state of N-doped carbon at different pH values12,13 (Fig. S4, see ESI†). To discover the crucial role of well-distributed iron on the molecular level in defining the structures of the catalysts, a physical mixture of N-methylimidazole and an iron salt in a molar ratio of 1:1 (MIm + Fe, Scheme 1) was also prepared as a control. In sharp contrast, the values of Eonset, E1/2, JK and the electron transfer number of MIm + Fe–C (obtained by carbonization of MIm + Fe) were inferior to those of IL-1-C (Fig. 1a), which evidently revealed the importance of a homogeneous distribution of iron ions in the precursors on the molecular level for the enhancement of the electrocatalytic activities of the resulting catalysts. In addition, IL-1-C exhibited high stability and little crossover effect from methanol oxidation compared with Pt/C (Fig. 1d). In this regard, one-step pyrolysis of imidazolium-based ILs bearing a halogen-coordinated iron anion without any supports or templates could be developed as a highly efficient way for preparing Fe–N/C catalysts for the ORR that even surpass commercially available Pt/C in alkaline electrolytes, thus providing a substantial platform with minimal influencing factors for studying the structure–activity relationships of Fe–N/C catalysts.
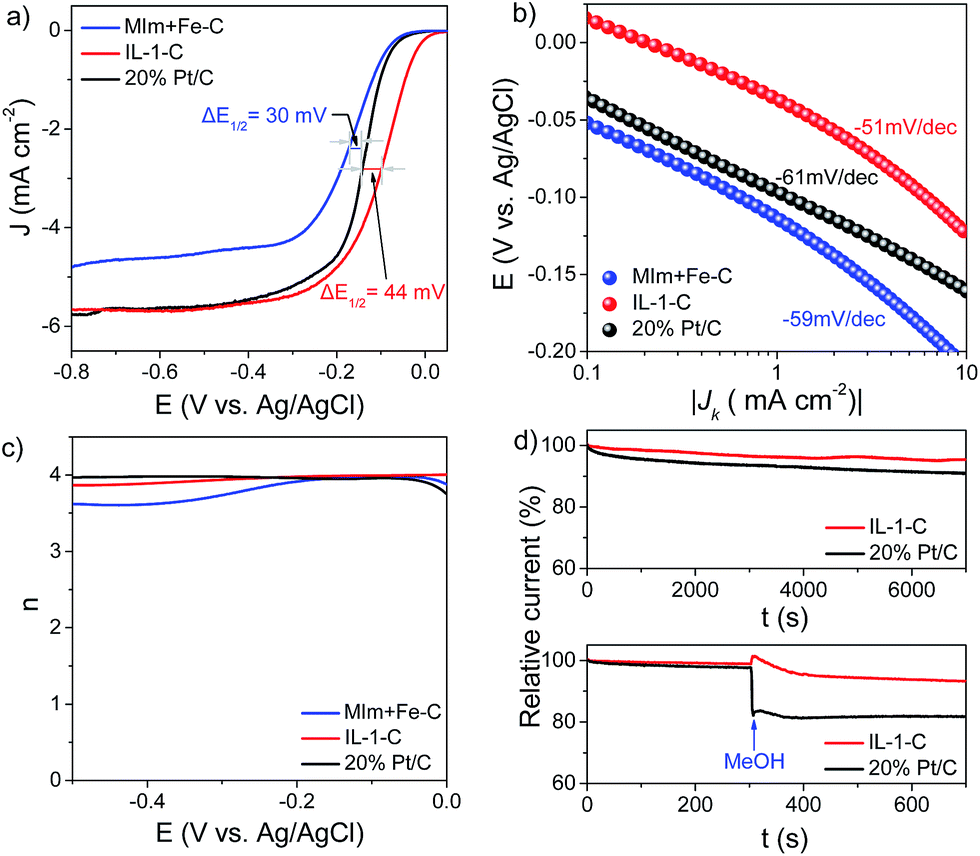 | ||
| Fig. 1 Performance in the ORR of catalysts. (a) LSV curves of MIm + Fe–C, IL-1-C and 20% Pt/C at 1600 rpm. (b) Tafel plots of MIm + Fe–C, IL-1-C and 20% Pt/C. (c) Electron transfer number of MIm + Fe–C, IL-1-C and 20% Pt/C based on RRDE data. (d) Current–time (I–t) response and evaluation of the crossover effect from methanol oxidation of IL-1-C and 20% Pt/C. RDE and RRDE data were recorded in O2-saturated 0.1 M KOH. Sweep rate: 10 mV s−1. | ||
The most attractive characteristic of ILs is that their properties can be easily manipulated by the free combination of numerous cations and anions. This feature allowed us to further investigate the general relationship between the molecular structures of IL-based precursors, the structures of catalysts, and the activities of catalysts, thus helping us to not only prepare more efficient Fe–N/C catalysts for the ORR but also to deepen our understanding of the fundamental mechanism. For this, IL precursors bearing a longer flexible alkyl chain substituent (IL-2), a more rigid cyanobenzyl substituent (IL-3), or a counter anion without any iron content (IL-3′) were synthesized and carbonized to give the final catalysts. (The rigidity and flexibility are from the point of view of organic chemistry. For instance, a benzene ring is more rigid than an alkane.) The hydrodynamic LSV curves in Fig. 2a show that their catalytic activity in the ORR evidently varied. Detailed values of Eonset, E1/2, JK, and the electron transfer number of the as-obtained catalysts for the ORR, together with typical textural information, are summarized in Table 1.
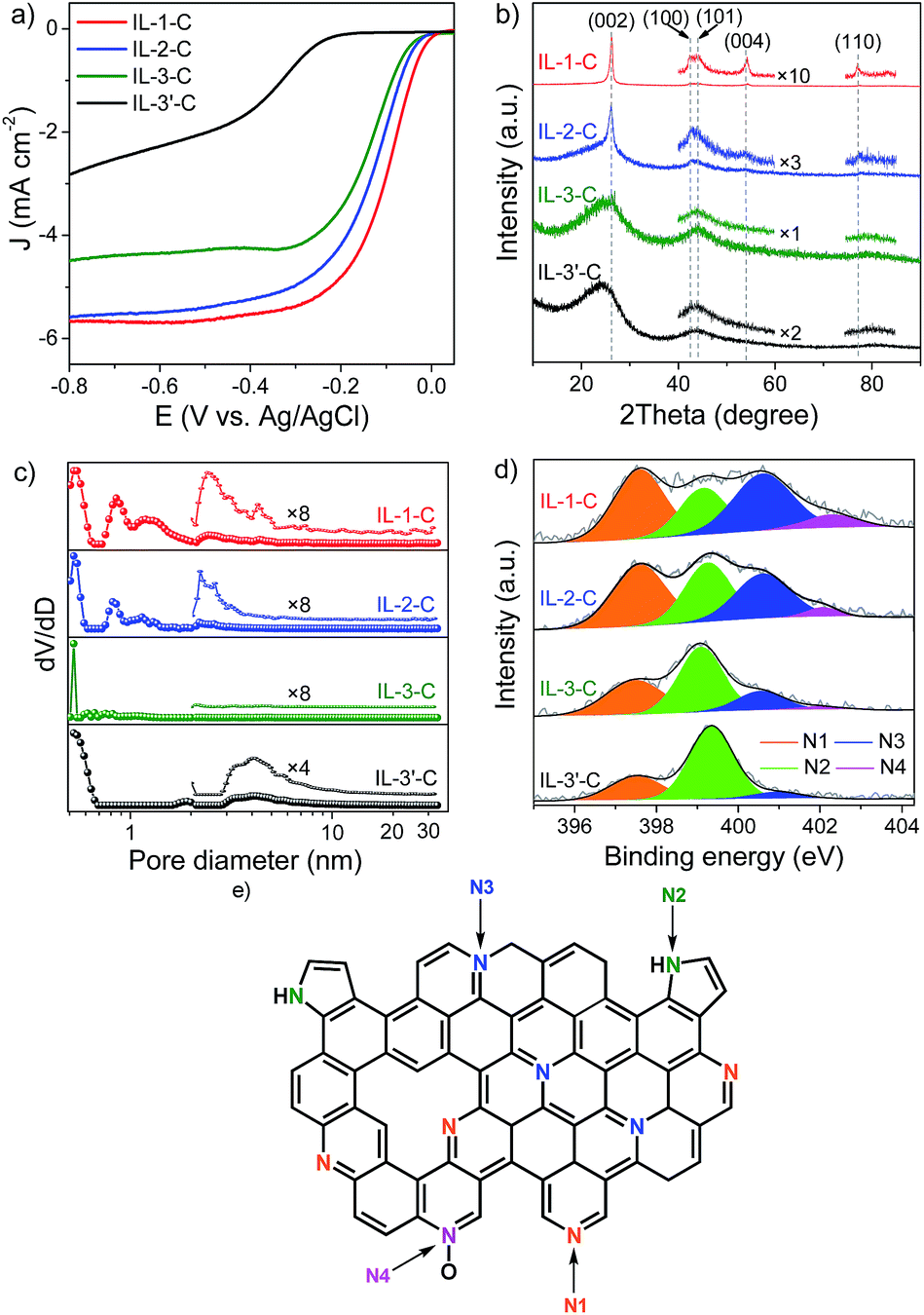 | ||
| Fig. 2 ORR activity in the ORR and structural information of catalysts. LSV curves (a), XRD patterns (b), pore size distribution curves (c) and high-resolution N 1s XPS spectra (d) of IL-1-C, IL-2-C, IL-3-C and IL-3′-C. N heterocyclic structures in catalysts (e). LSV was performed at 1600 rpm at a sweep rate of 10 mV s−1 in O2-saturated 0.1 M KOH. N1, N2, N3 and N4 represent pyridinic, pyrrolic, graphitic, and oxidized N+–O− nitrogen species, respectively. | ||
| Catalyst | E onset mV | E 1/2 mV | J K mA cm−2 | n | (N1 + N3)/Ntotale (at%) | Relative specific resistancef | SAg m2 g−1 | MSAh m2 g−1 | MCi cm3 g−1 |
|---|---|---|---|---|---|---|---|---|---|
| a E onset was defined as the potential at which the current density reached 5% of the diffusion-limiting current density. b E 1/2 was defined as the potential at which the current density reached 50% of the diffusion-limiting current density. c J K was calculated at −0.3 V by the Koutecky–Levich (K–L) equation. d The electron transfer number (n) was calculated at −0.3 V by RRDE measurements. e N1, N3 and Ntotal represent pyridinic N, graphitic N, and total N, respectively. The total N content of all catalysts was estimated to the range from 2.22% to 5.01% by XPS. f The relative specific resistance of catalysts was calculated by normalizing the measured specific resistance with that of a graphite reference. g SA: surface area. h MSA: mesopore surface area. i MC: mesopore capacity. | |||||||||
| MIm + Fe–C | −72 | −173 | 27.4 | 3.74 | 59.0 | 1.10 | 276 | 207 | 0.65 |
| IL-1-C | −11 | −99 | 35.1 | 3.96 | 70.3 | 0.92 | 448 | 82 | 0.16 |
| IL-2-C | −31 | −131 | 32.8 | 3.94 | 65.6 | 1.04 | 1010 | 91 | 0.16 |
| IL-3-C | −34 | −137 | 20.4 | 3.90 | 49.2 | 1.4 | 1242 | 98 | 0.09 |
| IL-3′-C | −212 | −379 | 0.9 | — | 33.6 | 1.12 | 253 | 148 | 0.11 |
At first glance, IL-3′-C exhibited much lower activity in the ORR than all its counterparts, which suggests that an even distribution of iron ions in precursors is rather helpful for improving activity in the ORR. To obtain more insight into the differences in catalytic activity in the ORR, the crystalline graphitic structures of the as-prepared Fe–N/C catalysts were first determined by X-ray diffraction (XRD). Fig. 2b shows that all catalysts contained graphitic carbon, as confirmed by the characteristic XRD peaks at ∼26.2°, 42.6°, and 44.4°, corresponding to the (002), (100) and (101) diffractions,37 respectively. Interestingly, the extent of graphitization decreased (IL-1-C > IL-2-C > IL-3-C) as the rigidity of the substituent on the imidazolium cation increased. For example, for IL-1, which only contained a H substituent, two additional (004) and (110) reflections, which indicated an even higher extent of graphitization, were observed after pyrolysis and were presumably due to a strong tendency towards graphitization for flexible substituents (TGA curves are in Fig. S5, see ESI†). Moreover, for MIm + Fe and IL-1, it was observed that the extent of graphitization of the latter was higher after pyrolysis, which implies that well-distributed iron on the molecular level also favoured graphitization. Accordingly, the TEM/SEM images in Fig. 3 show that IL-1-C was more characterized by a sheet-like texture in comparison with the other Fe–N/C catalysts. The Raman spectra also confirmed the different extent of graphitization and were highly consistent with the XRD results (Fig. S6, see ESI†). Hence, both the flexibility of the substituent in the cation and the even molecular distribution of iron in the anion of the IL precursors played an important role in increasing the extent of graphitization of the final Fe–N/C catalysts, which will be discussed below in turn regarding the facilitation of electron transfer in the ORR.
 | ||
| Fig. 3 SEM and TEM images of MIm + Fe–C (a and e), IL-1-C (b and f), IL-2-C (c and g) and IL-3-C (d and h). The scale bars in the SEM and TEM images represent 1 μm and 50 nm, respectively. | ||
Because the surface area of catalysts largely determines the number of exposed active sites,24 the N2 adsorption–desorption isotherms of IL-1-C, IL-2-C, IL-3-C and IL-3′-C were investigated. Fig. S7 (see ESI†) shows a large uptake of N2 and a hysteresis loop for all samples at a relative pressure (p/p0) of ca. 0.001 and 0.48–1.0, respectively, which reveals the presence of both micro- and mesoporosity. Their BET surface areas were as high as 253 to 1242 m2 g−1. Pore size distributions were further analyzed by the density functional theory (DFT) method using the adsorption branch of the isotherm (Fig. 2c). For IL-1-C, IL-2-C and IL-3-C, it was observed that both micropores and mesopores exhibited a slight shift toward smaller sizes and the relative content of mesopores decreased gradually when the rigidity of the substituent on the imidazolium cation increased. Thus, without any templates, supports or multiple pyrolysis steps, we could regulate the porosity and surface area of Fe–N/C catalysts by manipulating the stiffness of the substituent in the cation.
It is also important to identify the types of N that are present to help confirm whether these N atoms can serve as active sites. Therefore, the detailed chemical environments of the N atoms on the surface of the catalysts were investigated using X-ray photoelectron spectroscopy (XPS). The N 1s XPS spectra of all samples (Fig. 2d) could generally be deconvolved into four peaks at 398.3 eV, 400.0 eV, 401.0 eV, and 402.3 eV, which were assigned to pyridinic (N1), pyrrolic (N2), graphitic (N3), and oxidized N+–O− (N4) nitrogen species28,38 (Fig. 2e), respectively. It was found that pyridinic N (36.2%) and graphitic N (34.1%) were the major N species in IL-1-C, and pyrrolic N species only accounted for 22.3%. However, when the H atom in the imidazolium cation was replaced by a rigid cyanobenzyl group (IL-3), the content of pyrrolic N species in IL-3-C greatly increased to 47.9%, which was more than double that of IL-1-C. It was suggested that the more rigid substituent in the imidazolium cation led to fewer pyridinic N and graphitic N species. In addition, fewer pyridinic and graphitic N species (total 59.0%) were observed in MIm + Fe–C than in IL-1-C (70.3%), which evidently confirmed that well-distributed iron on the molecular level had a positive effect in increasing the content of pyridinic and graphitic N species during pyrolysis. Thus, different types of N species were produced selectively by changing the substituent in the cation and the distribution of iron in the anion of ILs.
In spite of extensive studies, the detailed contributions of the extent of graphitization, surface area/microstructure, type/amount of N dopants, and Fe species in doped carbon to its catalytic activity in the ORR are still controversial.24,39–42 This was presumably due to the complexity of previous research processes, which often involved multiple interfaces between precursors, supports and templates and sophisticated pyrolysis steps such as secondary high-temperature annealing,43,44 resulting in a lack of sufficient consistency for a fair comparison. As has been shown, in our study the proposed preparation of Fe–N/C catalysts almost entirely relied on the facile manipulation of the molecular structures of IL precursors, which made the system simple. This advantage is supposedly ascribed to the unique molecular structure of the imidazolium-based IL precursors: (1) the imidazolium core group acted as the major feedstock of C and N; (2) the substituent on the imidazolium ring largely determined the porosity and influenced the type of N dopant; and (3) a homogeneous distribution of Fe ion on the molecular level via cation–anion interactions would promote C–C and C–N bonding in a honeycomb manner. Moreover, a series of Fe–N/C catalysts with tunable structures and activities could also be obtained by the appropriate selection of the substituent and the distribution of Fe ions in imidazolium-based ILs. Both of these two features are much preferred for studies of the correlation between the structures and activities of the catalysts as follows.
Electrical conductivity is a vital parameter for determining the feasibility of electron transfer across interfaces between the substrate electrode, electrocatalyst, and oxygen in the ORR. In general, a large extent of graphitization corresponds to high conductivity. Moreover, the doping of electron-rich active N species, which generates a donor state above the Fermi energy level45 and increases the electron density,46 also favors fast electron transfer in a catalytic ORR. For this reason, the specific electrical resistances of the as-prepared Fe–N/C catalysts were close to or even superior to that of graphite. It was observed that as the value of IG/ID in the Raman spectra and the contents of pyridinic and graphitic N increased, the conductivity increased (Fig. 4a). For instance, IL-1-C, which had the highest content of pyridinic and graphitic N and value of IG/ID in this study, exhibited the highest electrical conductivity. Accordingly, the values of JK of the as-prepared catalysts in the kinetic limiting regions increased approximately with the electrical conductivity (Fig. 4b). The type and amount of N dopants, which are further key factors for high catalytic activity in the ORR, were supposed to influence the values of E1/2 and JK, respectively. It was found that pyridinic N (N1) and graphitic N (N3) were more active in the catalytic reduction of oxygen, and a higher content of them generally corresponded to a higher value of JK, as shown in Fig. 4c.
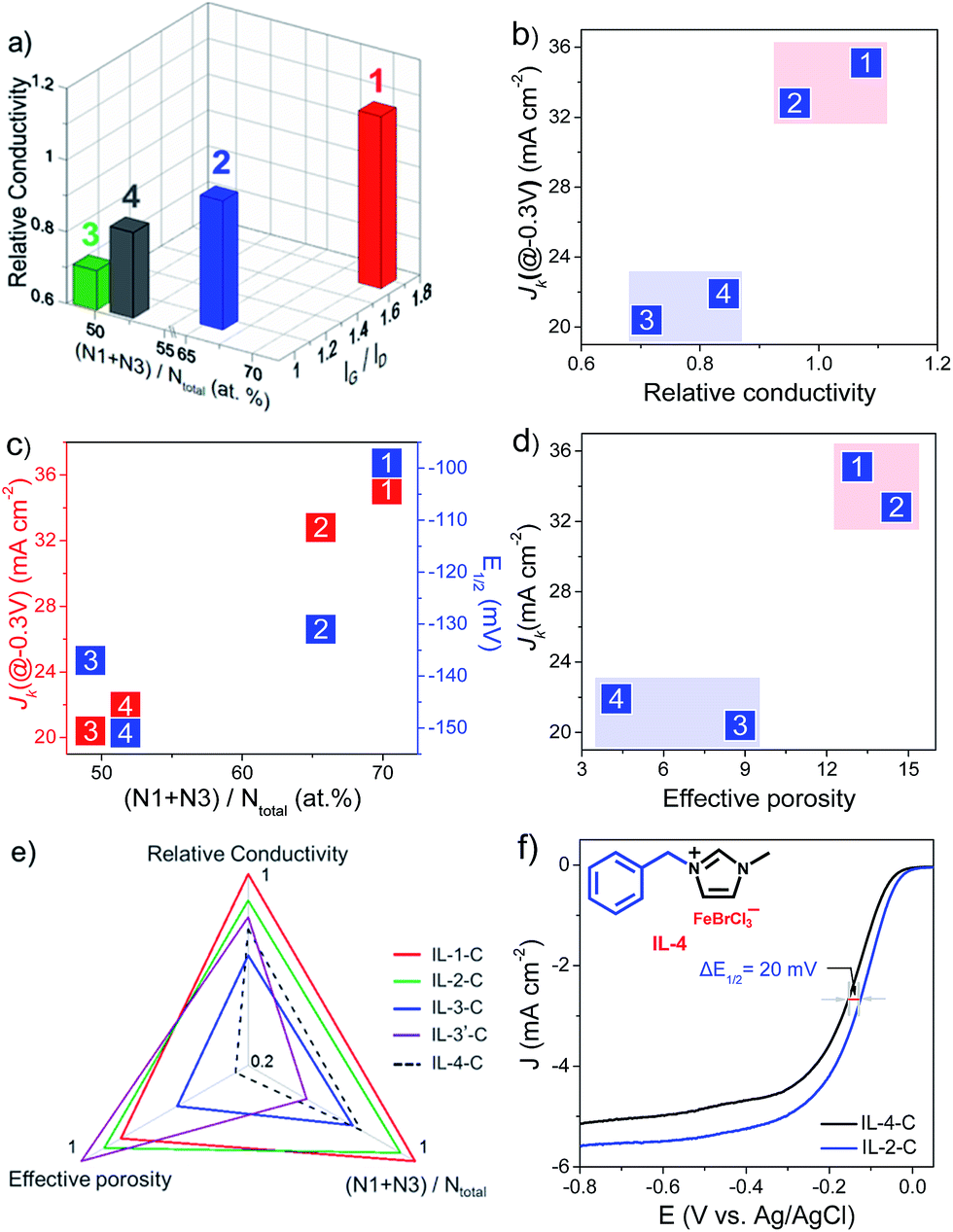 | ||
| Fig. 4 Structure–activity relationships of catalysts. (a) Conductivity of catalysts versus IG/ID and N1/N3 content. (b) Relationship between the conductivity of catalysts and JK. (c) JK and E1/2 of catalysts plotted against N1/N3 content. (d) Relationship between the effective porosity and JK. (e) Proposed radar plot showing the dependence of activity in the ORR on conductivity, N1/N3 content, and effective porosity. 1, 2, 3 and 4 represent IL-1-C, IL-2-C, IL-3-C and IL-4-C, respectively. (f) LSV curves of IL-2-C and IL-4-C at 1600 rpm at a sweep rate of 10 mV s−1 in O2-saturated 0.1 M KOH. JK was calculated at −0.3 V. | ||
The performance of Fe–N/C catalysts was also largely dependent on their surface area and pore structure, which determined the amount of accessible active sites and the mass transport properties of species relevant to the ORR (H+/OH−, O2, and H2O).47 However, activity in the ORR was not simply dependent on the total BET surface area, most of which consisted of micropores. It was suggested that mesopores of larger size would be more preferred for the migration of species relevant to the ORR, as well as higher electrochemical and chemical accessibility of catalytic active sites. Thus, the mesopore surface area (MSA) and mesopore capacity (MC) jointly affected activity in the ORR. In this sense, a single quantitative criterion that encompassed both of these textural factors was roughly proposed by multiplying MSA by MC and defined as “effective porosity” to correlate the performance in the ORR of Fe–N/C catalysts. It was noted that IL-1-C and IL-2-C, which had a high “effective porosity” (Table 1), displayed high values of JK (Fig. 4d). In contrast, IL-3-C, despite possessing the largest total BET surface area in this study and a high mesopore surface area, exhibited a low value of JK, which could be ascribed to its low mesopore capacity.
Nevertheless, it should be noted that the catalytic activity in the ORR of the as-synthesized Fe–N/C catalysts was jointly influenced by their electrical conductivity, type/amount of N dopants, and “effective porosity”, but not by one of these factors alone. For instance, compared with IL-1-C, although MIm + Fe–C had a high “effective porosity”, it exhibited a low value of JK. This phenomenon was attributed to the uneven dispersion of iron in the precursor MIm + Fe, which led to the agglomeration of iron species and loss of active N species (N1 + N3) during pyrolysis (Table 1). In this respect, a radar chart, which consisted of normalized electrical conductivity, type/amount of N dopants, and “effective porosity”, is proposed in Fig. 4e. It was generally revealed that catalysts that lay on the outer line exhibited superior catalytic activity in the ORR.
To confirm the correspondence between the electrical conductivity, type/amount of N dopants, and “effective porosity” for the performance in the ORR of Fe–N/C catalysts, the precursor IL-4 with a benzyl substituent, of which the rigidity was greater than that of IL-2, was prepared. Structural characterization of the carbonized product (IL-4-C) demonstrated a normalized content of pyridinic and graphitic N of 0.74, an electrical conductivity of 0.77, and an “effective porosity” of 0.26, all of which were less than those of IL-2-C (Fig. 4e). According to a preliminary inspection of the proposed radar chart, we could predict that the catalytic activity of IL-4-C would be inferior to that of IL-2-C. Interestingly, an electrochemical study showed that the activity in the ORR of IL-4-C (Fig. 4f), including the values of JK and E1/2 (Fig. 4c), was inferior to that of IL-2-C, which was in agreement with the proposed correspondence between the structural features and the performance in the ORR of Fe–N/C catalysts. Moreover, it is highly foreseeable that by the further modulation of Fe–N/C catalyst structures via the facile combination of an unlimited range of cations/anions in IL-based precursors, and evaluation of the corresponding activities, the reliability of the correlations between the structure and activity of catalysts, which was demonstrated by using the radar plot, would be improved. Thus, by using an IL platform, the proposed radar chart (Fig. 4e) provides a feasible way of predicting the activity in the ORR of Fe–N/C catalysts merely from their textural features, which would greatly facilitate the design and preparation of highly active catalysts for the ORR.
Conclusion
In summary, highly efficient Fe–N/C electrocatalysts were successfully developed from versatile imidazolium-based ILs with halogen-coordinated iron anions. Owing to the unique molecular structure of the IL precursors, no supports, templates or multi-step pyrolysis were required for the preparation of the catalysts. Moreover, by taking advantage of the free combination of cations and anions in the IL precursors, a diverse range of Fe–N/C catalysts with tunable electrical conductivity, type/amount of N dopants, and “effective porosity” (mesopore surface area and mesopore capacity, instead of total surface area) were obtained. Based on this, it was revealed that the electrocatalytic activity depended jointly on all of these textural characteristics. Accordingly, a radar chart was also further proposed to provide a feasible way of predicting the activity in the ORR of Fe–N/C catalysts merely from their structural features. The proposed strategy for the preparation of Fe–N/C catalysts would not only be promising in the development of high-performance catalysts for practical applications of the ORR, but also offer a novel platform for studying the relationships between processing, structure and activity.Acknowledgements
This study was financially supported in part by the National Natural Science Foundation of China (21203023, 91333110, and 21305065), Natural Science Foundation of Jiangsu Province (BK2012317, BK20130788), and the Fundamental Research Funds for the Central Universities.References
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. D. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- X. J. Zhou, J. L. Qiao, L. Yang and J. J. Zhang, Adv. Energy Mater., 2014, 4, 130123 Search PubMed.
- H. Shi, Y. F. Shen, F. He, Y. Li, A. R. Liu, S. Q. Liu and Y. J. Zhang, J. Mater. Chem. A, 2014, 2, 15704–15716 CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- L. B. Lv, T. N. Ye, L. H. Gong, K. X. Wang, J. Su, X. H. Li and J. S. Chen, Chem. Mater., 2015, 27, 544–549 CrossRef CAS.
- J. Wang, K. Wang, F. B. Wang and X. H. Xia, Nat. Commun., 2014, 5, 5258 CrossRef PubMed.
- M. L. Xiao, J. B. Zhu, L. G. Feng, C. P. Liu and W. Xing, Adv. Mater., 2015, 27, 2521–2527 CrossRef CAS PubMed.
- Y. C. Wang, Y. J. Lai, L. Song, Z. Y. Zhou, J. G. Liu, Q. Wang, X. D. Yang, C. Chen, W. Shi, Y. P. Zheng, M. Rauf and S. G. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS.
- J. S. Lee, G. S. Park, S. T. Kim, M. L. Liu and J. Cho, Angew. Chem., Int. Ed., 2013, 52, 1026–1030 CrossRef CAS PubMed.
- W. H. Niu, L. G. Li, X. J. Liu, N. Wang, J. Liu, W. J. Zhou, Z. H. Tang and S. W. Chen, J. Am. Chem. Soc., 2015, 137, 5555–5562 CrossRef CAS PubMed.
- J. Liang, R. F. Zhou, X. M. Chen, Y. H. Tang and S. Z. Qiao, Adv. Mater., 2014, 26, 6074–6079 CrossRef CAS PubMed.
- Z. S. Wu, L. Chen, J. Z. Liu, K. Parvez, H. W. Liang, J. Shu, H. Sachdev, R. Graf, X. L. Feng and K. Mullen, Adv. Mater., 2014, 26, 1450–1455 CrossRef CAS PubMed.
- Z. H. Xiang, Y. H. Xue, D. P. Cao, L. Huang, J. F. Chen and L. M. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433–2437 CrossRef CAS PubMed.
- Z. H. Wen, S. Q. Ci, Y. Hou and J. H. Chen, Angew. Chem., Int. Ed., 2014, 53, 6496–6500 CrossRef CAS PubMed.
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- Q. Wang, Z. Y. Zhou, Y. J. Lai, Y. You, J. G. Liu, X. L. Wu, E. Terefe, C. Chen, L. Song, M. Rauf, N. Tian and S. G. Sun, J. Am. Chem. Soc., 2014, 136, 10882–10885 CrossRef CAS PubMed.
- G. G. Eshetu, M. Armand, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2014, 53, 13342–13359 CrossRef PubMed.
- S. G. Zhang, M. S. Miran, A. Ikoma, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2014, 136, 1690–1693 CrossRef CAS PubMed.
- Y. J. Sa, C. Park, H. Y. Jeong, S. H. Park, Z. Lee, K. T. Kim, G. G. Park and S. H. Joo, Angew. Chem., Int. Ed., 2014, 53, 4102–4106 CrossRef CAS PubMed.
- N. R. Sahraie, J. P. Paraknowitsch, C. Gobel, A. Thomas and P. Strasser, J. Am. Chem. Soc., 2014, 136, 14486–14497 CrossRef PubMed.
- Z. L. Li, G. L. Li, L. H. Jiang, J. L. Li, G. Q. Sun, C. G. Xia and F. W. Li, Angew. Chem., Int. Ed., 2015, 54, 1494–1498 CrossRef CAS PubMed.
- W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- M. Dobbelin, V. Jovanovski, I. Llarena, L. J. C. Marfil, G. Cabanero, J. Rodriguez and D. Mecerreyes, Polym. Chem., 2011, 2, 1275–1278 RSC.
- J. Gao, J. Q. Wang, Q. W. Song and L. N. He, Green Chem., 2011, 13, 1182–1186 RSC.
- Y. J. Zhang, K. Fugane, T. Mori, L. Niu and J. H. Ye, J. Mater. Chem., 2012, 22, 6575–6580 RSC.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- R. Arrigo, M. Havecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlogl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- W. Ding, Z. D. Wei, S. G. Chen, X. Q. Qi, T. Yang, J. S. Hu, D. Wang, L. J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. M. Huang and Y. Chen, ACS Nano, 2014, 8, 6856–6862 CrossRef CAS PubMed.
- J. Wu, W. M. Li, D. Higgins and Z. W. Chen, J. Phys. Chem. C, 2011, 115, 18856–18862 CAS.
- K. Strickland, M. W. Elise, Q. Y. Jia, U. Tylus, N. Ramaswamy, W. T. Liang, M. T. Sougrati, F. Jaouen and S. Mukerjee, Nat. Commun., 2015, 6, 7343 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- L. F. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. H. Tang, H. Gong, Z. X. Shen, L. Y. Jianyi and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- F. G. Sun, J. T. Wang, H. C. Chen, W. C. Li, W. M. Qiao, D. H. Long and L. C. Ling, ACS Appl. Mater. Interfaces, 2013, 5, 5630–5638 CAS.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00648e |
| This journal is © The Royal Society of Chemistry 2016 |