
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic switching of ionic conductivity by cooperative interaction of polyviologen and liquid crystals for efficient charge storage†
Kan
Sato
,
Takaaki
Yamasaki
,
Takahiro
Mizuma
,
Kenichi
Oyaizu
* and
Hiroyuki
Nishide
*
Department of Applied Chemistry, Waseda University, Tokyo 169-8555, Japan. E-mail: nishide@waseda.jp; Fax: +81-3-3209-5522; Tel: +81-3-3200-2669
First published on 15th February 2016
Abstract
The ionic conductivity of a liquid crystal electrolyte was switched along with redox reactions of polyviologen. The surface charge of the polymer determined the alignment and the bulk conductivity of the electrolyte. The cooperative switching firstly enabled the suppression of self-discharging and quick-response, giving rise to facile and persistent charge storage.
Organic redox-active polymers have attracted substantial attention as charge storage materials for secondary batteries, electrochromic devices, sensors, and memories.1–7 The devices exhibit fast charging/discharging performances in lightweight and flexible or even stretchable forms due to the chemical and mechanical robustness of the organic polymers.8–11 In order to improve battery performances, a series of studies with different concepts of polymer design were reported.12–17 The use of unconjugated polymer backbones enabled successful charging/discharging with theoretical capacities whilst conjugated ones typically tended to suffer from chemical instability along with the high ratio of doping.18,19 Cross-linking structures suppressed the elution of polymers into electrolytes which may cause the loss of the capacity and even self-discharging induced by the shuttle process.20,21 Multi-electron redox-active moieties such as anthraquinones increased the theoretical capacity as electrodes,22,23 and composite electrodes with optimized compositions of redox-polymers and conductive carbons were also essential to maintain both power and energy density.24
Apart from electrode-active materials, emerging functional electrolytes such as ionic liquids have also attracted considerable attention for further favourable performances.25,26 Appropriate combinations of ionic species as electrolyte layers enabled the desirable properties such as ionic conductivity, viscosity, potential window, vapour pressure, and other important functions.25,27 In particular, liquid crystal electrolytes containing ionic liquids have been reported as a new class of functional electrolytes, owing to their anisotropic ionic conductivity in bulk scales.28–30 We anticipate a substantial potential of using them for charge storage applications because their ionic conductivity can be, in theory, reversibly tuned in favoured directions28,31 along with charge/discharge reactions, and it will contribute to both the quick response of the devices and the suppression of unfavourable self-discharging.
Herein we report a new technique of dynamically switching the ionic conductivity of liquid crystal electrolytes by cooperatively controlling the redox reaction of polyviologen 1 (ref. 32) to induce the alignment of the liquid crystal electrolyte containing nematic liquid crystal 4-cyano-4′-pentylbiphenyl (5CB) and ionic liquid 1-ethyl-3-methylimidazolium tetracyanoborate (EMIM TCB) which exhibits the anisotropic ionic conductivity. The bulk ionic conductivity of the device was reversibly tuned along with the charge/discharge reactions by changing the alignment of the liquid crystals. High conductivity was achieved during the charging/discharging reactions, but low conductivity was obtained only after the completion of the charge (Fig. 1). The highly conductive state during the redox reaction should result in quick responses of the devices, and the lower conductivity that emerges only in the fully charged state should suppress the undesired self-discharging induced by the shuttle processes of the ionic species.33,34
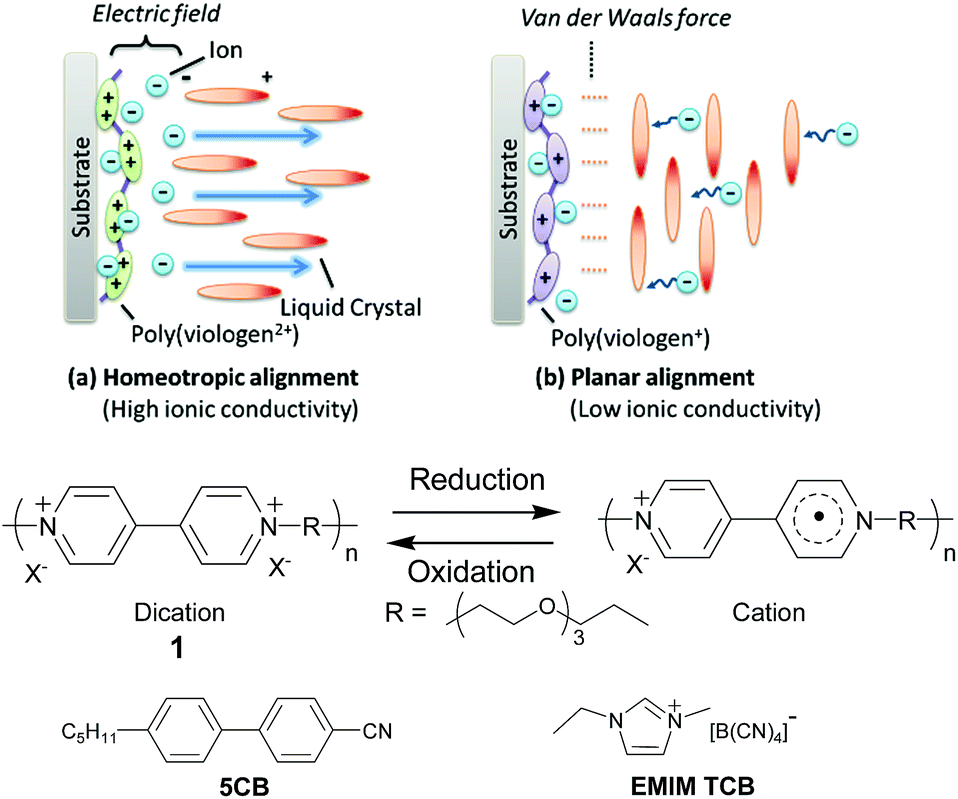 | ||
| Fig. 1 Dynamic switching of ionic conductivity by cooperative interaction of polyviologen and nematic liquid crystals. | ||
We expected that the alignment of 5CB could be in principle controlled by adjusting the force balances of (a) the electric field of the double layer in the vicinity of the electrode and (b) the Van der Waals force between the polyviologen and 5CB. When polyviologen 1 is in the dication state, perpendicular alignment of 5CB should be induced because of the electrostatic interaction between the dipole moment of 5CB (dielectric anisotropy Δε = +13)31 and the diffusion layer's electric field originating from the densely populated charge on the polyelectrolyte. On the other hand, when polyviologen 1 was one-electron reduced with each redox moiety, the Van der Waals force between the 1 and 5CB would become dominant and then induce the planar alignment of liquid crystals. The surface driven switching of liquid crystals by tuning the balance of the electric double layer and Van der Waals force described above was preliminary studied by Abbot's group, but they examined only the cases and mechanisms of self-assembled monolayers of ferrocene or redox-inactive compounds for optical applications.31,35 Our present report will be the first to successfully use the redox polymers and the liquid crystal electrolytes for the dynamic tuning of ionic conductivity enabling the efficient charge storage.
The electrochromic device was fabricated using a spin-coated 1 layer on an ITO substrate as the electrochromic electrode, ATO nanoparticles on an ITO as the capacitive counter, and 35 mM EMIM TCB in 5CB as the electrolyte solution. We selected EMIM TCB because it exhibited higher tendency for homeotropic alignment compared to former reported common electrolytes including tetrabutylammonium tetrafluoroborate (TBABF4) (Table S1, further discussion in the ESI†).31 Hydrophobic ionic liquids exhibited more favourable miscibility and higher degrees of dissociation in 5CB which should contribute to the strong electric field of the double layer in the vicinity of the charged polymer and induce homeotropic alignment (Fig. S1 and S2†). The reversible electrochromic response was observed by applying constant voltages to the device (Fig. 2). The polymer layer in the discharged dicationic state was transparent at 0 and 2.5 V vs. the capacitor electrode but turned to red at −2.5 V by reduction into the charged cationic state. The results suggested the reversible redox reactions of 1 between the dication and cation states. Simultaneously, we observed the dynamic alignment changes of 5CB accompanied with the redox reactions (Fig. 3). Polarized light microscopy observation showed the homeotropic alignment in the dication state and planar or tilted alignment in the cation state. In the former state, the strongly charged surfaces of both 1 and the metallic oxide (ATO, counter electrode)30 have certainly induced the strong electric field of the double layer at the surfaces, and caused homeotropic alignment. On the other hand, the planar or tilted alignment in the cation state should be induced by dominantly affecting the Van der Waals force between 1 and liquid crystals after the decrease in the polymer charge by the reduction of the dication to the cation. 5CB molecules close to the counter electrode seemed to always align perpendicularly against the substrate as expected in the absence of substantial redox reactions at the surface.31
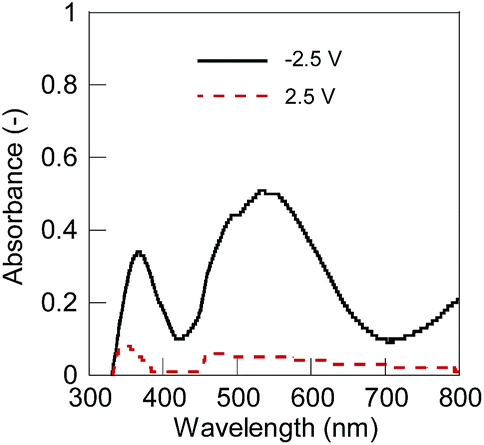 | ||
| Fig. 2 UV-vis spectra of the electrochromic device. Solid line (black): −2.5 V, and dashed line (red): 2.5 V. | ||
 | ||
| Fig. 3 Polarized light microscopy images of the electrochromic device. Inset: conoscope observation. The cross-shape indicates the homeotropic alignment of liquid crystals. | ||
Dynamically switching the alignment of the liquid crystal along with the redox reactions enabled both quick charging/discharging and suppression of self-discharging. For comparison, we used a 1000 mT external magnetic field induced by neodymium magnets to always homeotropically align 5CB against substrates.36 We observed the significant difference in ionic conductivity between the planar (8.25 × 10−6 S cm without magnet) and homeotropically aligned (8.70× 10−6 S cm with magnet) conditions after charging (Fig. 4(a)). The difference in the resistivity was calculated to be 6300 Ω cm and the ratio was 1.05, which was slightly lower than the original anisotropic ratio of the electrolyte (1.13 in Fig. S3†) presumably because 5CB always aligned perpendicularly in the vicinity of the counter ATO electrode. The rate of self-discharging was successfully reduced under the planar alignment conditions compared to the homeotropic state (Fig. 4(b)). Open circuit potentials differed up to 200 seconds and the result suggested that the low ionic conduction of the electrolyte with the planar alignment efficiently suppressed the unfavourable self-discharging reactions with the ionic species.
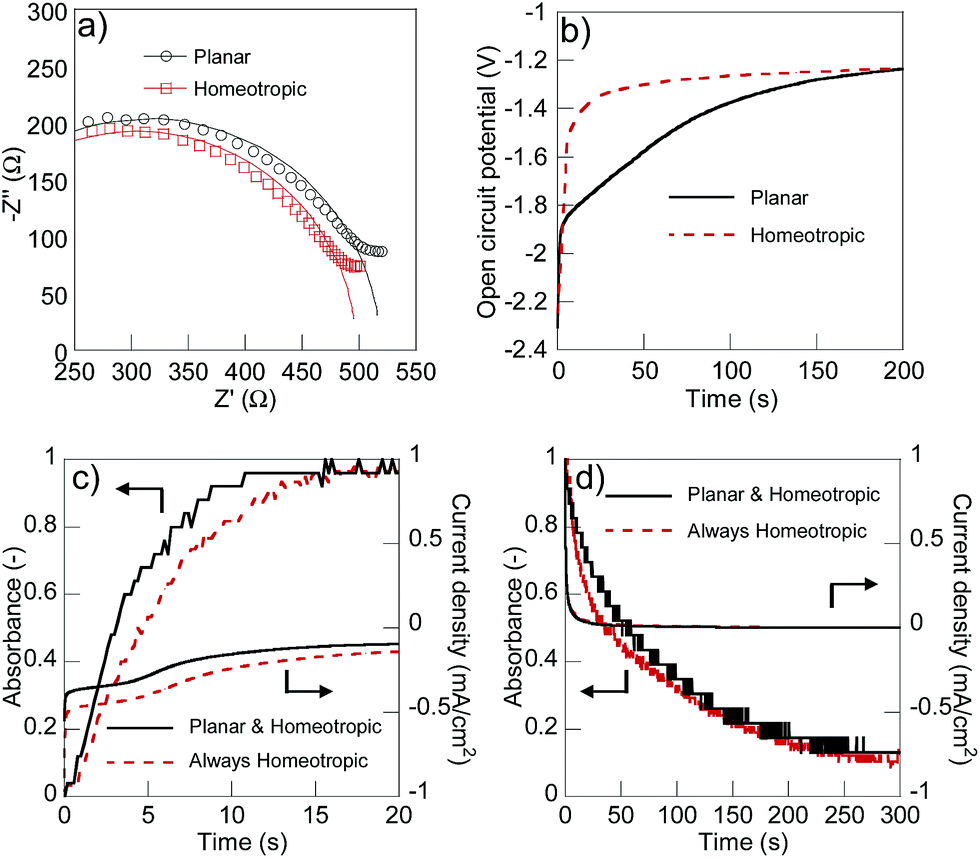 | ||
| Fig. 4 (a) Electrochemical impedance spectra of the electrochromic device at −2.5 V (106 to 5 × 104 Hz, AC amplitude 10 mV). (b) Open circuit potential of the device after charging at −2.5 V. (c) Time dependences of the absorbance at 530 nm and current density applied at −2.5 V, and (d) 2.5 V. The magnetic field (1000 mT) was applied to induce the homeotropic alignment (dotted line). | ||
After 200 seconds, the alignment of 5CB changed from planar to homeotropic, and no significant difference was observed between the conditions with or without the external magnetic field. The alignment change from homeotropic to planar should be caused by the self-discharging of the polymer surface. The use of liquid crystal electrolytes with even higher anisotropic ionic conductivity will more efficiently suppress the unfavourable self-discharging reactions. We observed no significant difference in the response rate for the charge/discharge reactions between the dynamically changed alignment in the absence of the magnet and permanently homeotropically aligned conditions using the magnet (Fig. 4(c) and (d)). Both devices turned on in 15 seconds, and decoloured in 300 seconds. Since the alignment of 5CB was homeotropic except for the fully charged state, the high ionic conductivity during the charge/discharge processes seemed to enable the fast response rate as high as that of the permanently homeotropic conditions with the magnet. The slower responses for decolouring than colouring should be caused partially by the dense stacking of viologen cations.37 Enhancing the switching ratio of ionic conductivity with a view to accomplish more significant co-operation is the topic of our continuous research.
Conclusions
We established a new technique of dynamically switching ionic conductivity of electrolytes with a difference of 6300 Ω cm utilizing the cooperative interaction of redox polymers and liquid crystals. The favourable change of the ionic conductivity along with the charge/discharge reactions enabled both the significant suppression of the self-discharging caused by ionic species and quick response of the device in 15 seconds. To the best of our knowledge, this is the first report on dynamic tuning of the ionic conductivity of electrolytes along with the charging/discharging by polymers which enables efficient charge storage. Our technique can be widely used for a series of reported liquid crystal electrolytes with higher regularity and anisotropic ionic conductivity,28,38 and a further efficient charge storage system can be realized by using them. The successful alignment will lead to facile and persistent charge storage with potential applications to electrochromic devices and batteries with dramatically enhanced performances.Acknowledgements
This work was supported by Grants-in-Aid for JSPS Fellows (No. 15J00888), Scientific Research (No. 24225003, 26620108, and 25288056), and the Leading Graduate Program in Science and Engineering, Waseda University from MEXT, Japan.Notes and references
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280 CAS.
- S. V. Vasilyeva, P. M. Beaujuge, S. Wang, J. E. Babiarz, V. W. Ballarotto and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1022–1032 CAS.
- Y. Yonekuta, K. Susuki, K. Oyaizu, K. Honda and H. Nishide, J. Am. Chem. Soc., 2007, 129, 14128–14129 CrossRef CAS PubMed.
- M. K. Park, S. Deng and R. C. Advincula, J. Am. Chem. Soc., 2004, 126, 13723–13731 CrossRef CAS PubMed.
- J. C. Forgie, A. L. Kanibolotsky, P. J. Skabara, S. J. Coles, M. B. Hursthouse, R. W. Harrington and W. Clegg, Macromolecules, 2009, 42, 2570–2580 CrossRef CAS.
- C. Hu, T. Sato, J. Zhang, S. Moriyama and M. Higuchi, ACS Appl. Mater. Interfaces, 2014, 6, 9118–9125 CAS.
- H. Nishide, K. Koshika and K. Oyaizu, Pure Appl. Chem., 2009, 81, 1961–1970 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS PubMed.
- N. Sano, W. Tomita, S. Hara, C.-M. Min, J.-S. Lee, K. Oyaizu and H. Nishide, ACS Appl. Mater. Interfaces, 2013, 5, 1355–1361 CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef.
- I. S. Chae, M. Koyano, T. Sukegawa, K. Oyaizu and H. Nishide, J. Mater. Chem. A, 2013, 1, 9608–9611 CAS.
- W. Choi, S. Endo, K. Oyaizu, H. Nishide and K. E. Geckeler, J. Mater. Chem. A, 2013, 1, 2999–3003 CAS.
- K. Oyaizu, A. Hatemata, W. Choi and H. Nishide, J. Mater. Chem., 2010, 20, 5404–5410 RSC.
- J. P. Blinco, J. L. Hodgson, B. J. Morrow, J. R. Walker, G. D. Will, M. L. Coote and S. E. Bottle, J. Org. Chem., 2008, 73, 6763–6771 CrossRef CAS PubMed.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- J. C. W. Chien and J. B. Schlenoff, Nature, 1984, 311, 362–363 CrossRef CAS.
- J. K. Feng, Y. L. Cao, X. P. Ai and H. X. Yang, J. Power Sources, 2008, 177, 199–204 CrossRef CAS.
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, J. Am. Chem. Soc., 2008, 130, 14459–14461 CrossRef CAS PubMed.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS PubMed.
- T. Sukegawa, K. Sato, K. Oyaizu and H. Nishide, RSC Adv., 2015, 5, 15448–15452 RSC.
- A. Lewandowski and A. Świderska-Mocek, J. Power Sources, 2009, 194, 601–609 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS PubMed.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2005, 45, 38–68 CrossRef PubMed.
- M. Yoshio, T. Mukai, K. Kanie, M. Yoshizawa, H. Ohno and T. Kato, Adv. Mater., 2002, 14, 351–354 CrossRef CAS.
- R. Rondla, J. C. Y. Lin, C. T. Yang and I. J. B. Lin, Langmuir, 2013, 29, 11779–11785 CrossRef CAS PubMed.
- Y.-Y. Luk and N. L. Abbott, Science, 2003, 301, 623–626 CrossRef CAS PubMed.
- R. H. Terrill, J. E. Hutchison and R. W. Murray, J. Phys. Chem. B, 1997, 101, 1535–1542 CrossRef CAS.
- A. Mirmohseni and R. Solhjo, Eur. Polym. J., 2003, 39, 219–223 CrossRef CAS.
- N. Oyama, N. Oki, H. Ohno, Y. Ohnuki, H. Matsuda and E. Tsuchida, J. Phys. Chem., 1983, 87, 3642–3647 CrossRef CAS.
- R. R. Shah and N. L. Abbott, J. Phys. Chem. B, 2001, 105, 4936–4950 CrossRef CAS.
- Y. Xiang, Z. Lin, T. Li, X. Liang, J. Zhang, J. W. Huang and L. N. Ji, Jpn. J. Appl. Phys., Part 1, 1999, 38, 6017–6018 CrossRef.
- T. M. Bockman and J. K. Kochi, J. Org. Chem., 1990, 55, 4127–4135 CrossRef CAS.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00320f |
| This journal is © The Royal Society of Chemistry 2016 |