
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Symmetric pseudocapacitors based on molybdenum disulfide (MoS2)-modified carbon nanospheres: correlating physicochemistry and synergistic interaction on energy storage†
Tobile N. Y.
Khawula
a,
Kumar
Raju
b,
Paul J.
Franklyn
c,
Iakovos
Sigalas
a and
Kenneth I.
Ozoemena
*bc
aSchool of Chemical and Metallurgical Engineering, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa
bEnergy Materials, Materials Science and Manufacturing, Council for Scientific and Industrial Research (CSIR), Pretoria 0001, South Africa. E-mail: kozoemena@csir.co.za
cMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa
First published on 24th March 2016
Abstract
Molybdenum disulfide-modified carbon nanospheres (MoS2/CNS) with two different morphologies (spherical and flower-like) have been synthesized using hydrothermal techniques and investigated as symmetric pseudocapacitors in an aqueous electrolyte. The physicochemical properties of these MoS2/CNS layered materials have been investigated using surface area analysis (BET), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), Raman, Fourier transform infrared (FTIR) spectroscopy, and advanced electrochemistry, including cyclic voltammetry (CV), galvanostatic cycling with potential limitation (GCPL), long-hour voltage-holding tests, and electrochemical impedance spectroscopy (EIS). The two different MoS2/CNS layered materials exhibit unique differences in morphology, surface area, and structural parameters, which have been correlated with their electrochemical capacitive properties. The flower-like morphology (f-MoS2/CNS) shows lattice expansion (XRD), large surface area (BET analysis), and small-sized nanostructures (corroborated by the larger FWHM of the Raman and XRD data). In contrast to the f-MoS2/CNS, the spherical morphology (s-MoS2/CNS) shows lattice contraction and small surface area with relatively large-sized nanostructures. The presence of CNS on the MoS2 structure leads to slight softening of the characteristic Raman bands (E12g and A1g modes) with larger FWHM. MoS2 and its CNS-based composites have been tested in symmetric electrochemical capacitors in an aqueous 1 M Na2SO4 solution. CNS improves the conductivity of the MoS2 and synergistically enhances the electrochemical capacitive properties of the materials, especially the f-MoS2/CNS-based symmetric cells (most notably, in terms of capacitance retention). The f-MoS2/CNS-based pseudocapacitor shows a maximum capacitance of 231 F g−1, with high energy density 26 W h kg−1 and power density 6443 W kg−1. For the s-MoS2/CNS-based pseudocapacitor, the equivalent values are 108 F g−1, 7.4 W h kg−1 and 3700 W kg−1. The high-performance of the f-MoS2/CNS is consistent with its physicochemical properties as determined by the spectroscopy and microscopy data. These findings have opened doors for further exploration of the synergistic effects between MoS2 graphene-like sheets and CNS for energy storage.
Introduction
Pseudocapacitors are redox-based electrochemical capacitors (ECs). Unlike their counterparts, the electrical double layer capacitors (EDLC) that only use carbon materials as electrode materials, pseudocapacitors employ redox-active materials such as conducting polymers, metal oxides and metal sulphides.1–4 Unlike batteries with high energy densities, ECs are characterized by their high power characteristics, which make them very attractive for several technologies and devices that require high-power applications (i.e., the ability to release energy pulses in a very short time, in a few seconds) such as in regenerative braking energy systems in vehicles and metro-rails, “stop–start” applications in modern cars, uninterrupted power supply (UPS), emergency doors in aircrafts, and escalators in buildings.5,6One of the emerging high-power supercapacitor electrode materials is molybdenum disulfide (MoS2), a member of the transition-metal dichalcogenides (TMDs). MoS2 has found applications in electrochemical devices, hydrogen storage, catalysis, capacitors, solid lubricants, and intercalation hosts.7,8 MoS2 is a layered-structured material with close relationship to graphene, characterized by a sheet-like morphology. The Mo layer is sandwiched between two S layers and the triple layers are stacked and held together by weak van der Waals interactions.2,9–11 Due to its higher intrinsic fast ionic conductivity (than oxides) and higher theoretical capacity (than graphite), MoS2 continues to attract a lot of attention, particularly in supercapacitors.2,7,12 Soon and Loh13 have pointed out the use of MoS2 as an electrode material for supercapacitors, and the results suggest that the supercapacitor performance of MoS2 is comparable to that of carbon nanotube (CNT) array electrodes. In addition to double-layer capacitance, diffusion of the ions into the MoS2 at slow scan rates obtains faradaic capacitance. Analogous to Ru in RuO2, the Mo central atom displays a range of oxidation states from +2 to +6. This plays an important role in enhancing charge storage capabilities.14 However, the electronic conductivity of MoS2 is still lower compared to graphite and the specific capacitance of MoS2 is still very limited when used alone for energy storage applications.12,13,15 As evident in several reports, there is the need to improve the capacitance of MoS2 with conductive materials such as CNT,12 polyaniline (PANI),2 polypyrrole (PPy),9 and reduced graphene (RGO).11 In a review by Márquez et al.,16 carbon nanospheres (CNS) were described as good candidates for catalytic and adsorption applications and their unclosed graphitic flakes provide the necessary ‘dangling bonds’ that could enhance surface reactions. CNS has also been used to enhance the conductivity of the battery cathode material, LiFePO4.17
To the best of our knowledge, the supercapacitive properties of MoS2 have only been investigated in half-cells (i.e., 3-electrode systems), which creates a huge knowledge gap on the true behavior of the electrode material when used in full-cells (2-electrode systems). Moreover, there is no literature on the effect of CNS on the supercapacitance of MoS2. Pseudocapacitors usually suffer from poor electrical conductivity and irreversible redox-activity, thereby leading to gradual loss of capacitance. To tackle the abovementioned challenges, this study adopted two different synthesis protocols to prepare MoS2 and MoS2/CNS composites with different physicochemistries (i.e., spherical and flower-like morphologies, structure, porous textures and electrochemistry) and then used them to fabricate symmetric pseudocapacitors. We clearly show that the pseudocapacitive properties of the MoS2/CNS (especially, in terms of cycling stability and electronic conductivity) are intrinsically linked to the presence of CNS.
Experimental procedure
Materials
Sodium molybdate dihydrate, Na2MoO4·2H2O (Analytical Reagent BDH chemicals Ltd, Poole England), thiourea (CH4N2S, Sigma-Aldrich Inc., USA), polyethylene glycol 1000 (PEG-1000, Fluka Analytical, Germany), L-cysteine (C3H7NO2S, Sigma-Aldrich Inc., USA). Hydrochloric acid (HCl, 32%), acetone (C3H6O) and absolute ethanol (C2H6O) were purchased from Associated Chemical Enterprises, in Johannesburg. All chemicals had purity higher than 98.99%. Distilled de-ionised water (18 MΩ cm) used in this study was obtained from the School of Chemistry at the University of the Witwatersrand.Synthesis
The CNS-modified spherical MoS2 (abbreviated herein as s-MoS2/CNS) composite was prepared as follows. First, 0.028 g CNS were ultrasonically dispersed in 20 mL deionized water. Then, 0.30 g Na2MoO4·2H2O were added and ultrasonically dispersed for 30 min. After adjusting the pH value to 6.5 with 12 M HCl, 0.80 g L-cysteine was added. The resultant mixture was diluted with water to 30 mL and rapidly stirred for about 1 h. The mixture was then transferred into a 40 mL Teflon cup and heated in a stainless steel autoclave at 180 °C for 36 h. Upon completion, the product was cooled naturally to room temperature, the s-MoS2/CNS composite was collected by filtration, washed with distilled water and acetone several times, and finally dried in the oven at 80 °C for 24 h.
The CNS-modified flower-like MoS2 composite (abbreviated herein as f-MoS2/CNS) was prepared as follows. 1.21 g Na2MoO4·2H2O were added to the sonicated CNS in 20 mL deionised water and further sonicated for 30 min. Then, 1.56 g thiourea and 0.28 g PEG-1000 were added to the solution and the mixture diluted to 30 mL before heating to 180 °C using a Teflon-lined stainless steel autoclave for 36 h. Upon completion, the product was cooled naturally to room temperature, the f-MoS2/CNS composite was collected by filtration, washed with distilled water and acetone several times, and finally dried in oven at 80 °C for 24 h. To homogenize the as-synthesized materials and further promote crystallization, the as-synthesized materials were annealed in a horizontal furnace with a quartz tube at 900 °C under a flow of nitrogen at a rate of 100 mL min−1 for 4 h.
Characterization techniques
The XRD patterns of the as-prepared MoS2 and MoS2/CNS nanopowders were obtained from a DMax/2500PC diffractometer using Co Kα radiation (K = 1.5418 Å) at 40 kV, 100 mA and a 2θ range of 10–90°. The FTIR spectra were obtained using a Bruker Tensor 27 FTIR spectrometer equipped with ZnSe crystal that absorbs strongly below 500 cm−1. FTIR spectra were obtained in the range of 550–4000 cm−1; a single beam measurement as the background spectrum was acquired prior to running the actual sample. Raman measurements were carried out in air using a Horiba Jobin Yvon spectrometer equipped with an Olympus BX40 microscope attachment to focus the laser beam on a small selected area of the sample, a 30 mW green argon laser (λ = 514 nm) as the excitation source, and a 1800 lines per mm grating monochromator with liquid nitrogen-cooled CCD. The BET (Brunauer, Emmett and Teller) measurements were performed to determine the specific surface area and pore size of the nano-sheets using a Micromeritics TriStar II instrument. The SEM images were obtained using a JEOL-JSM 7500F scanning electron microscope operated at 2.0 kV and a FEI Nova Nanolab 600 SEM. TEM images were obtained from a FEI Tecnai T12 microscope operated at an acceleration voltage of 120 kV. Elemental composition was obtained using the Oxford INCA EDS software on the SEM.Fabrication of the symmetric pseudocapacitor and electrochemical measurements
Symmetric pseudocapacitive properties of the materials were investigated using Swagelok cells (MTI, Inc., USA). Nickel foam (Celmet: thickness = 1.6 mm, surface area 7500 m2, cell size = 0.5 mm, 48–52 cells per inch) was used as substrate and current collector in the fabrication of the symmetric pseudocapacitors. Before use, the nickel foam was thoroughly cleaned by sonicating in 1 M HCl solution for 30 min, washing with copious amount of distilled water, and finally drying under vacuum. A 1 M Na2SO4 solution was used as the electrolyte, whereas a porous filter paper (Whatman®) served as the separator. The electrode materials were prepared by first thoroughly mixing the active materials, either MoS2/CNS or MoS2, carbon black as the conducting agent, and polyvinylidene fluoride (PVDF) as the binder (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:5 weight ratio) with a few drops of anhydrous N-methyl-2-pyrrolidone (NMP) using pestle and mortar, to produce a homogeneous paste. The resulting slurry was coated onto the nickel foam substrate with a spatula. The electrode was then dried at 60 °C overnight in a vacuum oven. The mass of the active material on the nickel foam was between 5 and 20 mg. For complete impregnation, the assembled cells were left for 12 hours prior to testing. The measurements were carried out in 1 M Na2SO4 aqueous electrolyte. All electrochemical measurements, cyclic voltammetry (CV), galvanostatic cycling with potential limitation (GCPL), and electrochemical impedance spectroscopy (EIS) were performed at room temperature using a computer-controlled multi-channel Potentiostat/Galvanostat Bio-Logic VMP3 work station driven by EC-Lab® v10.40 software with Z-fit tool for EIS data analysis. EIS measurements were carried out in the frequency ranging from 10 kHz to 10 mHz at the open circuit voltage, with AC voltage amplitude of 1.5 mV.
:15:5 weight ratio) with a few drops of anhydrous N-methyl-2-pyrrolidone (NMP) using pestle and mortar, to produce a homogeneous paste. The resulting slurry was coated onto the nickel foam substrate with a spatula. The electrode was then dried at 60 °C overnight in a vacuum oven. The mass of the active material on the nickel foam was between 5 and 20 mg. For complete impregnation, the assembled cells were left for 12 hours prior to testing. The measurements were carried out in 1 M Na2SO4 aqueous electrolyte. All electrochemical measurements, cyclic voltammetry (CV), galvanostatic cycling with potential limitation (GCPL), and electrochemical impedance spectroscopy (EIS) were performed at room temperature using a computer-controlled multi-channel Potentiostat/Galvanostat Bio-Logic VMP3 work station driven by EC-Lab® v10.40 software with Z-fit tool for EIS data analysis. EIS measurements were carried out in the frequency ranging from 10 kHz to 10 mHz at the open circuit voltage, with AC voltage amplitude of 1.5 mV.
The specific capacitance (Csp), maximum specific power density (Pmax) and specific energy density (Esp) were evaluated using the conventional eqn (1)–(5):21,22
 | (1) |
 | (2) |
 | (3) |
 | (4) |
 | (5) |
Results and discussion
Material characterization
A one-pot hydrothermal route was used for the synthesis of MoS2, CNS and MoS2/CNS. Fig. 1 shows the SEM and TEM images of the CNS (Fig. 1c and d), confirming the spherical nature of the CNS in agreement with literature. The micrographs show the interconnected uniform amorphous CNS in the particle size diameter range of 100–200 nm. Fig. 1 compares the SEM and TEM images of the s-MoS2 and s-MoS2/CNS. The SEM and TEM micrographs of the s-MoS2 (Fig. 1a and b) show the formation of a sphere-like morphology consisting of several agglomerated clusters of s-MoS2 sheets. | ||
| Fig. 1 SEM (a, c and e) and TEM (b, d, f, h and i) micrographs of s-MoS2 (a and b), CNS (c and d), s-MoS2/CNS (e and f); magnified views of s-MoS2/CNS (g and h), and d-spacing of s-MoS2 (i). | ||
A closer examination of the TEM images of the CNS (Fig. 1d) and s-MoS2/CNS (Fig. 1f–h) clearly suggests a uniform dispersion and excellent integration of the MoS2 with the CNS. The value of the d-spacing of the s-MoS2 shown in the TEM image (Fig. 1i) is 0.62 nm, which is in agreement with literature.23 In this synthesis method, it seems that the CNS particles acted as substrates for nucleation, wherein MoO4− ions reacted with sulphur ions from L-cysteine to form the MoS2 sheets on the CNS. The presence of CNS prevented MoS2 stacking and thus the formation of a porous 3-D sphere-like architecture with highly dispersed wrinkled and fluffy MoS2 sheets on CNS particles. This unique structure is also apparent in the TEM images (Fig. 1f and h). In the s-MoS2/CNS composite, the MoS2 nano-sheet edges are readily exposed and not entangled as in MoS2. This loose structure is desirable for superior charge storage. However, the MoS2 sheets in the composite are not visible as layered; instead, they grew in various orientations and were intertwined around the spheres. An important consequence is the small accessible surface area coupled with the small micropores. This could potentially cause an obstruction for ion transportation and thus limit the capacitance. The elemental composition of as-synthesized s-MoS2, obtained from the EDS (see ESI, Fig. S1†) obtained Na (3.35), S (66.11) and Mo (30.55) by weight percentage, clearly confirming the stoichiometry of the MoS2. The impurity ‘Na’ is attributed to the starting precursor Na2MoO4·2H2O.
Similarly, the flower-like nanostructures (f-MoS2) were obtained using a one-pot hydrothermal method. This method relies on sodium molybdate and thiourea to provide MoO42− ions and sulphur atoms, respectively. Interestingly, the addition of small amounts of PEG in our synthesis protocol assisted in the efficient dispersion of the MoS2 to generate the flower-like morphology. The starting precursor materials in the MoS2 formation play a vital role, especially in the achieved morphology; a slight change, even in the reducing agent, can bring about enormous value. In this hydrothermal process, the reaction involves three steps: (a) the hydrolysis of sulfur precursor to form H2S, followed by (b) the reduction of Mo and (c) finally the formation of MoS2.19
| CS(NH2)2 + 2H2O → 2NH3 + CO2 + H2S | (6) |
| 4Na2MoO4 + 15CS(NH2)2 + 6H2O → 4MoS2 + Na2SO4 + 6NaSCN + 24NH2 + 9CO | (7) |
The SEM and TEM images of as-synthesized MoS2 in Fig. 2 clearly show flower-like hierarchical 3-D structures. The f-MoS2 sheets self-assemble into a highly porous structure (Fig. 2a–c and e). The composite structure (Fig. 2d) also exhibits a clear flower-like morphology. The occurrence of this morphology may be associated with the presence of a surfactant (PEG-1000). Remarkably, after coating CNS with MoS2 sheets, the surface appears rough and wrinkled by the MoS2 sheets (Fig. 2d, f and g). The value of the d-spacing of the f-MoS2 (Fig. 2h) is 0.61 nm, which is in agreement with literature.23 The EDS data (see ESI, Fig. S2†) depict strong Mo and S overlapping signals, the MoS2 contains Na 1.3 atomic%, S 62.09 atomic% and Mo 36.61 atomic%, confirming the stoichiometry of the MoS2.
 | ||
| Fig. 2 SEM (a–d) and TEM (e–h) micrographs of f-MoS2 (a–c), f-MoS2/CNS (d), f-MoS2 (e); magnified views of f-MoS2/CNS (f and g) and d-spacing of f-MoS2 (h). | ||
MoS2 has a hexagonal crystal system and layer-structured D46h crystal system and P63 space group. Fig. 3 compares the XRD patterns of the CNS and its MoS2 spherical and flower-like composites. There is no significant difference between the MoS2 and MoS2/CNS patterns, confirming good integration of the CNS with the MoS2 structure. The diffraction peaks at 2θ = 15.8°, 37.9° and 41.5° are indexed to the hexagonal phase of MoS2 (002), (100) and (201), respectively. For the spherical morphology (s-MoS2/CNS composite), the diffraction peaks are similar to the individual s-MoS2, meaning that CNS fully interacts with the MoS2 and its presence does not interfere with the structure of the MoS2. For the spherical material, s-MoS2/CNS, the incorporation of CNS into the MoS2 nanosheets decreases the intensity of the peaks, in particular the (002). The result indicates the formation of few layers of the MoS2 in the composite; thus, CNS impedes the growth of the MoS2 layer in a hexagonal array. Importantly, upon incorporation of the CNS, there is a slight shift in the diffraction lines of the MoS2 to higher 2θ (see Fig. 3a–c), which is an indication of a lattice contraction. However, for the flower-like morphology (f-MoS2/CNS), the incorporation of the CNS results in a slight shift to the lower 2θ (see Fig. 3d–f), which is an indication of lattice expansion in the S–Mo–S interlayer spacing and formation of a few layers of MoS2. In addition, unlike the spherical composite (Fig. 3a–f), the presence of the CNS did not negatively impact the peak intensity of (002). In fact, the (002) peak became more intense and sharper, indicating a higher degree of crystallinity, comparable to that of the bulk MoS2. This finding may be due to strong integration between the MoS2 and CNS arising from the synthesis protocol adopted in this study, which allowed for the two materials to undergo chemical reaction rather than just physical mixing. Furthermore, the peak (103) is more apparent, with a new broad peak (006) of hexagonal MoS2 at 46° (Fig. 3d and e).
 | ||
| Fig. 3 XRD patterns of MoS2, CNS and MoS2/CNS composites for spherical (a–c) and flower-like (d–f) products and their magnified views. | ||
BET (five-point analysis) was used to measure the specific surface area and porosity of the as-synthesized materials. As shown in Table 1, the s-MoS2 has a specific surface area of 17.8 m2 g−1; on the other hand, the specific surface area of the s-MoS2/CNS decreased to 9.17 m2 g−1. This dramatic reduction is explained by the hindrance to the growth of the MoS2, due to the presence of the CNS (also confirmed by the XRD patterns). In fact, the CNS makes the surface area of MoS2 inaccessible. Moreover, the pore sizes of MoS2 and MoS2/CNS were revealed to be 22.9 and 18.61 nm, respectively. The f-MoS2/CNS composite had a higher surface area with value of 61 m2 g−1 compared to 25 m2 g−1 recorded for the flower-like MoS2. This means that incorporation of CNS prevented the agglomeration of MoS2 sheets (also suggested from the XRD data). Surprisingly, the BET data follow the XRD data, especially the (002) peak height. Considering that capacitance is a function of the surface area, one would expect the materials with higher surface area (such as the f-MoS2/CNS) to provide the best capacitance value. Moreover, despite the fact that the pores do not directly contribute to the surface area, they provide accessible pathways for easy diffusion of ions and reversible charge storage.
| Sample | SABET (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| s-MoS2 | 17.80 | 0.100 | 22.93 |
| s-MoS2/CNS | 9.17 | 0.040 | 18.61 |
| f-MoS2 | 25.00 | 0.018 | 36.13 |
| f-MoS2/CNS | 61.00 | 0.020 | 14.68 |
Raman spectroscopy was used to provide more insight into the structure and topology of the as-synthesized MoS2-based nanocomposites. Fig. 4 shows the Raman spectra of as-synthesized MoS2 nanosheets, CNS and MoS2/CNS nanocomposites. The CNS exhibited the signature D and G peaks of carbon-based materials at 1347 and 1590 cm−1, respectively. As we expected, the MoS2/CNS composites also showed the D and G peaks close to the regions wherein they were observed for the CNS alone, confirming the successful integration of the CNS into the two MoS2-based composites. There was no detectable difference in the intensity ratios of the D to G band (ID:IG) of the CNS (0.95), s-MoS2/CNS (0.94) and f-MoS2/CNS (0.94), which implies that the CNS essentially retained its pristine structure even after integration with the MoS2. The characteristic Raman bands for bulk s-MoS2 were observed at 375.69 and 402.49 cm−1 due to E12g and A1g modes with full-widths at half maximum (FWHM) of 8.47 and 8.23 cm−1, respectively. The E12g mode describes the in-layer displacement of the Mo and S atoms, whereas the A1g mode relates to the out-of-layer symmetric displacements of S atoms along the c axis.24,25 Interestingly, the incorporation of the CNS into the MoS2 resulted in the slight softening of these two Raman bands compared to those of the bulk MoS2; the spherical MoS2/CNS appeared at 375.07 cm−1 (E12g) and 400.84 cm−1 (A1g), while the flower-like morphology appeared at 374.91 cm−1 (E12g) and 401.78 cm−1 (A1g). Moreover, the increase in the FWHM values were more pronounced for the flower-like morphology than for the spherical morphology.
 | ||
| Fig. 4 Raman spectra of spherical (a–c) and flower-like (d–f) MoS2, CNS and MoS2/CNS nanocomposites and their magnified views. | ||
In the flower-like morphology, FWHM values are larger in the f-MoS2/CNS than in the bulk f-MoS2 (cf. E12g = 13.55 vs. 9.29 cm−1 of the bulk f-MoS2 or A1g = 10.88 vs. 8.72 cm−1 of the bulk f-MoS2). The broadening of the Raman bands is related to phonon confinement and also indicates that the lateral dimensions of these MoS2 layers are in the nano-dimension.25 The larger FWHM for the f-MoS2/CNS compared to its s-MoS2/CNS counterpart is indicative of the smaller particle sizes and larger surface area, which corroborates the BET analysis. In a recent study by Lee et al.,26 the authors showed that the frequency difference between E12g and A1g modes could serve as a convenient and robust diagnosis of the layer thickness of MoS2 samples. From our results in Table 2, the frequency difference (i.e., |E12g − A1g|) decreases as follows: bulk MoS2 (ca. 27 cm−1) > f-MoS2/CNS (ca. 26 cm−1) = s-MoS2/CNS (ca. 26 cm−1). Thus, our results indicate that there is no significant difference in the layer thicknesses of the two MoS2/CNS composites and the two synthesis methods we adopted in this study further confirm the change in the physicochemical properties of the MoS2 complexes.
| Raman shift/cm−1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| s-MoS2 | s-MoS2/CNS | f-MoS2 | f-MoS2/CNS | |||||
| E12g | A1g | E12g | A1g | E12g | A1g | E12g | A1g | |
| Peak position | 375.69 | 402.49 | 375.07 | 400.84 | 374.91 | 401.78 | 375.23 | 400.95 |
| |E12g − A1g| | 26.8 | 25.77 | 26.87 | 25.72 | ||||
| FWHM | 8.47 | 8.23 | 8.19 | 8.71 | 9.29 | 8.72 | 13.55 | 10.88 |
Fig. 5 shows FT-IR spectra of the MoS2, CNS and MoS2/CNS nanocomposites. In MoS2, the peaks at 1583 and 1518 cm−1 are consistent with NH2 in-group deformation (arising from the thiourea or L-cysteine used in the synthesis), and the peak at 1466 cm−1 is assigned to the N–C–N asymmetric stretching mode. The band at 1231 cm−1 is assigned to stretching vibrations of O–H bonds. The C–O–H stretching mode occurred at 1092 cm−1. The C–S stretching vibrations are observed at 729 cm−1. The weak peak around 600 cm−1 is assigned to Mo–S vibration. Similar peaks are observed for the MoS2/CNS nanocomposite with the additional peaks at 2892 cm−1 and 2971 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C symmetric and asymmetric stretching vibrations as explained elsewhere.27 Indeed, the presence of the functional groups on the CNS could have contributed to the efficient integration of the CNS with the MoS2 (as observed in the TEM images).
C symmetric and asymmetric stretching vibrations as explained elsewhere.27 Indeed, the presence of the functional groups on the CNS could have contributed to the efficient integration of the CNS with the MoS2 (as observed in the TEM images).
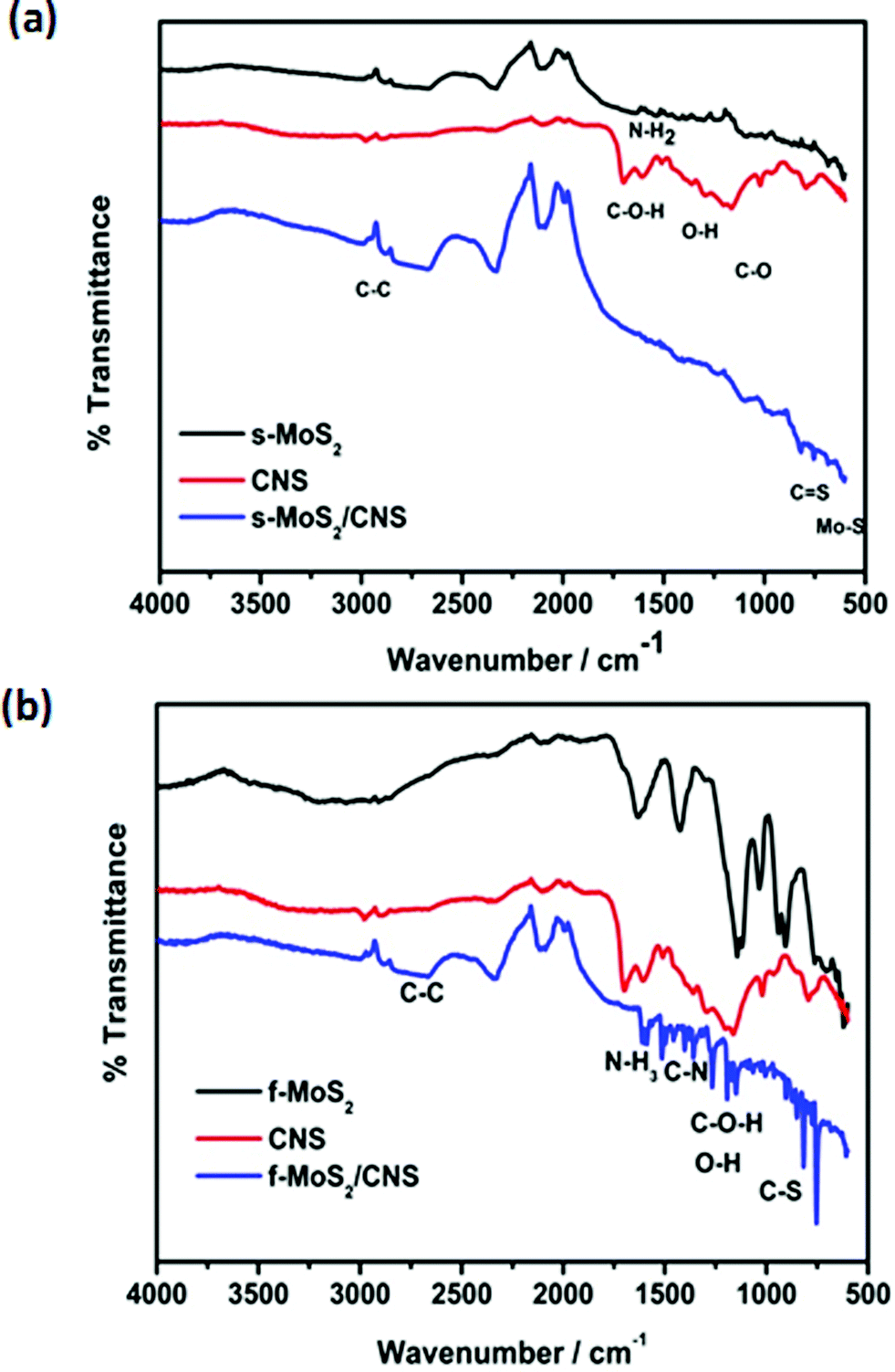 | ||
| Fig. 5 FT-IR spectra of spherical (a) and flower-like (b) MoS2, CNS and MoS2/CNS nanocomposites. | ||
Fig. 6 compares the XPS data of s-MoS2/CNS (Fig. 6a and b) and f-MoS2/CNS (Fig. 6c and d). XPS analysis reveals the predominant Mo 3d and S 2p characteristic peaks for both s-MoS2/CNS and f-MoS2/CNS. The two materials clearly show that the only elements present are Mo, S, adventitious C and O, and Na ‘impurity’ (Table 3).
 | ||
| Fig. 6 High resolution XPS analysis of s-MoS2/CNS (a and b) and f-MoS2/CNS (c and d). Mo 3d (a and c) and S 2p (b and d). | ||
| Sample | Binding energy/eV | Atomic/%, det. limit of 0.1 at% | ||||||
|---|---|---|---|---|---|---|---|---|
| Mo 3d5/2 | Mo 3d3/2 | Mo 3d5/2 | Mo 3d | S 2p | C 1s | O 1s | Na | |
| s-MoS2/CNS | 229.33 | 232.70 | 235.75 | 25.5 | 31.6 | 29.8 | 12.1 | 1.0 |
| f-MoS2/CNS | 229.00 | 232.80 | 236.50 | 22.8 | 28.4 | 15.8 | 23.1 | 9.9 |
Both materials show the typical spectral signature of MoS2, with the co-existence of metallic (1T) and semiconducting (2H) phases.28–30 There is a clear difference between the two complexes, notably the S 2p splits into two peaks in the s-MoS2/CNS (162.42 and 163.67 eV for S 2p3/2 and S 2p1/2, respectively), whereas the f-MoS2/CNS splits into four peaks (located at 162.31, 163.34, 164.63 and 169.23, which are assigned to S 2p3/2, S 2p1/2, S22− or S2− and S4+, respectively), suggesting that the S atoms exist in two different chemical states. This finding is indicative of f-MoS2/CNS undergoing partial oxidation into MoSxOy at the edges and defect sites, which effectively leads to enhanced redox processes. The molybdenum spectra (Fig. 6a and c) show the expected Mo 3d5/2 and Mo 3d3/2, including the Mo6+ species (ca. 236 eV) usually observed in partially oxidized MoS2 complexes. In addition to the significant changes in the S 2p of the f-MoS2/CNS, its surface atoms, with the exception of O and Na, are lower than observed for the s-MoS2/CNS. Considering that surface Na species are involved in the storage mechanism, one may conclude that Na will be more accessible to the f-MoS2/CNS in an aqueous solution for enhanced energy storage than the s-MoS2/CNS.
Electrochemical properties
Fig. 7a shows the cyclic voltammetric (CV) evolutions of the individual CNS, s-MoS2 and f-MoS2 obtained from the 3-electrode configuration in 1 M Na2SO4 at 5 mV s−1. These results show that s-MoS2 is more capacitive than the f-MoS2 but, as it will be shown later, the capacitance retention of the s-MoS2 is worse than that of the f-MoS2. From Fig. 7a, it is evident that CNS do not show any significant current response. Fig. 7b compares the CVs of the composite materials, showing that the f-MoS2/CNS has better performance (high current response and slightly wider voltage window) compared to the s-MoS2/CNS. Fig. 7c and d exemplify typical GCPL curves of the various symmetric cells obtained in 1 M Na2SO4 at 0.5 A g−1. The performance of the s-MoS2 is better than its composite, whereas the f-MoS2/CNS is better than its precursor f-MoS2. As will be shown later, the nanocomposite materials (both spherical and flower-like) show better capacitance retention and cycling stability than their MoS2 materials. As observed in Fig. 7, the CV and GCPL curves of the composite materials, notably the flower-like materials, show quasi-rectangular shapes, which is a strong deviation from the ideal rectangular shape that was expected from the EDLC. Furthermore, the GCPL of the flower-like composite (Fig. 7d) shows a broad peak at around 0.3 V. These results clearly confirm the pseudocapacitive behavior of these MoS2-based composite materials. | ||
| Fig. 7 CV comparison of (a) s-MoS2, f-MoS2 and CNS; (b) s-MoS2/CNS and f-MoS2/CNS. GCPL comparison of (c) s-MoS2/CNS and s-MoS2, and (d) f-MoS2/CNS and f-MoS2. Conditions: CVs obtained at 5 mV s−1, while the GCPL was obtained at 0.5 A g−1; all data were acquired in 1 M Na2SO4. | ||
Fig. 8 shows the plots of specific capacitance versus current densities. With the exception of the s-MoS2/CNS-based cell, the capacitance values of all the cells decrease with increasing gravimetric currents. The capacitance of the s-MoS2/CNS-based cell remains essentially the same (cf. 108 F g−1 at 0.1 A g−1versus 94 F g−1 at 1 A g−1, which is about 13% capacitance loss). This ability of the s-MoS2/CNS to maintain high capacitance even at high current density is indicative of high-power performance.
 | ||
| Fig. 8 GCPL results for specific capacitance at various current densities of spherical and flower-like MoS2 and MoS2/CNS. | ||
Table 4 compares the values of capacitance, maximum energy and power density of the symmetric cells; unfortunately, there is no related literature with which to compare our results, except for 3-electrode systems. The maximum specific capacitance for the flower-like composite f-MoS2/CNS electrodes is 231 F g−1, with maximum energy density 26 W h kg−1 and power density 6443 W kg−1. For the sphere-like morphologies, the equivalent values obtained were 108 F g−1, 7.4 W h kg−1 and 3700 W kg−1. The inferior performance of the s-MoS2/CNS electrodes can be explained by the inaccessible surface area for charge storage in the MoS2/CNS composite due to the presence of CNS, as shown by the Raman spectroscopic data. The maximum energy density achievable with f-MoS2/CNS (26 W h kg−1) is more than twice that of MoS2 (10 W h kg−1) alone. The high energy density is owed to the favorable porous nanostructure of the composite, in which MoS2 sheets serve as active sites for redox reactions, coupled with CNS interaction with electrolyte and the synergistic effects of MoS2/CNS composites.
| Aqueous electrolyte | Electrode material | Device configuration | Voltage (V) | C sp/F g−1 | E sp/W h kg−1 | P max/W kg−1 | Ref. |
|---|---|---|---|---|---|---|---|
| a Key: PANI = polyamine; PPy = polypyrrole; RGO = reduced graphene oxide; MWCNT = multi-walled carbon nanotubes. | |||||||
| 1 M Na2SO4 | f-MoS2/CNS | Symmetric | 0.9 | 231 | 26 | 6443 | This work |
| 1 M Na2SO4 | s-MoS2/CNS | Symmetric | 0.7 | 108 | 7.4 | 3700 | This work |
| 1 M Na2SO4 | s-MoS2 | Symmetric | 1.0 | 195 | 27 | 8750 | This work |
| 1 M Na2SO4 | f-MoS2 | Symmetric | 0.8 | 96 | 8.59 | 4000 | This work |
| 1 M Na2SO4 | MoS2 | 3-Electrode | 0.7 | 92.85 | 7.25 | 186.5 | 14 |
| 1 M H2SO4 | PANI/MoS2 | 3-Electrode | 1.0 | 575 | 265 | 18000 |
2 |
| 1 M KCl | PPy/MoS2 | 3-Electrode | 0.8 | 553.7 | 49 | 400 | 8 |
| 1 M Na2SO4 | MoS2–graphene composite | 3-Electrode | 1.0 | 243 | 85 | 7600 | 6 |
| 1 M Na2SO4 | MoS2/RGO | 3-Electrode | 0.6 | 253 | 12.65 | 300 | 10 |
| 1 M Na2SO4 | MoS2/MWCNT | 3-Electrode | 1.0 | 452.7 | — | — | 12 |
| 3 M KOH | Porous tubular C/MoS2 | 3-Electrode | 0.5 | 210 | — | — | 14 |
In theory, the specific capacitance of the MoS2 may be estimated from the amount of power needed to carry out the electrolysis of 1 mol of active material (i.e., 1 F = 96485 C) and the molar mass of the material (MoS2 = 160.07 g mol−1) using the eqn (8):31
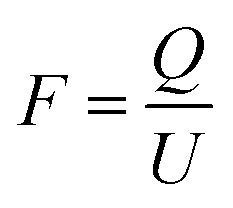 | (8) |
Voltage-holding cycling experiments, complemented with EIS experiments, were performed to provide insight into the cycling stability of the four symmetric cells. The zeroth hour cycling capacitance (brown data points in Fig. 9 and 10) for each of the four symmetric cells was found to be lower than that at the 10th hour cycling, which suggests that MoS2-based cells require a significant amount of time to equilibrate prior to recording data from voltage-holding tests. Thus, the best voltage-holding test data were obtained from the 10th hour. Fig. 9 exemplifies the typical voltage-holding test performed at 0.7 A g−1 for the spherical morphology. At the 10th hour, the s-MoS2 started with about 140 F g−1 and obtained 20 F g−1 at the end of the 50th hour (Fig. 9a), which is about 86% loss of capacitance. The composite s-MoS2/CNS started with about 90 F g−1, but maintained 55 F g−1 at the end of the 50th hour (Fig. 9c), which is about 39% loss of capacitance. The fitted EIS data for both the s-MoS2 (Fig. 9b) and its composite form (s-MoS2/CNS, Fig. 9d) obtained before and after the 50 hour long-cycling experiments are summarized in Table 5. For the flower-like materials (Fig. 10), the capacitance of the f-MoS2 at the 10th hour was about 24 F g−1 and obtained 14 F g−1 at the end of the 50th hour (Fig. 10a), which is about 42% loss of capacitance. The composite f-MoS2/CNS started with about 90 F g−1, but maintained 84 F g−1 at the end of the 50th hour (Fig. 10c), which is a mere 6% loss of capacity. The fitted EIS data for both the f-MoS2 (Fig. 10b) and its composite form (f-MoS2/CNS, Fig. 10d), obtained before and after the 50 hour long-cycling experiments are summarized in Table 6.
 | ||
| Fig. 9 Typical GCPL plots at 0.7 A g−1 (a and c) and Nyquist plots (b and d) obtained before and after 50 hour voltage experiments for the spherical (a and b) MoS2 and MoS2/CNS-based (c and d) symmetric pseudocapacitors. The electrical equivalent circuit used in fitting the Nyquist plots is shown in (e). | ||
 | ||
| Fig. 10 Typical GCPL plots at 1 A g−1 for f-MoS2 (a) and 1.5 A g−1 for f-MoS2/CNS (c); Nyquist plots for f-MoS2 (b) and f-MoS2/CNS (d) obtained before and after 50 hour voltage experiments. The electrical equivalent circuit used in fitting the Nyquist plots is shown in Fig. 9e. | ||
| Parameter | s-MoS2//s-MoS2 | s-MoS2/CNS//s-MoS2/CNS | ||
|---|---|---|---|---|
| 0th hour cycle | 50th hour cycle | 0th hour cycle | 50th hour cycle | |
| R s/Ω | 0.23 ± 0.05 | 0.49 ± 0.06 | 0.33 ± 0.19 | 1.14 ± 0.87 |
| Q 1/mF s(α − 1) | 0.33 ± 0.08 | 0.47 ± 0.03 | 0.2 ± 0.03 | 2.21 ± 0.11 |
| n 1 | 0.40 ± 0.09 | 0.77 ± 0.15 | 0.82 ± 0.17 | 0.64 ± 0.18 |
| R ct1/Ω | 3.75 ± 0.42 | 10.08 ± 2.7 | 0.72 ± 0.21 | 1.39 ± 0.13 |
| Q 2/mF s(α − 1) | 8.10 ± 0.34 | 8.70 ± 0.02 | 13.32 ± 1.32 | 9.38 ± 0.02 |
| n 2 | 0.80 ± 0.25 | 0.80 ± 0.22 | 0.47 ± 0.29 | 0.83 ± 0.17 |
| R ct2/Ω | 12.51 ± 0.07 | 7.20 ± 1.95 | 1.73 ± 0.55 | 3.23 ± 0.29 |
| Phase angle | −73° | −53° | −80° | −80° |
| Knee frequency | 1 Hz | 1 Hz | 50 Hz | 5 Hz |
From the results of the cycling performances of the spherical (Fig. 9) and flower-like (Fig. 10) materials, the following important findings should be emphasized. First, the zeroth hour cycling capacitance for each of the cells was found to be lower than that at the 10th hour cycling, which suggests that MoS2-based cells or related layered materials require a significant amount of time to equilibrate prior to recording data from voltage-holding tests. Second, the cycling stability for MoS2 is very poor, but can be greatly improved by integrating it with conductive carbon materials such as the CNS. Third, the cycling stability of the cells from the MoS2 or its carbon composite is strictly dependent on its morphology; the flower-like morphology shows enhanced electrochemistry compared to the spherical morphology.
Finally, to obtain some insight into the capacitive properties of the cells, we were able to satisfactorily fit the raw EIS data with the electrical equivalent circuit (EEC). The EEC consists of Voigt RC elements (Fig. 9e), involving series resistance (Rs), charge-transfer resistance (Rct) and constant-phase elements (CPE or Q). For the cell with the spherical morphology (Table 5), the Rs values of the s-MoS2 before and after the 50th hour were 0.23 and 0.49 Ω, respectively. For the s-MoS2/CNS, the Rs values before and after the 50th hour were 0.33 and 1.14 Ω, respectively. The combined Rct values before and after the 50th hour were 16.26 and 17.28 Ω, respectively. For the s-MoS2/CNS it was 2.45 Ω before and 4.62 Ω after the 50th hour cycling. The result clearly proves that the presence of the CNS enhanced the conductivity of the s-MoS2-based cells. However, the data for the cells fabricated from the flower-like morphology (Table 6), the Rs values of the f-MoS2 before and after the 50th hour, were 0.21 and 0.40 Ω, respectively. For the f-MoS2/CNS, the Rs values before and after the 50th hour were 0.26 and 0.31 Ω, respectively. The combined Rct values for the f-MoS2 before and after the 50th hour were 3.43 and 4.14 Ω, respectively. For the f-MoS2/CNS, it was 3.37 before and 5.09 Ω after the 50th hour cycling. Like the s-MoS2-based cells, the results for the f-MoS2-based cells suggest that the CNS component serves to decrease the internal resistance of the f-MoS2, thereby improving the conductivity and capacitance of the f-MoS2/CNS-based symmetric pseudocapacitor. The impedance of the CPE (ZCPE) is related to the frequency-independent constant (Q) and radial frequency (w) according to the eqn (9):32
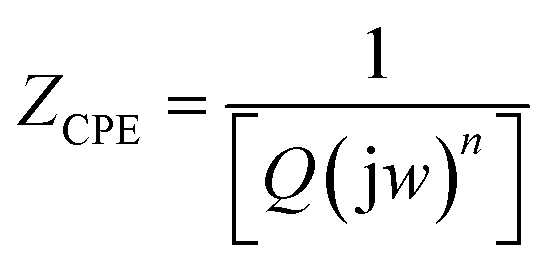 | (9) |
Z versus logf. When n = 0, 1, −1 or 0.5, the CPE describes a pure resistor, a pure capacitor, an inductor, or Warburg impedance (Zw), respectively, due to the diffusion of the ions. For all the cells, the n values observed for these electrodes are generally greater than 0.5, which confirms the pseudocapacitive properties of the MoS2-based symmetric cells, corroborating the CV data in Fig. 7a and b.
| Parameter | f-MoS2//f-MoS2 | f-MoS2/CNS//f-MoS2/CNS | ||
|---|---|---|---|---|
| 0th hour cycle | 50th hour cycle | 0th hour cycle | 50th hour cycle | |
| R s/Ω | 0.21 ± 0.102 | 0.4 ± 0.1 | 0.26 ± 0.08 | 0.31 ± 0.17 |
| Q 1/mF s(α − 1) | 0.2 ± 0.14 | 0.16 ± 0.03 | 0.12 ± 0.07 | 0.17 ± 0.02 |
| n 1 | 0.67 ± 0.19 | 0.73 ± 0.1 | 0.84 ± 0.14 | 0.90 ± 0.13 |
| R ct1/Ω | 1.70 ± 0.62 | 1.57 ± 0.16 | 0.32 ± 0.13 | 1.28 ± 0.74 |
| Q 2/mF s(α − 1) | 21.12 ± 0.07 | 9.4 ± 0.08 | 12.24 ± 0.68 | 9.64 ± 0.36 |
| n 2 | 0.85 ± 0.15 | 0.62 ± 0.13 | 0.68 ± 0.21 | 0.47 ± 0.29 |
| R ct2/Ω | 1.73 ± 0.18 | 2.57 ± 1.24 | 3.05 ± 0.18 | 3.81 ± 0.41 |
| Phase angle | −78° | −70° | −75° | −80° |
| Knee frequency | 10 Hz | 10 Hz | 50 Hz | 50 Hz |
The data for the Bode plots (see ESI, Fig. S3†) summarised in Tables 5 and 6 show that the phase angle for each of the systems before and after 50 hour cycling was greater than −75°, but lower than the −90° expected of an ideal EDLC system. The phase angle result is a further confirmation of the pseudocapacitive behaviour of these MoS2-based systems. In addition, the knee frequency (fo, ϕ = 45°), which is the maximum frequency at which the dominant behaviour of the supercapacitor (power density), can be observed. The knee frequency relates to the rate or power capability of the supercapacitor; the higher the fo value is, the more rapidly such a supercapacitor can be charged and discharged. The fo values for the s-MoS2/CNS are 50 Hz (time constant = 0.02 s) before cycling and 5 Hz (time constant = 0.2 s) after the 50th hour voltage-holding testing. However, for the f-MoS2/CNS system, the fo value remained at 50 Hz before and after the voltage-holding test, which is a further confirmation of the high electrochemical cycling stability of the f-MoS2/CNS system.
The energy-storage mechanism of MoS2 in aqueous supercapacitors is well described in the literature;29,31 it is the combined phenomena involving the transition from EDLC to the pseudocapacitive process (faradaic/redox process) and increased active surface area due to possible exfoliation. First, there is the accumulation of ions at the double layer interface between the MoS2 flakes and the electrolyte. This is subsequently accompanied by a redox process: upon charging (reduction), the alkali metal ions in the electrolyte (Na+) adsorb onto the surface and intercalate between the MoS2 layers, followed by deintercalation upon discharging (oxidation), as shown in eqn (10).
| MoS2 + Na+ + e− ⇄ MoS–SNa | (10) |
The repeating intercalation–deintercalation process of the sodium ions over several cycles leads to partial exfoliation of the MoS2 layers, resulting in an increased surface area and enhanced specific capacitance. The evidence for the possible partial exfoliation of the MoS2 during the repeated intercalation–deintercalation process of electrolyte ions can be observed from the zeroth hour cycling capacitance (brown data points in Fig. 9 and 10 described above), which was lower than the 10th hour cycling before stabilizing.
Conclusions
Two variants of carbon nanosphere-modified molybdenum disulphide (MoS2/CNS) nanostructures were successfully synthesized by a simple one-pot hydrothermal method. The two different synthetic methods obtained two different morphologies: flower-like (f-MoS2/CNS) and spherical (s-MoS2/CNS) morphologies. The physical and chemical characterisations reveal that the two materials were properly integrated into the CNS surface. The addition of CNS impedes the growth of the s-MoS2 crystals in the composite, but enhances the growth of the f-MoS2, particularly in the (002) plane of hexagonal MoS2. The f-MoS2/CNS shows lattice expansion and large surface area, whereas the s-MoS2/CNS shows lattice contraction and smaller surface area. The presence of CNS on the MoS2 structure leads to slight softening of the characteristic Raman bands (E12g and A1g modes) with larger FWHM. The electrochemical capacitive behaviour of the MoS2-based materials was evaluated in symmetric cells. The electrochemical performance of the composites demonstrates that the MoS2/CNS composite from flower-like MoS2 exhibits better capacitance, energy and power densities (231 F g−1, 26 W h kg−1 and 6443 W kg−1) compared to the spherical morphology (108 F g−1, 7.4 W h kg−1 and 3700 W kg−1). CNS play a vital role in improving the electrochemical properties of the MoS2-based electrode materials, especially with respect to improving the capacitance retention (i.e., stable electrochemical cycling). The excellent electrochemical performance of MoS2/CNS was accredited to the morphology of the composite and synergistic effects between MoS2 sheets and CNS. These findings show great promise for future studies of MoS2 modified with other conducting carbon nanostructures for the development of high-performance electrochemical energy storage systems.Acknowledgements
This study was supported by the University of the Witwatersrand (Wits) and CSIR, South Africa. The support of the Wits Materials for Energy Research Group (MERG) towards this research is gratefully acknowledged. Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to MERG. TNY Khawula thanks MERG for postgraduate bursary. K. Raju thanks the CSIR for postdoctoral fellowships. CSIR is a partner in the CREATe-Network project being funded by the European Commission under the Marie Skłodowska-Curie Actions Research and Innovation Staff Exchange (RISE).References
- B. E. Conway, Electrochemical Supercapacitors, Scientific Fundamentals and Technological Applications, Kluwer Acadamic/Plenum Publishers, New York, 1999 Search PubMed.
- K. J. Huang, L. Wang, Y. J. Liu, H. B. Wang, Y. M. Liu and L. L. Wang, Electrochim. Acta, 2013, 109, 587 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1 CrossRef CAS.
- J. M. Miller, Energy storage technology markets and applications: Ultracapacitors in combination with lithium-ion, IEEE Xplore DOI:10.1109/ICPE.2007.4692343: 7th International Conference on Power Electronics, April 2007.
- Mazda ‘i-ELOOP’ Capacitor-Based Brake Energy Regeneration System, http://www.mazda.com/publicity/release/2011/201111/111125a.html.
- K. J. Huang, L. Wang, Y. J. Liu, H. B. Wang, Y. M. Liu and L. L. Wang, Int. J. Hydrogen Energy, 2013, 38, 14027 CrossRef CAS.
- S. Wang, C. An and J. Yuan, Materials, 2010, 3, 401 CrossRef CAS.
- G. Ma, H. Peng, J. Mu, H. Huang, X. Z. Zhou and Z. Lei, J. Power Sources, 2013, 229, 72 CrossRef CAS.
- M. Mandal, D. Ghosh, S. S. Kalra and C. K. Das, Recent Res. Sci. Technol., 2014, 3, 65 Search PubMed.
- K. Changa and W. Chen, Chem. Commun., 2011, 47, 4252 RSC.
- K. J. Huang, L. Wang, J. Z. Zhang, L. L. Wang and Y. P. Mo, Energy, 2014, 67, 234 CrossRef CAS.
- J. M. Soon and K. P. Loh, Electrochem. Solid-State Lett., 2007, 10, A250 CrossRef CAS.
- B. Hu, X. Qin, A. M. Asiri, K. A. Alamry, A. O. Al-Youbi and X. Sun, Electrochim. Acta, 2013, 100, 24 CrossRef CAS.
- K. K. Moorthy, G. K. V. Subramani, S. R. Krishnan and S. J. Kim, Mater. Res. Bull., 2014, 50, 499 CrossRef.
- A. N. Márquez, R. Romero, A. Romero and J. L. Valverde, J. Mater. Chem., 2011, 21, 1664 RSC.
- T. Jiang, W. Pan, J. Wang, X. Bie, F. Du, Y. Wei, C. Wang and G. Chen, Electrochim. Acta, 2010, 55, 3864 CrossRef CAS.
- K. J. Huang, L. Wang, J. Li and Y. M. Liu, Sens. Actuators, B, 2013, 178, 671 CrossRef CAS.
- S. Wang, G. Li, G. Du, X. Jiang, C. Feng, Z. Guo and S. J. Kim, Chinese J. Chem. Eng., 2010, 18, 910 CrossRef CAS.
- M. W. Dlamini, D. O. Kumi, T. N. Phaahlamohlaka, A. S. Lyadov, D. G. Billing, L. L. Jewell and N. J. Coville, ChemCatChem, 2015, 7, 3000 CrossRef CAS.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219 CrossRef PubMed.
- K. Makgopa, P. M. Ejikeme, C. J. Jafta, K. Raju, M. Zeiger, V. Presser and K. I. Ozoemena, J. Mater. Chem. A, 2015, 3, 3480 CAS.
- C. Wang, W. Wan, Y. Huang, J. Chen, H. H. Zhou and X. X. Zhang, Nanoscale, 2014, 6, 5351 CAS.
- H. Li, Q. Zhang, C. C. Ray Yap, B. K. Tay, T. H. Tong Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385 CrossRef CAS.
- G. L. Frey, R. Tenne, M. J. Matthews, M. S. Dresselhaus and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 2883 CrossRef CAS.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695 CrossRef CAS PubMed.
- A. N. Mohan and B. Manoj, Int. J. Electrochem. Sci., 2012, 7, 9537 CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313 CrossRef CAS PubMed.
- M. A. Bissett, I. A. Kinloch and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2015, 7, 17388–17398 CAS.
- D. Voiry, A. Goswami, R. Kappera, C. C. C. Silva, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 CrossRef CAS PubMed.
- C. Peng, D. Hu and G. Z. Chen, Chem. Commun., 2011, 47, 4105 RSC.
- J. B. Jorcin, M. E. Orazem, N. Pébère and B. Tribollet, Electrochim. Acta, 2006, 51, 1473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00114a |
| This journal is © The Royal Society of Chemistry 2016 |