
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust nano-architectured composite thin films for a low-temperature solid oxide fuel cell cathode†
Han Gil
Seo
,
Yoonseok
Choi
,
Bonjae
Koo
,
Ahreum
Jang
and
WooChul
Jung
*
Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: wcjung@kaist.ac.kr; Fax: +82-42-350-3310; Tel: +82-42-350-3314
First published on 7th April 2016
Abstract
Thin-film based, low temperature solid oxide fuel cells (LT-SOFCs) have gained much attention due to their capability to reduce the operating temperature (<450 °C) and the ensuing extended life and reduced cost, compared to conventional SOFCs. While highly porous and continuously connected Pt thin films are viewed as an attractive cathode, they are readily apt to sinter and grow into larger, isolated crystallites upon annealing, leading to severe degradation of the cathode performance over time. Here, we address this issue by fabricating a post-encapsulated composite structure, in which gas-permeable and catalytically active solid electrolytes surround porous Pt electrodes. Nanoporous Pt thin films deposited onto a yttria-stabilized ZrO2 (YSZ) single-crystal substrate are uniformly coated with Sm-doped CeO2 (SDC) via a simple, cost-effective and scalable coating method known as cathodic electrochemical deposition (CELD). Physical characterization of the nanostructured Pt/SDC composite thin films is done using a range of tools, in this case SEM, TEM, XRD and ICP-MS. AC impedance spectroscopy (ACIS) of symmetric cells (cathode|electrolyte|cathode) with SDC coatings reveals that the enhanced Pt–SDC interactions led to exceptionally high ORR activity and outstanding thermal stability. These observations provide new directions for the achievement of a robust and catalytically active LT-SOFC cathode through modification of the metal surface.
1. Introduction
Solid oxide fuel cells (SOFCs) have gained a considerable amount of attention as a promising next-generation power system due to their high energy conversion efficiency and fuel flexibility.1 The main issue with regard to SOFCs is their high operating temperature (700–1000 °C) and the resulting material and cost limitations and operating complexities.2 In this regard, thin-film based, low-temperature solid oxide fuel cells (LT-SOFCs), manufactured using micro-fabrication techniques, have recently been investigated.3–7 The thin film membrane, downscaled to several hundreds of nanometers, allows lower operating temperatures, e.g., less than 450 °C by reducing the ohmic resistance of a solid electrolyte (SE). This helps to solve the main technical issues associated with conventional SOFCs running at elevated temperatures. This approach also has the merits of rapid start-up and shutdown cycles and reduced system cost.2As of now, the key challenge that has limited the development and deployment of LT-SOFCs is the lack of suitable cathode materials with the requisite combination of thermal and chemical stability, electrical conductivity and catalytic activity toward oxygen reduction reaction (ORR). While perovskite-based conducting oxides such as (La,Sr)MnO3, (La,Sr)(Co,Fe)O3 and (Ba,Sr)(Co,Fe)O3 are widely used as cathode materials for the conventional SOFCs, the poor ORR activity at reduced temperatures (<450 °C) and the ensuing large cathodic polarization resistance significantly hinder their utilization for LT-SOFCs.8–11 Moreover, the use of such multi-component oxides typically requires a high-temperature sintering process to obtain a single-phase crystallite, which may lead to thermo-mechanical failures in thin-film components, particularly when the fabrication protocols are not carefully controlled.
In this regard, platinum (Pt) is viewed as an attractive choice as a cathode material due to its process compatibility with micro-fabrication techniques, excellent ORR activity, and good electronic conductivity. Furthermore, in contrast to other metals (e.g., Ni, Al, Cu), Pt has excellent great oxidation tolerance in air, with none of its oxides (e.g., amorphous PtOx, PtO and PtO2) known to be thermodynamically stable above 350 °C.12,13 For these reasons, despite the high cost of Pt, essentially most successful demonstrations in the literature of thin-film based LT-SOFCs, where success is defined as a peak power density ranging from several tens to several hundreds of milliwatts per cm2 at around 400 °C, involve Pt thin films as an electrode.14–18 Considering that the ORR at SOFC cathodes is generally assumed to take place only within a limited region near the triple phase boundary (TPB), where the electronic conductor, the ion conductor, and the gas phase are in simultaneous contact, it is necessary to fabricate a highly porous but interconnected Pt structure with a high site density of TPBs. However, porous nanoscale Pt films are apt to sinter and grow into larger, isolated crystallites upon annealing, leading to a severe loss of catalytic activity and current collecting ability.19–24 Thus, the poor thermal stability of nanoporous Pt film limits its technical application to LT-SOFCs.
Here, we address this issue by fabricating a post-encapsulated composite structure, in which gas-permeable and catalytically active SE layers surround porous Pt films. This electrode structure enables the achievement of decent long-term durability with exceptionally high kinetic performance as LT-SOFC cathodes. There have been several reports in the literature regarding Pt/SE composite thin film electrodes, where SE oxide serves as a physical protection layer against Pt agglomeration and/or an oxygen ion current pathway so as to maximize the TPB site density. For example, Buyukaksoy et al. attempted to coat the surfaces of Pt thin film electrodes by the infiltration of yttria-stabilized ZrO2 (YSZ), showing improved thermal stability at extremely high temperatures (>800 °C) for over 200 h.25 On the other hand, Hertz et al. fabricated nanocomposite Pt/YSZ thin film electrodes by means of magnetron co-sputtering, exhibiting an area-specific resistance (ASR) of the resulting composite films of less than 500 Ω cm2 at 400 °C.26 Chang et al. also reported an enhanced power density by 2.5 times through the application of ALD-coated thin YSZ layers onto porous Pt electrodes.24 It should be noted, however, that most relevant studies focused only on Pt/YSZ composite structures, and the fabrication methods employed appear tedious and costly. Given that acceptor-doped CeO2 (ceria) has higher ionic conductivity and more favorable catalytic activity for ORR27–29 as compared to YSZ, the combination of Pt and ceria may be feasible for a high-performance SOFC cathode. To the best of our knowledge, however, very few studies have made an attempt to fabricate Pt/ceria composites,30 which fewer yet utilizing this combination in LT-SOFCs.
In this work, nanoporous Pt thin films deposited onto a YSZ single-crystal substrate are coated with Sm-doped CeO2 (SDC) via a simple, cost-effective and scalable coating method known as cathodic electrochemical deposition (CELD). While it is widely used for various industrial applications, CELD has rarely been investigated as a manufacturing tool for SOFC electrodes. In previous work, we demonstrated that the Ni-surface modification of SDC layers via CELD showed remarkably enhanced activity toward the fuel oxidation reaction and carbon coking stability as a SOFC anode.31 As continuing work, we apply this coating method to achieve a robust and high-performance LT-SOFC cathode. Electron microscopy reveals that nanostructured SDC layers are uniformly coated onto both surfaces of a Pt electrode and a YSZ SE. The electrochemical characteristics of the composite films were investigated via AC impedance spectroscopy (ACIS) as a function of the deposit time, temperature and oxygen partial pressure. The enhanced Pt–SDC interactions lead to exceptionally high ORR activity and outstanding thermal stability. We show that a CELD treatment of only 5 min creates an oxide coating that increases the electrode conductance by more than two orders of magnitude compared to a bare Pt electrode, while also simultaneously maintaining the conductance level for more than 100 h at 600 °C without any degradation.
2. Experimental
2.1 Fabrication of nanoporous Pt thin films
Highly porous and interconnected Pt thin films with a thickness of 100 nm were fabricated on both (100) single-crystal YSZ (8 mol%, 10 × 10 × 0.5 mm3, MTI Corp.) and (100) single-crystal Si (10 × 10 × 0.5 mm3, Siltron) substrates by means of oxygen reactive DC magnetron sputtering. The sputtering system was operated with a three-inch Pt target (99.98%, Wooilmetal) at a DC power of 50 W in a mixed argon and oxygen atmosphere (Ar/O2 = 1), while maintaining a working pressure of 10 mTorr. After the reactive sputtering process, we decomposed the as-deposited PtOx films by quickly putting them into a tube furnace, which had been maintained at 600 °C in air. And after heat treatment for 1 h, they were quenched to room temperature (RT) and finally porous Pt nanostructures were obtained.322.2 Electrochemical deposition of Sm0.16Ce0.84O1.92−δ films
An aqueous electrolyte solution was prepared with nitrate-based cation precursors, Ce(NO3)3·6H2O and Sm(NO3)3·6H2O, (99.99%, Alfa Aesar) and then saturated with O2 gas with a flow rate of 300 sccm for 30 min. Cation molar concentrations of 0.0475 M and 0.0025 M for Ce3+ and Sm3+, respectively, were chosen to create the resulting coating layer of Sm0.16Ce0.84O1.92−δ. A traditional three-electrode cell was used with a standard calomel electrode (SCE, +0.24 V vs. SHE, Koslow) for a reference electrode, and a carbon rod was used as a counter (anodic) electrode. Electrical contact with the porous Pt films, deposited on both sides of a YSZ substrate, was made using tailor-made aluminum clips, which served as a working electrode. Electrochemical deposition was conducted at room temperature in the potentiostat mode, in which a fixed electric potential of −0.7 V (vs. SCE) between the working electrode and the reference electrode was applied and the resulting current from the counter electrode to the working electrode was measured. The deposition time varied from 15 s to 15 min.2.3 Physical characterization of Pt/SDC composite films
The microstructures of both bare and CELD-coated Pt films were examined by scanning electron microscopy (SEM, Hitachi S-4800). The pore size and TPB density of the bare Pt films were estimated through an image analysis using ImageJ software (U.S. National Institutes of Health, 1.48v). Transmission electron microscopy (TEM, 300 kV-Tecnai G2 F30 S-Twin) was used for close monitoring of the interfaces in the vicinity of the TPBs, and the elemental compositions of the Pt/SDC films were line-scanned using energy-dispersive X-ray spectroscopy (EDX). An inductively coupled plasma mass spectrometry (ICP-MS, Agilent IPC-MS 7700S) analysis was undertaken with a mixture of nitric and hydrochloric acid (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) at 200 °C for 30 min by microwave digestion to measure the chemical composition of the coated SDC layers precisely. X-ray diffraction (XRD, Rigaku Ultima IV) with Cu-Kα radiation (λ = 1.5418 Å) at 40 kV and 40 mA with a scan rate of 4° per min from 20° to 60° was used to identify the crystal structures of the films.
:3) at 200 °C for 30 min by microwave digestion to measure the chemical composition of the coated SDC layers precisely. X-ray diffraction (XRD, Rigaku Ultima IV) with Cu-Kα radiation (λ = 1.5418 Å) at 40 kV and 40 mA with a scan rate of 4° per min from 20° to 60° was used to identify the crystal structures of the films.
2.4 Electrochemical analysis
A symmetric cell structure with identically sized (10 × 10 mm2) bare and CELD-coated Pt electrodes on both sides of the YSZ electrolyte was used for AC impedance spectroscopy (ACIS, VSP-300, Biologic) measurements. AC amplitude of 20 mV at a frequency range of 4 mHz to 2 MHz was used. The perturbation voltage of 20 mV was chosen in this work so as to lie within the linear regime of the sample's current–voltage response. The cells were placed inside a continuous-flow alumina tube into which mixtures of O2 and Ar were delivered via digital mass flow controllers. The total flow rate was kept constant at 100 sccm (standard temperature and pressure), implying a gas velocity of 19.7 cm min−1. Impedance measurements were carried out at temperatures between 300 and 450 °C and at an oxygen partial pressure between 0.05 and 1 atm. Fig. S1† shows a detailed temperature profile of the measurements. The long-term stability of bare Pt and CELD-coated Pt electrodes was then evaluated at 600 °C and 0.21 atm O2 for 40 h and 100 h, respectively.3. Results and discussion
3.1 Overall fabrication processes
Fig. 1 describes the entire fabrication process used to obtain the Pt/SDC composite films. It involves the formation of nanoporous Pt films by the thermal decomposition of Pt oxidic compounds (PtOx) followed by the deposition of SDC layers onto them. The porous Pt structure was obtained when meta-stable PtOx formed under Ar/O2 plasma conditions and was then reduced upon a heat treatment. The resulting films exhibited percolating Pt networks infiltrated with interconnected nano-sized pores, critical for the proper performance of solid oxide fuel cell cathode. For a more detailed description of fabrication process of the Pt films, the reader is referred to a previous publication by the authors.32 | ||
| Fig. 1 Schematic diagram of the overall fabrication procedures (top) and the targeted structure of composite films in this work, compared to bare Pt (bottom). SDC: Sm0.16Ce0.84O1.92−δ. | ||
After decomposition, the nanoporous Pt films served as a working electrode for the subsequent electrochemical deposition of ceria. A description of the deposition of ceria films by CELD is available in the literature.33,34 In short, the CELD process involves three steps: (1) the electro-generation of hydroxyl ions (OH−) by the reduction of species such as H2O, O2, H+ or NO3− at the Pt working electrode, (2) the chemical precipitation of Ce(OH)3 through an increase in the solution pH in the vicinity of the Pt electrode, and (3) the complete oxidation of hydrate to form CeO2 upon dry and/or annealing. Given that the thermodynamic stability of Sm is similar to that of Ce (e.g., Pourbaix diagrams of both Ce–H2O and Sm–H2O are nearly identical.35), one can expect Sm to be incorporated into ceria films by the same mechanism during the electrochemical deposition step. The important point here is that the SDC coating layers must serve as an ionic current pathway to maximize the TPB site density and as a physical protector against Pt agglomeration. Accordingly, the deposited layers must ensure simultaneous contact between the Pt electrode and the YSZ electrolyte while maintaining sufficient oxygen gas accessibility. In contrast to metal electrodeposition, in which a cationic species is reduced to an insoluble metallic form directly on a growing film, CELD involves oxide precipitation reactions within a solution phase. Hence, the process can be used to coat the surfaces of composite anode structures in which both electronically insulating and electronically conductive phases are present.31,34 The targeted structure of the composite film in this work is shown in Fig. 1 (bottom images) with the maximum number of TPBs.
3.2 Physical and chemical characterization
Fig. 2 exhibits SEM images of both bare and CELD-coated Pt films as a function of the deposition time. The bare Pt film provided a highly porous and continuously connected geometry, comparable to those of other Pt electrodes for LT-SOFCs in literatures, with an estimated pore size of 45 ± 24 nm and a TPB density of 15 μm μm−2 according to the SEM image analysis.18,36,37 This nanoporous Pt film structure serves as a simple platform on which to evaluate the impacts of CELD coating layers on the cathode performance. With the electrochemical deposition, the Pt films are uniformly covered by a dark layer which infiltrates into the nanoscale pores in the Pt network. As the deposition time increases, the CELD-coating layers thickens, eventually covering the entire Pt surface (Fig. 2(b)–(d) and (f)–(h)). Only 1 min of deposition creates coating layers sufficiently thick to observe a reduction in the pore size visually compared to the bare Pt films. Cross-sectional SEM images showed that the layer thicknesses are 21 ± 4 and 76 ± 13 nm for deposition times of 3 and 5 min, respectively. It should be noted that the deposited SDC films typically exhibit nano/micro-scale cracks due to the volume change when transforming from a hydrated cerium oxy-hydroxide to CeO2 or to the possible shrinkage of ceria,34,38 conceivably facilitating gas-phase diffusion through the SDC film as well. Upon annealing at 450 °C in air for 10 h, the SDC film becomes more porous, with nano-scale cracks, compared to an as-deposited film, as shown in Fig. S2.† Importantly, the coated layers make simultaneous contact between the Pt electrode and the YSZ substrate, enabling continuous ionic current flows from the YSZ electrolyte through the layers. Furthermore, as discussed later, this structure prevents the pre-deposited Pt films from sintering. | ||
| Fig. 2 Top-view and cross-section SEM images of CELD-coated Pt films on a YSZ single-crystal substrate at applied potential of −0.7 V vs. SCE as a function of the deposition time; (a) and (e) for bare Pt (0 s), (b) and (f) for 1 min, (c) and (g) for 3 min, (d) and (h) for 5 min. | ||
The chemical composition of the deposits, analyzed by ICP-MS, shows that the concentration of Sm in the film is about 16%, which is higher than that of the starting solution (5%), as shown in Table 1. This behaviour may stem from that fact that the standard reduction potential of Ce3+ is more negative than that of Sm3+39 or from the fact that Sm(OH)3 is less soluble than Ce(OH)3.40 XRD patterns of both the bare and the CELD-coated Pt films are presented in Fig. 3. Here, to avoid shadow effects from a substantially large YSZ (100) peak in the single-crystal substrate, the composite film was fabricated on a Si (100) substrate. The CELD-coated layer, after annealing at 600 °C for 10 h in air, shows only a cubic fluorite structure without any secondary phase, indicating that Sm0.16Ce0.84O1.92−δ is the primary phase in this work. The calculated value of the lattice constant of the SDC film is 5.427 Å, which as in good agreement with the reported value of 5.427 Å for Sm0.15Ce0.85O1.925−δ.41 This, in fact, is the most widely used composition of ceria in SOFCs.
| Sample | Concentration of Ce, ppm | Concentration of Sm, ppm | Atomic percent of Sm, at% |
|---|---|---|---|
| −0.7 V, 10 min | 585.1 | 116.9 | 15.7 |
 | ||
| Fig. 3 X-ray patterns of bare and CELD-coated Pt films on a (100) single-crystal Si substrate after annealing at 600 °C for 10 h in air. | ||
The atomic-scale features of the YSZ|SDC and Pt|SDC interfaces are shown in Fig. 4. The SDC layers, grown at −0.7 V for 5 min, are uniformly coated onto the Pt surfaces (Fig. 4(a)), providing a good match to the line scan profile data (Fig. 4(c)). The imaged regions indicate good contact, suggesting easy transport of ionic species between the two oxides and ready electron transfer from the Pt phases to the Pt|SDC interfaces, as shown in Fig. 4(b). As discussed later in more details, these features are essential for expanding the active reaction sites from Pt|YSZ|air to Pt|SDC|air.
 | ||
| Fig. 4 Cross-section TEM images of (a) CELD-coated Pt films deposited at applied potential of −0.7 V vs. SCE for 5 min, (b) an atomic-scale feature of the YSZ|SDC and Pt|SDC interfaces and (c) a line scan profile at Pt|SDC interface (the yellow arrow marked in (a)). | ||
3.3 AC impedance spectroscopy
Selected AC impedance results obtained at 375 °C with pO2 = 0.21 atm are presented in Fig. 5. The raw impedance spectra for symmetric cells including both bare and CELD-coated Pt electrodes generally showed two adjacent semicircles, which are distinguished as a nearly ideal high-frequency (HF) and a slightly distorted low-frequency (LF) semicircle, respectively. While the HF arc remains nearly identical for all samples regardless of the CELD treatment, the LF arc decreases substantially after the SDC layers are coated onto the Pt electrodes. These impedance spectra were modelled by a series of two parallel RC sub-circuits. More generally, the capacitor was replaced by a constant phase element (CPE), for which the overall impedance was determined by ZQ = 1/Q(iω)n. The effective capacitance could be extracted from the expression, C = (R1−nQ)1/n. Several features of the HF impedance spectra are consistent with the source being the bulk impedance of the YSZ electrolyte single-crystal substrate. These include the activation energy of resistance of 1 eV and its pO2 independence. Furthermore, the magnitude of both the conductivity (1.2 × 10−5 S cm−1 at 350 °C) and dielectric constant (47–66 at T = 300–450 °C), calculated based on the present cell geometry and measured values of R and C, are in good agreement with the literature data for YSZ (3.1 × 10−5 S cm−1 and 60 at 350 °C).42 This is also supported by the fact that the CELD coating does not change the HF impedance spectra. | ||
| Fig. 5 Typical impedance spectra of bare (black) and CELD-coated Pt (blue) electrodes on both sides of a YSZ single-crystal electrolyte measured at 375 °C with pO2 = 0.21 atm. A magnified semicircle of CELD-coated Pt electrode at low-frequency is indicated. The impedance spectra were modelled by a series of two parallel RC sub-circuits (insert). Solid line: the fitting results. | ||
Accordingly, the remaining LF spectra are attributed to the electrode response. The R and C values derived from this part of the impedance spectra are designated as Relectrode and Celectrode, respectively. The dependences of the resistance terms of both the bare and CELD-coated Pt electrodes on the temperature and oxygen partial pressure are summarized in Fig. 6. Here, as an indicator of the electrode activity toward the ORR, the values of the electrode conductance normalized with regard to the nominal electrode area were used. In general, the electrode conductance of both the bare and CELD-coated Pt films increases with the temperature with a linear slope in an Arrhenius plot, typical electrode behaviour when thermally activated processes are involved. Here, two factors should be mentioned. First, the electrode conductance increased substantially when the SDC layers are coated. For example, a CELD treatment of only 5 min creates an oxide coating that increases the values of the electrode conductance by more than two orders of magnitude compared to a bare Pt electrode. This implies that modification of the Pt surface by CELD is highly favorable for the ORR, suggesting it as a suitable method to enhance the kinetic performance of LT-SOFC cathodes. Second, despite the significant difference in the magnitudes of the conductance levels between the bare and the coated films, they show nearly identical activation energy and pO2 dependence values within a reasonable amount of error (<7%). Table 2 summarizes the dependences of the electrode conductance on the temperature and pO2 as a function of the deposition time. The values of the activation energy range from 1.16 to 1.36 eV for all deposition times (0–15 min) at T = 300–450 °C and pO2 = 0.21 atm. Furthermore, the power-law dependence, pO2m, of the electrode conductance on the oxygen partial pressure exhibits similar m values between 0.27 and 0.40. These observations indicate that the dominant reaction sites in all cases are identical, likely attributable to the TPBs between Pt, SE and air. Accordingly, the different conductance levels among the samples are considered to be the result of the differing TPB site density levels.
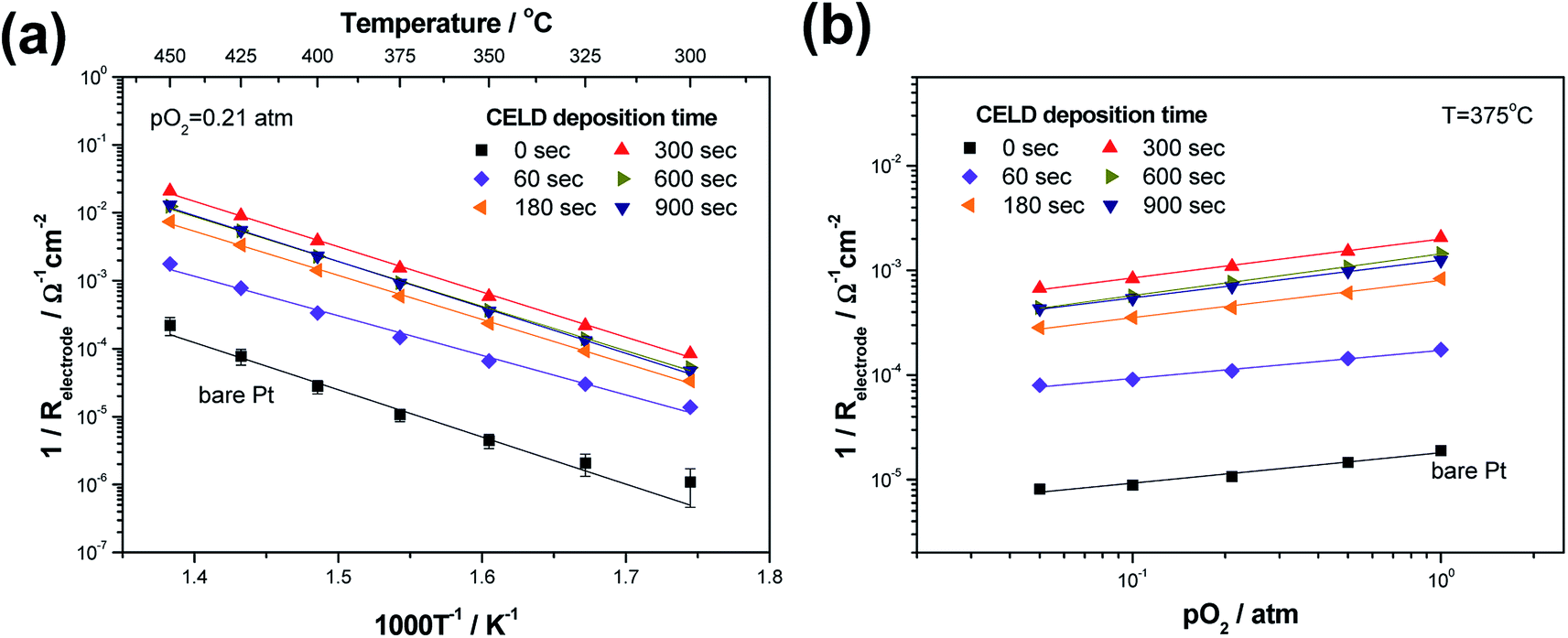 | ||
| Fig. 6 (a) Temperature and (b) pO2 dependences of the electrode conductance values of CELD-coated Pt electrodes as a function of deposition time (0–15 min). | ||
| Deposition time, s | Activation energy (Ea), eV | pO2 dependence |
|---|---|---|
| 0 | 1.36 ± 0.08 | 0.29 ± 0.03 |
| 60 | 1.16 ± 0.04 | 0.27 ± 0.01 |
| 180 | 1.29 ± 0.02 | 0.36 ± 0.02 |
| 300 | 1.32 ± 0.02 | 0.37 ± 0.01 |
| 600 | 1.30 ± 0.02 | 0.40 ± 0.01 |
| 900 | 1.34 ± 0.02 | 0.36 ± 0.01 |
Focusing on the coated samples, the electrode conductance increases considerably with the deposition time (and thereby the deposited thickness) during the initial 5 min of deposition (see Fig. S3† for the detailed relationship between the deposition time and film thickness). However, the electrode conductance levels remain relatively constant after reaching a maximum value at a deposition time of 5 min (Fig. 7(a)). The increase in the conductance is attributed to the fact that the number of percolated SDC nanoparticles in the vicinity of Pt and YSZ tended to increase over time, leading to an extension in the number of active reaction sites from Pt|YSZ|air to Pt|SDC|air. On the other hand, upon a longer deposition time (>5 min), the porous Pt surfaces are sufficiently covered by SDC layers without a further increase in the number of TPB sites. One may expect that thicker SDC coating layers inhibit oxygen gas-phase diffusion, but this is not observed, confirming that the nano/micro-scale cracks which form during the drying and/or annealing processes provided sufficient gas permeability in the composite films.
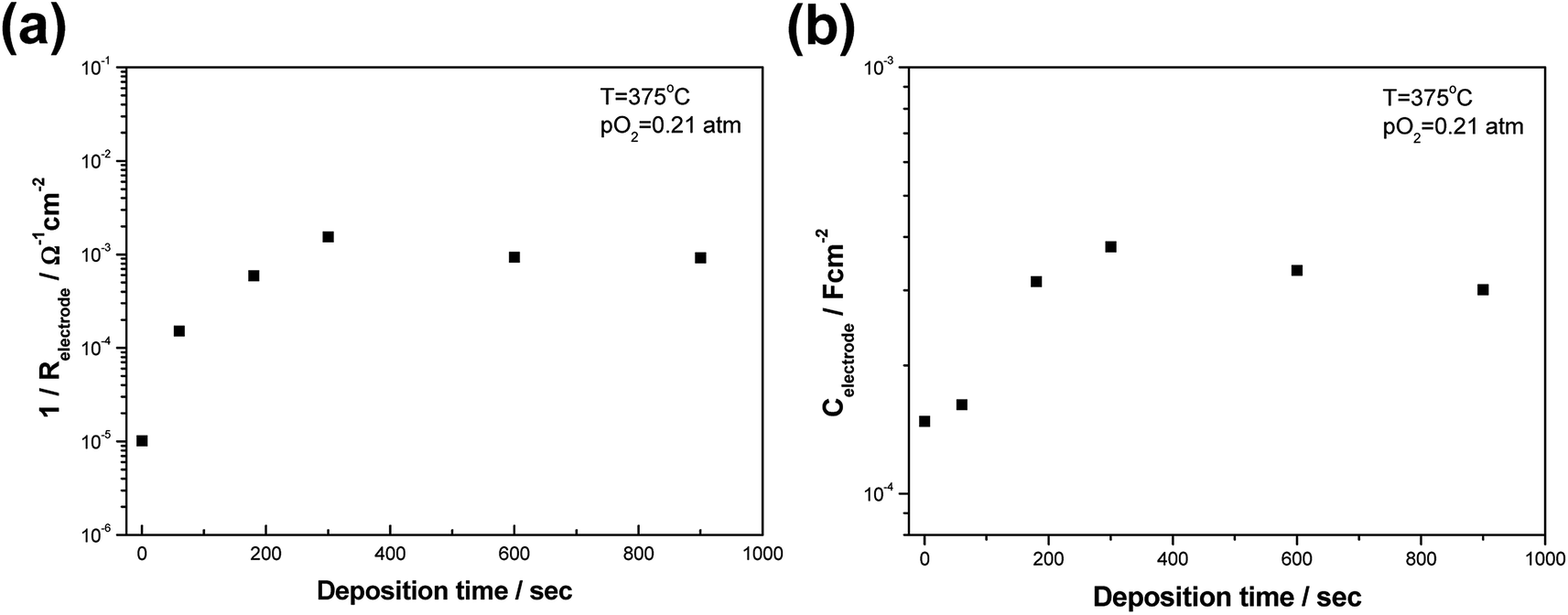 | ||
| Fig. 7 The values of (a) electrode conductance and (b) capacitance of CELD-coated Pt electrodes as a function of deposition time (0–15 min). | ||
Turning to the capacitive part of the electrode impedances, all sample show typical double-layer capacitance behavior with reference to magnitude (∼10−4 F cm−2) and pO2 independence.43,44 Interestingly, the magnitude of Celectrode also increases with the deposition time, reaching a stable value when it exceeds 5 min, as in the case for Relectrode (Fig. 7(b)). This behavior indicates that electrochemically active interface area increases upon SDC deposition, strongly supporting the previous conclusion that the ORR active reaction sites, TPBs, extend from Pt|YSZ|air to Pt|SDC|air.
While the detailed reaction mechanisms of the ORR on the Pt/SE composite electrodes remain not fully understood, several elemental processes have been suggested as the acceptable rate-determining step (RDS).9,45–49 These typically include (1) the adsorption/dissociation of O2 involving an electron transfer, (2) the surface diffusion of adsorbed oxygen species on Pt surface to the TPBs, and (3) the incorporation of oxygen species into the SE lattice near the TPBs. If the first process (1) is RDS in this work, the ORR rate must then increase with the exposed surface area of Pt, resulting in a decrease in the total electrode conductance caused by the CELD coating because the SDC deposits partly cover the Pt surfaces. Accordingly, the possibility that process (1) is the RDS is eliminated. To narrow down the dominant process governing the ORR activity in the composite electrodes, we inserted a dense interlayer of SDC with a thickness of 200 nm by pulsed layer deposition (PLD) in between YSZ and Pt and analyzed the impedance results as a function of the temperature and pO2 (see Fig. S4†). This was done to convert the dominant reaction sites from Pt|YSZ|air to Pt|SDC|air, so as to observe the role of the SE composition on the ORR activity. In fact, we find that both Pt|YSZ|Pt and Pt|SDC|YSZ|SDC|Pt cells exhibit nearly identical values of Relectrode, Ea and the pO2 power dependence, as shown in Fig. S5.† Such independence of the electrode impedance on the SE composition likely indicates that process (3) is not the RDS, but (2) is in this work. Because the effective diffusion lengths from the Pt surface to the TPB (Pt|SDC|air) of the CELD-coated Pt electrodes are expected to be shorter than that of the bare Pt electrodes, the increase in the electrode conductance upon the CELD treatment can be explained. This is also supported by earlier work. For example, Wang et al. reported that the slow surface diffusion of oxygen could govern the electrode conductance with an activation energy (Ea) in the range of 1.1–1.3 eV and pO2 dependence (m) of 3/8, where m is defined as a power-law dependence, pO2m, of electrode conductance, which are comparable to our results (Ea values of 1.16–1.36 eV and m values of 0.27–0.40, respectively), at 500 °C in Pt electrodes on solid electrolyte.49,50 These similar values may support the important role of oxygen diffusion on top of Pt electrodes regarding the overall electrode conductance levels. However, it should be noted that the possible role of SDC itself in catalysing cathode reactions should not be perfectly eliminated in our study due to inherent activity of ceria toward ORR. Fig. S6† shows the reported activities of ceria and other materials as a function of the temperature, in which the activity is defined by the surface oxygen exchange coefficient (ks). In fact, ceria is as active as (La,Sr)(Co,Fe)O3 – a state-of-the-art SOFC cathode. Further investigation regarding the detailed ORR mechanism on the composite electrodes will be needed.
3.4 Long-term stability
Of potentially greater importance than the initial performance, particularly for a nanostructured component intended to be utilized at high temperatures for long periods of time, is the retention of the activity during the prolonged exposure to operational conditions. In this regard, the long-term durability levels of both the bare and the CELD-coated (deposition time = 5 min) Pt electrodes were tested at 600 °C, as shown in Fig. 8(a) and (b), respectively. A temperature of 600 °C was chosen for this assessment to accelerate the agglomeration of Pt films,22 although the targeted operation temperature in this study is less than 450 °C. Temperatures higher than 600 °C were not used, given that the goal of this study was to find a process compatible with typical micro-fabrication schemes that operate under restricted thermal budgets. The stability of the CELD-coated Pt is exceptional. Whereas the bare Pt shows a dramatic increase in both the HF and LF resistance levels, the CELD films display nearly constant resistance levels for more than 100 h (Fig. 8). After only 40 h of annealing, SEM reveals that the bare Pt film is considerably sintered, thus forming isolated particles and thereby disconnecting the lateral current flows (see Fig. S7†). This may be why the HF impedance, which was originally attributed to the bulk resistance of the YSZ electrolyte, increases over time, as the disconnected Pt film reduces the active cross-sectional area for the electrochemical cells effectively. On the other hand, the CELD-coated films exhibit outstanding long-term stability while maintaining nearly constant levels of both the HF and LF resistance over time (Fig. 8(c) and (d)). The enhanced thermal stability of the CELD-coated Pt films can be attributed to the existence of SDC deposits, which may serve as a physical shielding layer from Pt agglomeration, as expected. Furthermore, several rear-earth oxides are known to form a strong bond with precious metals under oxidizing conditions, thereby giving rise to high sintering resistance.22,51 For example, Nagai et al. reported that Pt particles on ceria-based oxides form robust Pt–O–Ce bonds, serving as an anchor and thereby significantly hindering the agglomeration of Pt particles at even 800 °C as compared to those on alumina.51 Therefore, one can conclude that the Pt–SDC interactions enhanced by the CELD treatment lead to robust LT-SOFC cathodes. | ||
| Fig. 8 Changes in the typical impedance spectra of (a) bare and (b) CELD-coated Pt electrodes over time (at 600 °C with pO2 = 0.21 atm for 40 h and 100 h, respectively). The time-dependent resistance levels of (c) high-frequency and (d) low-frequency impedance spectra obtained from samples with bare (black) and CELD-coated Pt (blue) films. | ||
Lastly, the material cost of Pt needs to be mentioned. We estimated the amount of Pt loading used in our work and compared this value with the 2020 target value from the Department of Energy (DOE) for proton exchange membrane fuel cells (PEMFCs).52 By assuming 30% porosity of the Pt thin films used in this work, the amount of Pt loading is calculated to be 0.150 mg cm−2, while the DOE target value is 0.125 mg cm−2. Therefore, one can say that the amount of Pt loading involved in this work is small enough such that there will be no significant impact on the cost.
4. Conclusions
Nanostructured precipitates of SDC are uniformly coated onto porous Pt thin films via a simple, cost-effective and scalable coating method known as CELD in an effort to realize a thermally stable composite cathode for low-temperature SOFCs. Several unique features of CELD enable the creation of gas-permeable and percolated SDC layers, showing simultaneous contact between a Pt electrode and YSZ electrolyte. This nano-architectured structure with SDC coatings extends the ORR reaction sites from Pt|YSZ|air to Pt|SDC|air, substantially increasing the number density of the TPBs and thereby the electrode conductance by more than two orders of magnitude compared with that of bare Pt. Furthermore, the enhanced Pt–SDC interactions provide outstanding thermal stability, maintaining higher electrode conductance even at 600 °C without any degradation for more than 100 h. In sum, the CELD method, as described here, is suggested a possible solution to achieve thin-film based cathodes capable of both high performance and long-term stability, key prerequisites for the practical development of LT-SOFCs.Acknowledgements
This work was supported by the Global Frontier R&D Program on Center for Multiscale Energy System funded by the National Research Foundation under the Ministry of Science, ICT & Future, Korea (2011-0031569). Additional support was provided by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (Ministry of Science, ICT & Future Planning) (2014R1A1A1003008).Notes and references
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- A. Evans, A. Bieberle-Hutter, J. L. M. Rupp and L. J. Gauckler, J. Power Sources, 2009, 194, 119–129 CrossRef CAS.
- H. L. Tuller, S. J. Litzelman and W. Jung, Phys. Chem. Chem. Phys., 2009, 11, 3023–3034 RSC.
- J. An, J. H. Shim, Y. B. Kim, J. S. Park, W. Lee, T. M. Gur and F. B. Prinz, MRS Bull., 2014, 39, 798–804 CrossRef CAS.
- J. M. Ralph, A. C. Schoeler and M. Krumpelt, J. Mater. Sci., 2001, 36, 1161–1172 CrossRef CAS.
- S. J. Litzelman, J. L. Hertz, W. Jung and H. L. Tuller, Fuel Cells, 2008, 8, 294–302 CrossRef CAS.
- N. P. Brandon, S. Skinner and B. C. H. Steele, Annu. Rev. Mater. Res., 2003, 33, 183–213 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4843 CrossRef CAS PubMed.
- S. Wang, T. Kato, S. Nagata, T. Kaneko, N. Iwashita, T. Honda and M. Dokiya, Solid State Ionics, 2002, 152, 477–484 CrossRef.
- F. S. Baumann, J. Fleig, G. Cristiani, B. Stuhlhofer, H. U. Habermeier and J. Maier, J. Electrochem. Soc., 2007, 154, B931–B941 CrossRef CAS.
- Y. Abe, M. Kawamura and K. Sasaki, Jpn. J. Appl. Phys., 1999, 38, 2092–2096 CrossRef CAS.
- Y. Abe, M. Kawamura and K. Sasaki, J. Vac. Sci. Technol., A, 2000, 18, 2608–2612 CAS.
- P. C. Su, C. C. Chao, J. H. Shim, R. Fasching and F. B. Prinz, Nano Lett., 2008, 8, 2289–2292 CrossRef CAS PubMed.
- C. C. Chao, C. M. Hsu, Y. Cui and F. B. Prinz, ACS Nano, 2011, 5, 5692–5696 CrossRef CAS PubMed.
- U. P. Muecke, D. Beckel, A. Bernard, A. Bieberle-Hutter, S. Graf, A. Infortuna, P. Muller, J. L. M. Rupp, J. Schneider and L. J. Gauckler, Adv. Funct. Mater., 2008, 18, 3158–3168 CrossRef CAS.
- C. W. Kwon, J. W. Son, J. H. Lee, H. M. Kim, H. W. Lee and K. B. Kim, Adv. Funct. Mater., 2011, 21, 1154–1159 CrossRef CAS.
- C. W. Kwon, J. I. Lee, K. B. Kim, H. W. Lee, J. H. Lee and J. W. Son, J. Power Sources, 2012, 210, 178–183 CrossRef CAS.
- N. Baumann, E. Mutoro and J. Janek, Solid State Ionics, 2010, 181, 7–15 CrossRef CAS.
- H. Galinski, T. Ryll, P. Elser, J. L. M. Rupp, A. Bieberle-Huetter and L. J. Gauckler, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 235415 CrossRef.
- T. Ryll, H. Galinski, L. Schlagenhauf, P. Elser, J. L. M. Rupp, A. Bieberle-Hutter and L. J. Gauckler, Adv. Funct. Mater., 2011, 21, 565–572 CrossRef CAS.
- C. C. Yu, S. Kim, J. D. Baek, Y. Li, P. C. Su and T. S. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 6036–6040 CAS.
- X. H. Wang, H. Huang, T. Holme, X. Tian and F. B. Prinz, J. Power Sources, 2008, 175, 75–81 CrossRef CAS.
- I. Chang, S. Ji, J. Park, M. H. Lee and S. W. Cha, Adv. Energy Mater., 2015, 5, 1402251 Search PubMed.
- A. Buyukaksoy, V. Petrovsky and F. Dogan, J. Electrochem. Soc., 2013, 160, F482–F486 CrossRef CAS.
- J. L. Hertz and H. L. Tuller, J. Electrochem. Soc., 2007, 154, B413–B418 CrossRef CAS.
- E. N. Armstrong, K. L. Duncan, D. J. Oh, J. F. Weaver and E. D. Wachsman, J. Electrochem. Soc., 2011, 158, B492–B499 CrossRef CAS.
- E. N. Armstrong, K. L. Duncan and E. D. Wachsman, J. Electrochem. Soc., 2011, 158, B283–B289 CrossRef CAS.
- C. B. Gopal and S. M. Haile, J. Mater. Chem. A, 2014, 2, 2405–2417 CAS.
- N. Ai, K. Chen and S. P. Jiang, Solid State Ionics, 2013, 233, 87–94 CrossRef CAS.
- Y. Choi, E. C. Brown, S. M. Haile and W. Jung, Nano Energy, 2016 DOI:10.1016/j.nanoen.2016.03.015.
- W. Jung, J. J. Kim and H. L. Tuller, J. Power Sources, 2015, 275, 860–865 CrossRef CAS.
- K. Kamada, N. Enomoto and J. Hojo, Electrochim. Acta, 2009, 54, 6996–7000 CrossRef CAS.
- E. C. Brown, Ph. D thesis, Caltech, 2011.
- M. Pourbaix, Atlas of electrochemical equilibiria in aqueous solutions, Pergamon Press Ltd., 1966 Search PubMed.
- C. C. Chao, M. Motoyama and F. B. Prinz, Adv. Energy Mater., 2012, 2, 651–654 CrossRef CAS.
- Y. B. Kim, C. M. Hsu, S. T. Connor, T. M. Gur, Y. Cui and F. B. Prinz, J. Electrochem. Soc., 2010, 157, B1269–B1274 CrossRef CAS.
- I. Zhitomirsky and A. Petric, Ceram. Int., 2001, 27, 149–155 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 87th edn, 2006 Search PubMed.
- T. Moeller and H. E. Kremers, J. Phys. Chem., 1944, 48, 395–406 CrossRef CAS.
- Y.-C. Wu, M.-J. Lee and X. Li, J. Eur. Ceram. Soc., 2015, 35, 4485–4495 CrossRef CAS.
- M. C. Steil, F. Thevenot and M. Kleitz, J. Electrochem. Soc., 1997, 144, 390–398 CrossRef CAS.
- N. L. Robertson and J. N. Michaels, J. Electrochem. Soc., 1991, 138, 1494–1499 CrossRef CAS.
- A. Mitterdorfer and L. J. Gauckler, Solid State Ionics, 1999, 117, 203–217 CrossRef CAS.
- J. Mizusaki, K. Amano, S. Yamauchi and K. Fueki, Solid State Ionics, 1987, 22, 313–322 CrossRef CAS.
- J. Mizusaki, K. Amano, S. Yamauchi and K. Fueki, Solid State Ionics, 1987, 22, 323–330 CrossRef CAS.
- D. Y. Wang and A. S. Nowick, J. Electrochem. Soc., 1979, 126, 1166–1172 CrossRef CAS.
- D. Y. Wang and A. S. Nowick, J. Electrochem. Soc., 1979, 126, 1155–1165 CrossRef CAS.
- D. Y. Wang, J. Electrochem. Soc., 1990, 137, 3660–3666 CrossRef CAS.
- D. Y. Wang and A. S. Nowick, J. Electrochem. Soc., 1981, 128, 55–63 CrossRef CAS.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- A. Steinbach, Annual Progress Report V. Fuel Cells, U.S. Department of Energy, 3M Company, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00052e |
| This journal is © The Royal Society of Chemistry 2016 |