
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of side chain length on the charge transport, morphology, and photovoltaic performance of conjugated polymers in bulk heterojunction solar cells†
Chunhui
Duan
a,
Robin E. M.
Willems
a,
Jacobus J.
van Franeker
ab,
Bardo J.
Bruijnaers
a,
Martijn M.
Wienk
ac and
René A. J.
Janssen
*ac
aMolecular Materials and Nanosystems, Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: r.a.j.janssen@tue.nl
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
cDutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ Eindhoven, The Netherlands
First published on 6th January 2016
Abstract
The effect of side chain length on the photovoltaic properties of conjugated polymers is systematically investigated with two sets of polymers that bear different alkyl side chain lengths based on benzodithiophene and benzo[2,1,3]thiadiazole or 5,6-difluorobenzo[2,1,3]thiadiazole. Characterization of the photovoltaic cells reveals a strong interdependency between the side chain length of conjugated polymers and photovoltaic performances (power conversion efficiency, short-circuit current, and fill factor) of the resulting bulk-heterojunction (BHJ) solar cells. Charge carrier transport and external quantum efficiency (EQE) measurements in combination with morphology characterization suggest that too long side chains lead to deteriorated charge transport, suboptimal BHJ morphology, considerable bimolecular recombination, and consequently poor photovoltaic performances. On the other hand, when the side chains are too short, they cannot afford a high enough solubility and molecular weight for the resulting polymers and produce poor solar cell performance as well. This study shows that side chain optimization is of significant importance to maximize the potential of photovoltaic active conjugated polymers, which indicates the fruitful molecular design rules toward highly efficient BHJ polymer solar cells.
1. Introduction
Polymer solar cells (PSCs) have received considerable attention in the past decade in the prospect of producing highly efficient, large-area, flexible photovoltaic modules via cheap roll-to-roll processing.1,2 Extensive research into bulk-heterojunction (BHJ) solar cells with photoactive layers composed of a conjugated polymer as the electron donor and a fullerene derivative as the electron acceptor has resulted in tremendous progress, with power conversion efficiencies (PCEs) exceeding 10% in single-junction devices.3–8 These achievements were largely driven by the development of new materials, especially state-of-the-art photoactive semiconducting conjugated polymers.4,8–12 Based on a fundamental understanding of the film microstructure13–17 and device physics,18–22 it has been possible to correlate the chemical structure of these novel materials with the device performance and thereby promote further advances in materials development and solar cell efficiencies. To pursue state-of-the-art new materials and to establish structure–property relationships, chemists have been manipulating the backbone structures of conjugated polymers.23–30 Implementing alternating donor–acceptor chain motifs,31,32 creating new heteroaromatic building blocks,33–36 and developing a reliable synthetic machinery for introducing heteroatoms (e.g. fluorine and sulfur)37,38 and functionalities (e.g. imide, acyl, and carboxylate groups)39–41 into the conjugated backbone have proved to be effective methods to create novel materials with improved performance.Recently, side chains of conjugated polymers are found to play a key role in polymer packing, backbone orientation, and BHJ morphology, in addition to serving primarily as solubilizing groups.42–44 The reported results show that the type, shape, bulkiness, length, position, and branching point of side chains all exert important effects on the intrinsic physical properties of neat polymers, on the microstructure of BHJ films, and even on the interaction between the polymers and fullerene acceptor.45–58 For instance, it was found that shifting the alkyl chain branching point away from the conjugated backbone in diketopyrrolopyrrole polymers can improve the hole mobility and enhance the short-circuit current density (Jsc) and PCE because of improved polymer crystallinity.49 Osaka et al.54 demonstrated that the backbone orientation of polymers with respect to the substrate can be tuned by altering the alkyl side chain composition. Wang's group55 compared two pairs of indacenodithiophene-based polymers with meta-hexylphenyl or para-hexylphenyl side chains on the indacenodithiophene unit and found that the meta-substituted polymers offer superior photovoltaic performances.
Despite these discoveries, it is still very difficult to draw simple and unambiguous correlations between a change in the side chain structure and an observed variation in device performance. Generally, subtle changes such as variation in molecular weight and processing history can lead to significant difference in both the film microstructure and device performance, making the establishment of a reliable structure–property relationship highly challenging. Moreover, most reported structure–property correlations have been established on the basis of a specific polymer system and necessarily a limited number of derivatives are studied. This limits the universality of the design rules. As making a decision on the length of side chains is a frequently encountered issue in the design of new polymers for chemists, we attempt in this contribution to establish a reliable and universal relationship between the side chain length of conjugated polymers and the photovoltaic performance by systematically investigating various factors that determine the final device performances of PSCs with a large number of polymers that belong to two different material systems.
Recently, we created a wide band gap donor–acceptor polymer, BDT-FBT-2T, based on benzo[1,2-b:4,5-b′]dithiophene (BDT) and 5,6-difluorobenzo[2,1,3]thiadiazole (FBT) toward multi-junction PSC applications.59 A shortcoming of BDT-FBT-2T is the relatively low open-circuit voltage (Voc) compared to the optical band gap (Eg) in single-junction PSCs. To solve this problem, a feasible method is removing the electron-rich thiophene rings from the conjugated backbone of BDT-FBT-2T (Scheme 1) to downshift the highest occupied molecular orbital (HOMO) energy level while keeping Eg unchanged. Two structurally related polymer systems, BDT-BT-Cn and BDT-FBT-Cn, thus emerge and were chosen for correlating the relationship between the side chain length and photovoltaic performance in this paper. Three polymers (BDT-BT-C40, BDT-BT-C32, and BDT-BT-C24) in the BDT-BT-Cn system and five polymers (BDT-FBT-C40, BDT-FBT-C32, BDT-FBT-C24, BDT-FBT-C20, and BDT-FBT-C16) in the BDT-FBT-Cn system were synthesized. The chemical structures of the polymers are shown in Scheme 2. The "Cn" in the abbreviations of the polymers denotes the statistical average number of carbon atoms in alkyl side chains of each repeat unit. Our results show that there are unambiguous trends between the length of side chains and photovoltaic properties of the polymers. The length of the side chains determines the photovoltaic properties of the polymers by influencing charge carrier transport and recombination, as well as BHJ morphology. Noticeably, during the course of this work, the research group in the University of Melbourne reported two structurally relevant polymers which belong to the polymer systems of BDT-BT-Cn and BDT-FBT-Cn, respectively, but featuring different alkyl side chains.60,61 The maximum PCE of the single-junction PSCs based on one of their polymers reached 9.4%. It is worth pointing out that the "Cn" of their state-of-the-art polymers is close to the optimal value observed in our polymer systems, which strongly validates our correlations.
 | ||
| Scheme 1 Molecular design rationales of BDT-BT-Cn and BDT-FBT-Cn polymers. | ||
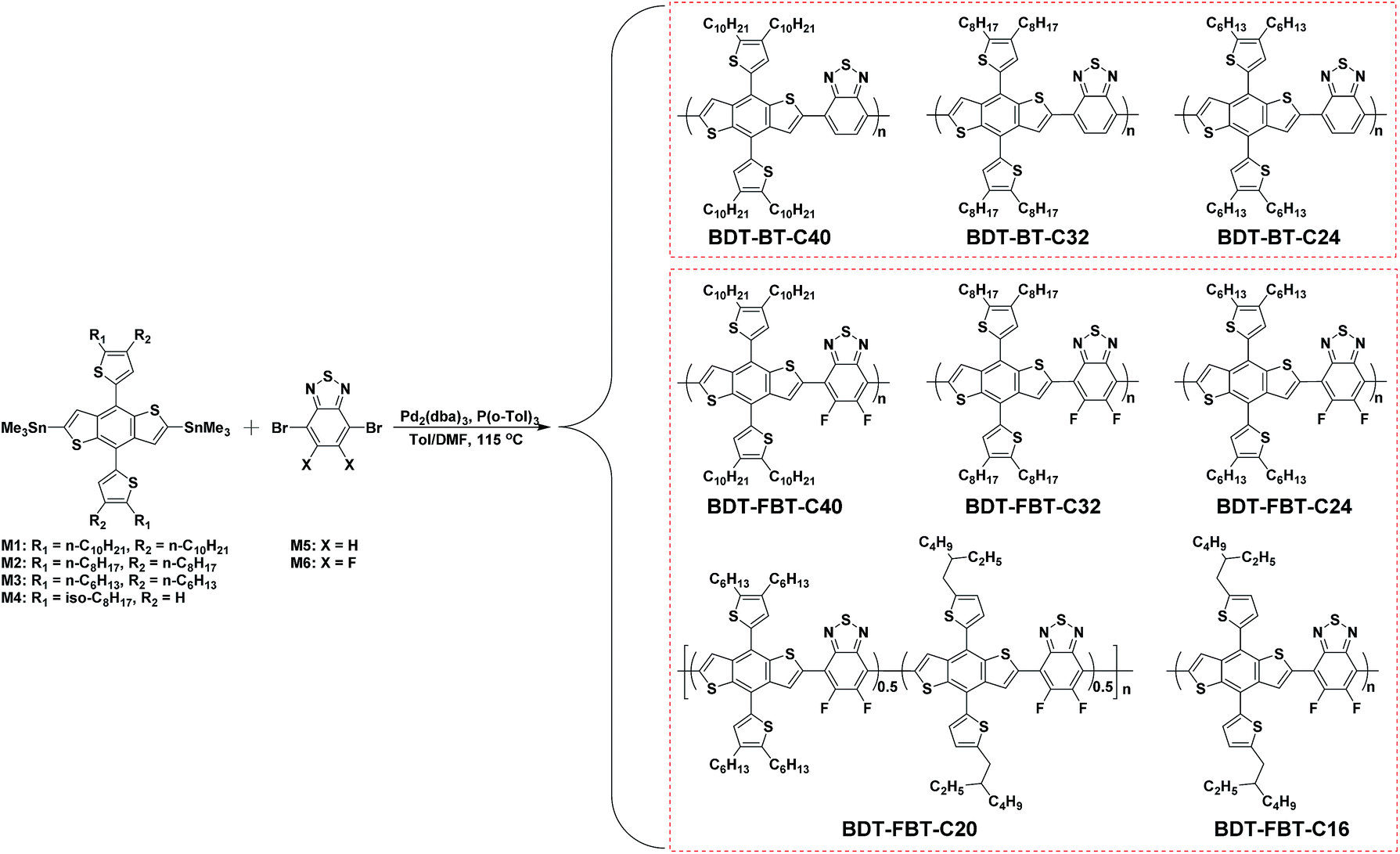 | ||
| Scheme 2 The synthesis route to BDT-BT-C40, BDT-BT-C32, BDT-BT-C24, BDT-FBT-C40, BDT-FBT-C32, BDT-FBT-C24, BDT-FBT-C20, and BDT-FBT-C16. | ||
2. Results and discussion
2.1. Molecular design and synthesis
To methodically investigate the effects of side chain length on photovoltaic properties of polymers in PSCs, "Cn" is systematically varied in the two polymer systems, BDT-BT-Cn and BDT-FBT-Cn, by using different BDT monomers in the polymerization. The polymers of BDT-BT-C40, BDT-BT-C32, and BDT-BT-C24 were synthesized for the BDT-BT-Cn system, and the polymers of BDT-FBT-C40, BDT-FBT-C32, BDT-FBT-C24, BDT-FBT-C20, and BDT-FBT-C16 were synthesized for the BDT-FBT-Cn system. Among these polymers, BDT-FBT-C16 is insoluble in any solvent and was thus not characterized further. “C20” in BDT-FBT-C20 denotes the statistical average value of the number of alkyl chain carbon atoms in each repeat unit of the polymer due to the random copolymerization. The chemical structures and synthetic scheme of the polymers are illustrated in Scheme 2. The polymers were synthesized by the Stille cross-coupling reaction with different bis(trimethyltin)-BDT monomers and dibrominated benzo[2,1,3]thiadiazole (BT) or FBT monomers. To properly establish structure–property relationships and make a fair comparison of a set of polymers, similar molecular weights are required.62 For example, different conclusions with respect to the effect of fluorination on solar cell performance were drawn for structurally similar polymers when their molecular weights differed.63–65 Gel permeation chromatography (GPC) measurements, performed at 140 °C using ortho-dichlorobenzene (o-DCB) as the eluent to reduce aggregation of polymer chains, show that good control of the polymerization reaction was achieved and similar molecular weights were obtained for the useful polymers, except for BDT-FBT-C20 (Table 1 and Fig. S1, ESI†). The low molecular weight of BDT-FBT-C20 is attributed to the lack of sufficient side chains, impeding the growth of polymer chains during polymerization. All the other useful polymers possess similar number-average molecular weights (Mn) of 30–40 kg mol−1 with similar polydispersity (PDI). All useful polymers are readily soluble in chlorinated aromatic solvents such as chlorobenzene (CB) and o-DCB.| Polymer | M n (kDa) | PDIa | Solution | Film | E optg (eV) | HOMO (eV) | LUMO (eV) | E CVg (eV) | ΔELUMOb (eV) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) | λ onset (nm) | λ max (nm) | λ onset (nm) | ||||||||
| a Determined by GPC at 140 °C using o-DCB as the eluent. b ΔELUMO = ELUMO (polymer) − ELUMO ([60]PCBM). | |||||||||||
| BDT-BT-C40 | 39.0 | 2.3 | 662 | 705 | 668 | 720 | 1.72 | −5.82 | −3.65 | 2.17 | 0.51 |
| BDT-BT-C32 | 29.8 | 3.1 | 662 | 706 | 669 | 730 | 1.70 | −5.79 | −3.64 | 2.15 | 0.52 |
| BDT-BT-C24 | 34.8 | 3.3 | 665 | 709 | 670 | 722 | 1.72 | −5.80 | −3.64 | 2.16 | 0.52 |
| BDT-FBT-C40 | 33.7 | 3.1 | 664 | 700 | 668 | 722 | 1.72 | −5.95 | −3.76 | 2.19 | 0.40 |
| BDT-FBT-C32 | 37.8 | 3.2 | 665 | 700 | 669 | 730 | 1.70 | −5.95 | −3.76 | 2.19 | 0.40 |
| BDT-FBT-C24 | 31.8 | 3.1 | 664 | 700 | 669 | 720 | 1.72 | −5.91 | −3.77 | 2.14 | 0.39 |
| BDT-FBT-C20 | 11.3 | 1.7 | 660 | 716 | 670 | 722 | 1.72 | −5.76 | −3.69 | 2.07 | 0.47 |
2.2. Optical properties and energy levels
UV-vis absorption spectra of the polymers in thin films are shown in Fig. 1 and relevant data are summarized in Table 1. The UV-vis absorption spectra in dilute solutions are presented in Fig. S2 (ESI†). All BDT-BT-Cn and BDT-FBT-Cn polymers exhibit similar absorption maxima (λmax ≈ 665 nm) in solution and in thin films (λmax ≈ 670 nm). The main difference is the stronger absorption at ∼490 nm of the BDT-FBT-Cn polymers compared to the BDT-BT-Cn polymers. In thin films, all polymers show an absorption onset between 720 and 730 nm, corresponding to an Eg of ∼1.7 eV, which is close to the ideal value for wide band gap absorbers toward multi-junction PSC applications.59 In the normalized absorption spectrum of BDT-FBT-C20, the intensity of the vibronic peak at ∼600 nm is substantially larger than those of the other BDT-FBT-Cn polymers. The normalized absorption spectra of the three other BDT-FBT-Cn polymers overlap each other almost completely. The small deviation of the absorption spectrum of BDT-FBT-C20 with respect to other BDT-FBT-Cn polymers may be caused by its low molecular weight and less ordered side chain pattern. Almost complete overlap of normalized absorption spectra is also observed for the three BDT-BT-Cn polymers. These similarities indicate that the effective conjugation length has been achieved and that the side chain length has little influence on the optical properties of the polymers.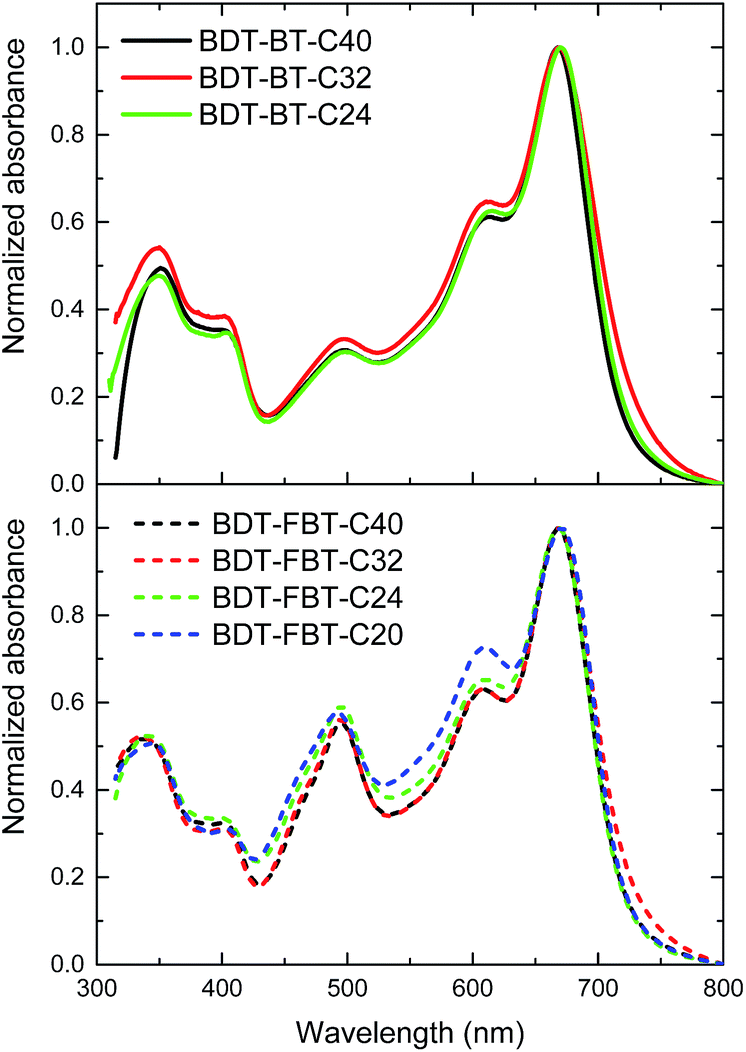 | ||
| Fig. 1 UV-vis absorption spectra of the polymers in thin films. Films were spin coated at 800 rpm from 8 mg mL−1 solutions in CB on glass substrates. | ||
The HOMO and lowest unoccupied molecular orbital (LUMO) energy levels of the polymers were determined by cyclic voltammetry (CV) experiments and are referenced to a work function of ferrocene of −5.23 eV, used as the internal standard (Fig. 2, Table 1, and Fig. S3, ESI†). The HOMO and LUMO levels of the BDT-FBT-Cn polymers are slightly deeper in energy than those of the BDT-BT-Cn polymers as a result of the electron-withdrawing effect of the fluorine atoms. The exception is BDT-FBT-C20 which shows higher-lying frontier orbitals, possibly being caused by its low molecular weight. Generally, polymers within the same system show very similar HOMO levels, signifying that the side chain length has little influence on the energy levels. The electrochemical band gaps of the polymers, which are estimated from the difference between the onsets of the oxidation and reduction waves in the voltammograms, are larger than their optical band gaps. Such a difference is not uncommon and may originate, e.g., from a difference in the relative permittivity in the two experiments and from the exciton binding energy that lowers the optical band gap.66,67 The offsets, ΔELUMO, between the LUMO levels of the polymers and that of [6,6]-phenyl-C61-butyric acid methyl ester ([60]PCBM) are quite similar within the same polymer system and significantly higher than the empirical threshold value of 0.30 eV for efficient exciton dissociation in BHJ solar cells for all polymers.68
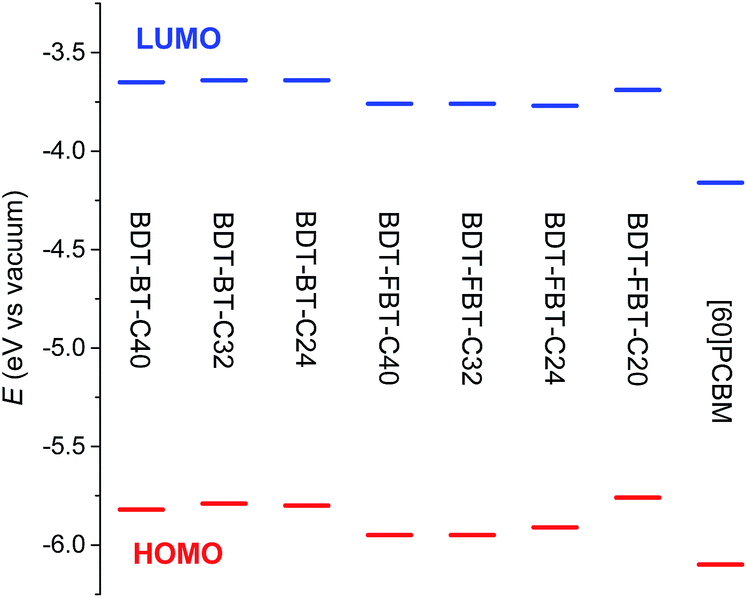 | ||
| Fig. 2 Energy levels of the polymers determined by cyclic voltammetry using a work function value of −5.23 eV for Fc/Fc+. | ||
2.3. Photovoltaic properties
The photovoltaic properties of the polymers were evaluated under simulated AM 1.5G illumination (100 mW cm−2) in single-junction PSCs with a conventional polarity device architecture consisting of an indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) hole collecting bottom contact, a polymer:fullerene photoactive layer, an electron collecting layer (ECL), and an Al back contact. For a systematic comparison of the photovoltaic performance, the device fabrication was first optimized for BDT-FBT-C24 in terms of the type of fullerene, polymer:fullerene weight ratio, solvent and type of co-solvent, thermal annealing, and ECL/Al contact (Table S1, ESI†). The best PSCs of BDT-FBT-C24 were fabricated from a CB solution containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 weight ratio mixture of BDT-FBT-C24 and[60]PCBM, using poly[(9,9)-bis(3'-(N,N-dimethylamino)propyl)-2,7-fluorene]-alt-2,7-(9,9-dioctylfluorene)] (PFN) as the ECL. The same conditions were then applied to all the polymers, with the exception of BDT-FBT-C20 based devices where an evaporated layer of LiF (∼1 nm) was used as the ECL because the photoactive layer cracked upon deposition of PFN. Typical current density–voltage (J–V) curves are presented in Fig. 3a and c. The external quantum efficiency (EQE) curves are shown in Fig. 3b and d, and device parameters are summarized in Table 2. The highest PCEs are achieved for BDT-BT-C24 (5.3%) and BDT-FBT-C24 (5.1%). All polymers afford a high Voc in PSCs, with the remarkable values of 0.98 and 0.99 V achieved by BDT-FBT-C32 and BDT-FBT-C24, respectively. This shows that the rationale of removing the electron-rich thiophene rings from the conjugated backbone of BDT-FBT-2T59 indeed raises the Voc. Generally, BDT-FBT-Cn polymers afford higher Voc than BDT-BT-Cn polymers as a result of the electron-withdrawing effect of fluorine atoms. It is worth pointing out that Voc values up to 0.95 V are rarely observed for the conjugated polymers with band gaps <1.75 eV.69–71
:2 weight ratio mixture of BDT-FBT-C24 and[60]PCBM, using poly[(9,9)-bis(3'-(N,N-dimethylamino)propyl)-2,7-fluorene]-alt-2,7-(9,9-dioctylfluorene)] (PFN) as the ECL. The same conditions were then applied to all the polymers, with the exception of BDT-FBT-C20 based devices where an evaporated layer of LiF (∼1 nm) was used as the ECL because the photoactive layer cracked upon deposition of PFN. Typical current density–voltage (J–V) curves are presented in Fig. 3a and c. The external quantum efficiency (EQE) curves are shown in Fig. 3b and d, and device parameters are summarized in Table 2. The highest PCEs are achieved for BDT-BT-C24 (5.3%) and BDT-FBT-C24 (5.1%). All polymers afford a high Voc in PSCs, with the remarkable values of 0.98 and 0.99 V achieved by BDT-FBT-C32 and BDT-FBT-C24, respectively. This shows that the rationale of removing the electron-rich thiophene rings from the conjugated backbone of BDT-FBT-2T59 indeed raises the Voc. Generally, BDT-FBT-Cn polymers afford higher Voc than BDT-BT-Cn polymers as a result of the electron-withdrawing effect of fluorine atoms. It is worth pointing out that Voc values up to 0.95 V are rarely observed for the conjugated polymers with band gaps <1.75 eV.69–71
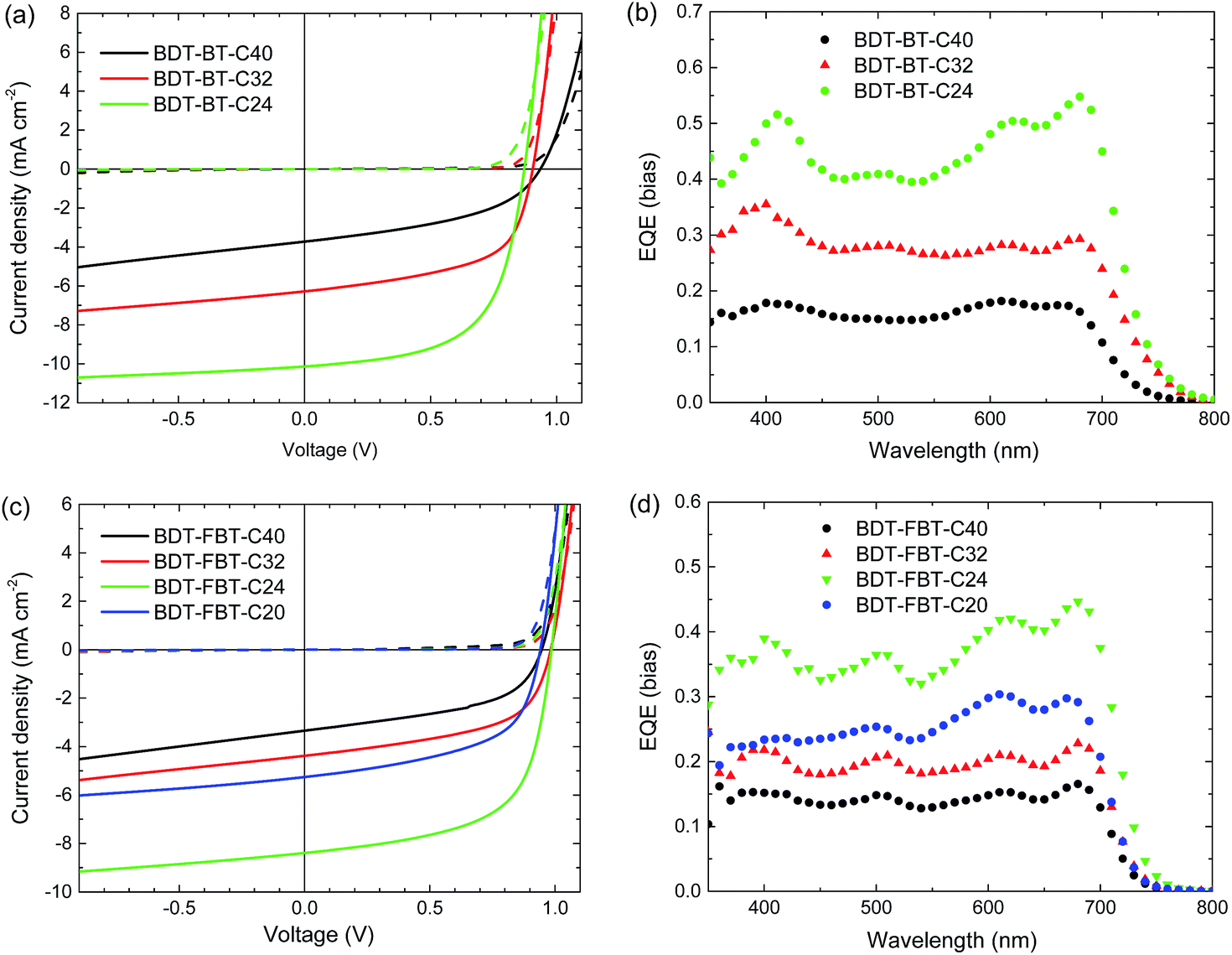 | ||
| Fig. 3 Current density–voltage (J–V) characteristics of the polymer:[60]PCBM solar cells based on (a) BDT-BT-C40, BDT-BT-C32, and BDT-BT-C24, and (c) BDT-FBT-C40, BDT-FBT-C32, BDT-FBT-C24, and BDT-FBT-C20 under AM1.5G illumination (100 mW cm−2). (b) and (d) EQE curves of the corresponding solar cells. | ||
| Polymer | ECL | J sc (mA cm−2) | J sc (EQE)b (mA cm−2) | V oc (V) | FFa | PCEa (%) | PCEc (%) | EQEmax | μ h (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined from J–V measurements under simulated solar irradiation. Average values and standard deviation of device statistics from 4 devices are given in parentheses. b Determined by convolution of the EQE with the AM1.5 G solar spectrum. c Calculated using Jsc (EQE). | |||||||||
| BDT-BT-C40 | PFN | 3.2 (3.3 ± 0.1) | 3.5 | 0.94 (0.93 ± 0.01) | 0.45 (0.44 ± 0.01) | 1.3 (1.3 ± 0.1) | 1.5 | 0.18 | 7.0 × 10−5 |
| BDT-BT-C32 | PFN | 5.6 (5.5 ± 0.1) | 6.3 | 0.91 (0.90 ± 0.01) | 0.57 (0.57 ± 0.01) | 2.9 (2.8 ± 0.1) | 3.3 | 0.35 | 7.8 × 10−4 |
| BDT-BT-C24 | PFN | 8.8 (8.7 ± 0.1) | 10.1 | 0.87 (0.87 ± 0.01) | 0.60 (0.59 ± 0.01) | 4.6 (4.5 ± 0.1) | 5.3 | 0.55 | 3.4 × 10−2 |
| BDT-FBT-C40 | PFN | 3.0 (3.0 ± 0.1) | 3.1 | 0.95 (0.93 ± 0.01) | 0.50 (0.49 ± 0.01) | 1.4 (1.4 ± 0.1) | 1.5 | 0.17 | 4.7 × 10−3 |
| BDT-FBT-C32 | PFN | 3.9 (3.8 ± 0.1) | 4.4 | 0.98 (0.98 ± 0.01) | 0.54 (0.54 ± 0.01) | 2.1 (2.0 ± 0.1) | 2.3 | 0.23 | 5.0 × 10−3 |
| BDT-FBT-C24 | PFN | 7.3 (7.2 ± 0.1) | 8.4 | 0.99 (0.99 ± 0.01) | 0.62 (0.61 ± 0.01) | 4.4 (4.3 ± 0.1) | 5.1 | 0.45 | 8.4 × 10−3 |
| BDT-FBT-C20 | LiF | 5.0 (4.9 ± 0.1) | 5.6 | 0.94 (0.94 ± 0.01) | 0.55 (0.54 ± 0.01) | 2.6 (2.5 ± 0.1) | 2.9 | 0.30 | 1.0 × 10−1 |
More interestingly, some trends in photovoltaic performances are clearly observable (Fig. 4). For BDT-BT-Cn polymers, the Jsc increases significantly from 3.5 to 6.3, and further to 10.1 mA cm−2 along with a decreasing length of the side chain. This trend in Jsc is verified by EQE spectra (Fig. 3b). The EQE maxima increase substantially from 0.18 for BDT-BT-C40 to 0.35 for BDT-BT-C32, and further to 0.55 for BDT-BT-C24. The fill factors (FFs) also increase considerably from 0.45 to 0.57, and further to 0.60 along with decreasing side chain length of the BDT-BT-Cn polymers. As a result, the PCEs of the PSCs display a similar trend to Jsc and FF, although the Voc drops slightly upon decreasing the side chain length. Similar trends in Jsc, FF, and PCE are also observed in the BDT-FBT-Cn system from the polymers of BDT-FBT-C40 to BDT-FBT-C24, but these polymers show opposite trends in Voc. BDT-FBT-C20 shows opposite trends in Jsc, FF and PCE. These are attributable to its lower molecular weight and the more disordered substitution pattern of linear and branched side chains. The EQE spectra (Fig. 3b and d) show the relative contribution of the polymer and the fullerene constituents to the photocurrent, which is clearly dependent on the side chain length. For example, BDT-BT-C40 and BDT-FBT-C40 based devices exhibit flat EQE curves though both at relatively low EQE values, suggesting balanced contributions of polymers and fullerene absorption to the Jsc. However, such a balance is broken upon decreasing the length of side chains with significant imbalance observed in BDT-BT-C24, BDT-FBT-C24, and BDT-FBT-C20 based devices. For instance, the BDT-BT-C24 based device shows an EQE maximum of 0.55 for the polymer at 680 nm but a significantly lower EQE of 0.43 for the fullerene at 440 nm (absorption valley for the polymer). The experiments on the two polymer systems, BDT-BT-Cn and BDT-FBT-Cn, consistently suggest that any excess of side chain length is detrimental to the photovoltaic performance. Although this study lacks a BDT-BT-C20 polymer, the results on BDT-FBT-C20 clearly indicate that insufficient solubilizing side chains cannot afford high enough molecular weights and thereby produce poor solar cell performance too. We admit that polymers with different side chains may require different processing methods to maximize their performance, solar cells with 1,8-diiodooctane as the solvent additive for active layer processing were also studied. As shown in Table S2,† we did observe changes in the solar cell performance of each polymer, but these results further confirmed the effect of side chain length as discussed above. In our sets of polymers, the incorporation of four hexyl chains is close to optimal. The next question is what causes this interesting effect of the side chain length?
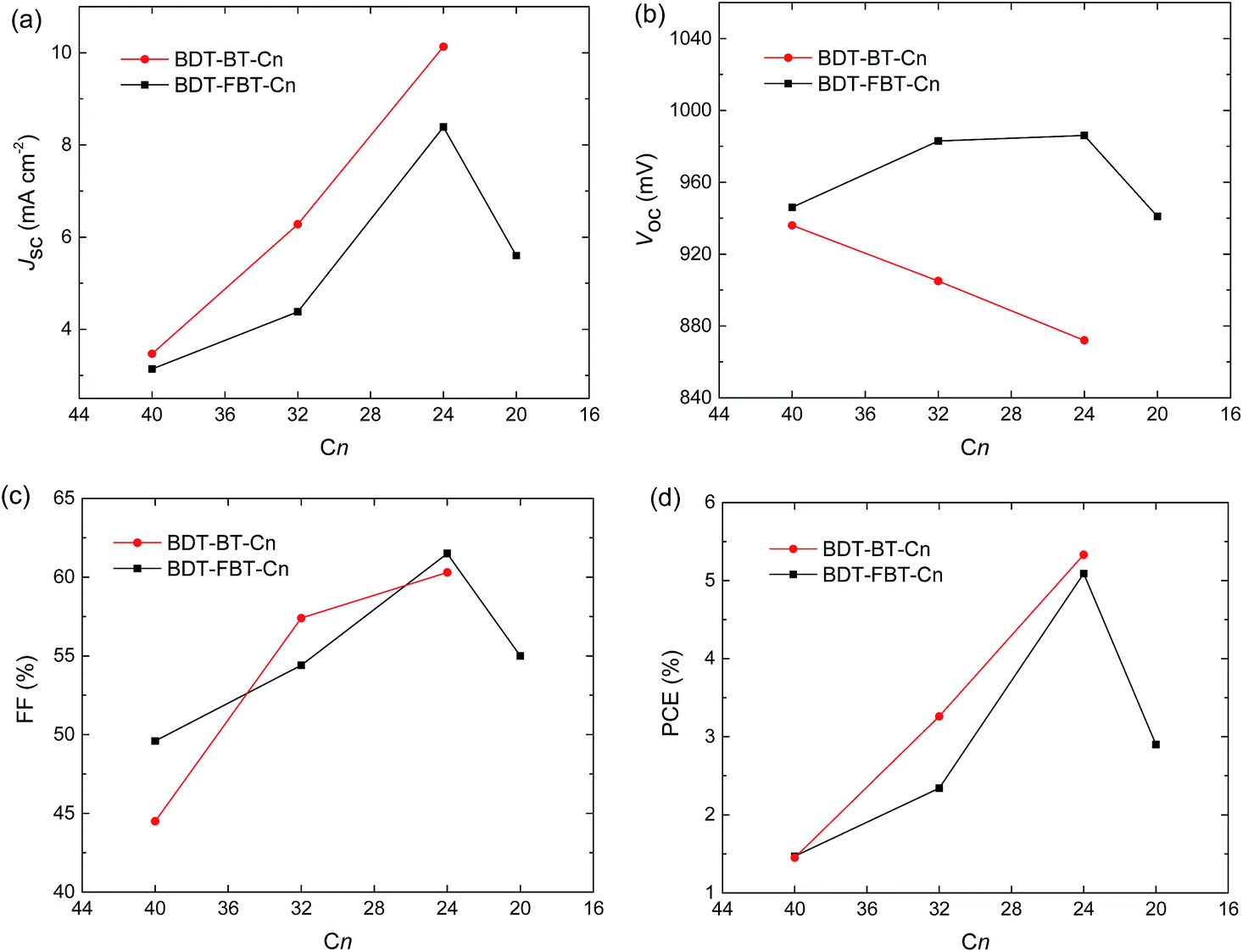 | ||
| Fig. 4 Solar cell performance parameters vs. number of carbons (Cn) in alkyl side chains of the polymers: (a) Jsc–Cn, (b) Voc–Cn, (c) FF–Cn, and (d) PCE–Cn. | ||
2.4. Factors influencing Jsc, FF, and PCE
The large variation in PCEs of the PSCs originates mainly from the difference in Jsc and FF. In principle, the photocurrent of PSCs depends on the product of the efficiencies of light absorption, exciton diffusion, exciton dissociation, charge transport, and charge collection. The FF of the PSCs depends on the competition between recombination and charge collection, which means that reducing recombination or enhancing charge collection will improve the FF. First, the length of side chains has a somewhat but limited influence on the absorption of the polymer:[60]PCBM blends (Fig. S4, ESI†). Second, as the measured LUMO levels for the polymers and the ΔELUMO within the same system are very similar (Fig. 2 and Table 1), the change of charge generation dynamics upon varying the side chain length can also be ruled out. Third, identical electrode contacts were adopted for all devices (except for BDT-FBT-C20 based devices where LiF/Al was used as the cathode contact), so charge collection efficiency at electrodes will not be responsible for the large difference in Jsc and FF. Therefore, the observed difference in device performance upon changing the side chain length of the polymers must be related to the factors that influence exciton diffusion and dissociation, charge carrier transport and recombination.Further investigation was first carried out by measuring the hole mobility of the polymers in field effect transistors (FETs) to investigate differences in charge transport (Fig. S5, ESI†). As shown in Table 2, the FET mobilities of both BDT-BT-Cn polymers and BDT-FBT-Cn polymers show a clear dependence on the length of side chain. In the BDT-BT-Cn system, the hole mobility is 7.0 × 10−5, 7.8 × 10−4, and 3.4 × 10−2 cm2 V−1 s−1 for BDT-BT-C40, BDT-BT-C32, and BDT-BT-C24, respectively. In the BDT-FBT-Cn system, the hole mobility is 4.7 × 10−3, 5.0 × 10−3, and 8.4 × 10−3 cm2 V−1 s−1 for BDT-FBT-C40, BDT-FBT-C32, and BDT-FBT-C24, respectively. This difference in charge transport abilities can thus, at least partially, account for the variation in Jsc and FF upon changing the length of side chains of the polymers. Notably, BDT-FBT-C20 exhibits the highest hole mobility of 1.0 × 10−1 cm2 V−1 s−1, which is probably because of less entanglement of polymer chains due to low molecular weight and suggests that there are other factors that affect photovoltaic properties of BDT-FBT-C20.
Keeping the large difference in the FFs of the PSCs in mind, we speculate that the side chain length in polymers has a substantial influence on the recombination losses as charge recombination losses manifest themselves most significantly in the FF.72 To investigate how the side chain length affects the recombination losses, EQEs of the solar cells measured under monochromatic light with or without bias light were analyzed. Under enhanced bias illumination of a 532 nm laser, bimolecular recombination becomes more considerable as the significant enhancement of charge carrier concentration. Because the EQE is measured by using a lock-in technique, it provides a measure of the differential photocurrent (dJph/dI) rather than of the photocurrent (Jph). For solar cells with a sublinear light-intensity dependence, Jph ∼ Iα (α < 1), this results in an EQE that is less than the actual EQE.73 Importantly, the ratio between the EQE measured under light bias and without light bias (denoted as ρ = EQEbias/EQEnobias) reflects the bimolecular recombination losses in the PSCs. We have previously reported that the bimolecular recombination efficiency (ηBR) at higher light intensities can be quantified by ηBR = 1 − ρ,74 implying that a smaller ρ implies more serious bimolecular recombination. Fig. 5 shows the average values of the ρ in the wavelength range from 400 to 750 nm of the PSCs. The original biased and unbiased EQEs of the devices are shown in Fig. S6 (ESI†). The average value of ρ increases drastically from 0.862 for BDT-BT-C40 to 0.929 for BDT-BT-C32, and further to 0.983 for BDT-BT-C24. A similar trend is also observed in BDT-FBT-Cn polymers (where ρ is 0.935 for BDT-FB4-C40, 0.945 for BDT-FBT-C32, and 0.970 for BDT-FBT-C24). Noticeably, BDT-FBT-C20 also has a low ρ value of 0.899, despite its relatively high hole mobility. We thus see that reducing the side chain length correlates with less bimolecular recombination losses in PSCs, except for BDT-FBT-C20. Moreover, the changes in the bimolecular recombination efficiency correlate with the variation in FF and in Jsc upon changing the side chain length (compare Fig. 4 and 5). It is worth pointing out that other recombination processes may also account for the difference in the Jsc and FF of the PSCs.75
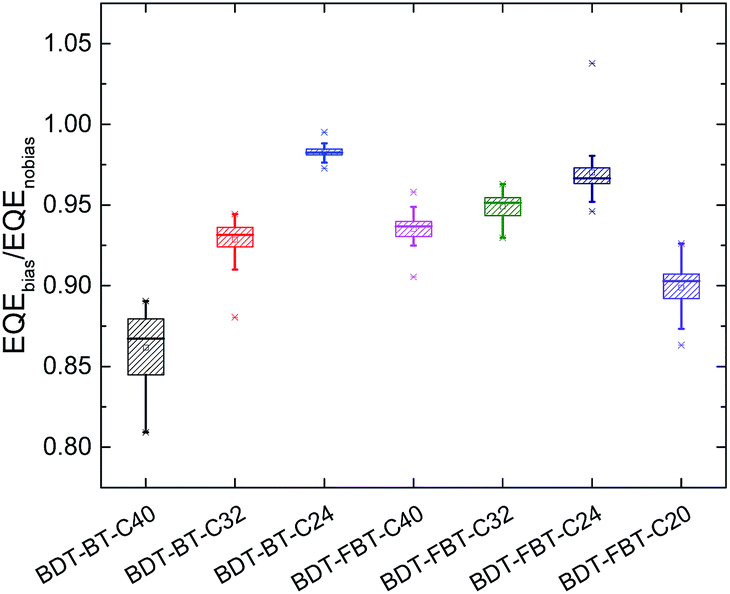 | ||
| Fig. 5 Average EQEbias/EQEno bias values of polymer:[60]PCBM solar cells based on BDT-BT-C40, BDT-BT-C32, BDT-BT-C24, BDT-FBT-C40, BDT-FBT-C32, BDT-FBT-C24, and BDT-FBT-C20. | ||
It is worth mentioning that no clear and consistent trend in the Voc of the two sets of polymers is observed. Generally, the Voc of organic solar cells is affected by several factors, such as HOMO and LUMO energy levels of the donor and acceptor, charge transport layers, charge generation and recombination, energetic disorder and the presence of defect states as well as the bulk heterojunction morphology and the crystallinity of the polymer. An insightful understanding of the change of Voc upon changing the side chain length would require further investigation.
2.5. Polymer crystallinity and BHJ morphology
Having established that the side chain length affects charge transport abilities and bimolecular recombination losses of the PSCs, we next attempt to correlate these observables with the morphological characteristics of the polymer:fullerene BHJ films, as the BHJ morphology significantly influences exciton diffusion, charge dissociation, and charge transport.The morphology of the polymer:[60]PCBM BHJ films was studied by atomic force microscopy (AFM) and transmission electron microscopy (TEM). AFM phase images (Fig. 6) indicate serious phase separation and large domain sizes in BDT-BT-C40 and BDT-FBT-C40 based BHJ films. Smaller nanoscale domains and a more continuous interpenetrating network are observed when the side chains of the polymers are shorter. Specifically, finely dispersed structures appear in the phase images of the BDT-BT-C24 and BDT-FBT-C24 based BHJ films. Further shortening the side chain leads to an almost featureless surface morphology for the BDT-FBT-C20 based BHJ film. In AFM height images (Fig. S7, ESI†), a gradual reduction of the root mean square roughness (Rq) of 4.94, 3.78, and 2.54 nm is recorded for the BHJ films of BDT-BT-C40, BDT-BT-C32, and BDT-BT-C24, respectively. A similar trend is observed in the BDT-FBT-Cn system, with Rq values of 6.10, 3.29, and 2.54 nm for the BHJ films of BDT-FBT-C40, BDT-FBT-C32, and BDT-FBT-C24, respectively. But the roughness of the BDT-FBT-C20:[60]PCBM film is increased to 4.12 nm.
 | ||
| Fig. 6 AFM phase images (5 × 5 μm2, vertical scale 20°) of the polymer:[60]PCBM blend films spin-coated from CB for BDT-BT-C40 (a), BDT-BT-C32 (b), BDT-BT-C24 (c), BDT-FBT-C40 (d), BDT-FBT-C32 (e), BDT-FBT-C24 (f), and BDT-FBT-C20 (g). | ||
The TEM images confirm the morphological changes of the BHJ films with the side chain length variation (Fig. 7). Serious phase separation and large domain size are clear in both BDT-BT-C40:[60]PCBM and BDT-FBT-C40:[60]PCBM films. Such large domains result from liquid–liquid phase separation during spin coating.76 Significantly, smaller domain size and better interconnectivity are observed in BDT-BT-C32:[60]PCBM and BDT-FBT-C32:[60]PCBM films. Moreover, finely dispersed structures are observable in BDT-BT-C24 and BDT-FBT-C24 based BHJ films. The transition from a liquid–liquid phase separated morphology to a finely dispersed structure when reducing the side chains and thereby solubility is consistent with the fact that liquid–liquid phase separation is suppressed when the polymer has an increased tendency to aggregate.77 No significant phase separation is observed in the BDT-FBT-C20 based BHJ film. Recently, it was shown that other low molecular weight conjugated polymers can form solid films with crystalline domains that are disconnected from each other.61,78,79 Interestingly, the high-magnification TEM-images (Fig. S8, ESI†) clearly show crystalline fringes for all polymers. In short, significant differences in the morphology of the BHJ films are found with changing the side chain length. These are consistent with the difference in the device performance of the polymers.
 | ||
| Fig. 7 Bright field TEM images of the polymer:[60]PCBM blend films spin-coated from CB for BDT-BT-C40 (a), BDT-BT-C32 (b), BDT-BT-C24 (c), BDT-FBT-C40 (d), BDT-FBT-C32 (e), BDT-FBT-C24 (f), and BDT-FBT-C20 (g). Image size: 1.5 × 1.5 μm2; scale bar: 200 nm. | ||
The data show that an insufficient side chain length yields low molecular weight polymers or even insoluble polymers that cannot be used. The low molecular weight results in poor phase interconnectivity, low domain purity, unfavorable structural orientation and so on, which have already been well studied.61,80,81 On the other hand, too long side chains produce too soluble polymers, which will affect drying kinetics and self-organization of the BHJ films during spin-coating as well as the compatibility between the polymers and fullerene. As a result, a suboptimal BHJ film morphology with serious phase separation and large domain size will be formed. In these liquid–liquid phase separated blends, charge generation mainly occurs in the continuous mixed polymer–fullerene phase, but because most fullerenes are assembled in the droplet domains, the electron transport in the mixed phase is hampered which results in significant bimolecular recombination.76,82 An optimal side chain length is thus required to balance solubility such that the right microstructure can be formed in BHJ films.
3. Conclusions
Two sets of conjugated polymers based on BDT and BT or FBT with different alkyl side chain lengths were designed and synthesized to systematically investigate the effect of side chain length on the photovoltaic properties of the polymers. Our results suggest strong interdependency between the side chain length of conjugated polymers and photovoltaic performances of the resulting BHJ solar cells. Too long side chains lead to deteriorated charge transport, a suboptimal BHJ morphology featuring serious phase separation, considerable charge carrier recombination, and consequently poor photovoltaic performances. On the other hand, too short side chains cannot afford high enough solubility and molecular weight for the resulting polymers and will definitely produce poor solar cell performance as well.80,81 Selecting a suitable side chain length is thus of significant importance to maximize the potential of conjugated polymers in BHJ solar cells.A recent statistical analysis revealed strong correlations between PCEs and morphology-dominated properties (Jsc and FF) and weak correlations between PCEs and energy level relevant properties (Voc).83 As demonstrated here, there is a strong interdependency between the side chain length and morphology-dominated properties (Jsc and FF) as well. Side chain optimization of conjugated polymers in combination with efficient optimization of the active layer microstructure and morphology will thus be highly fruitful to future PSC efficiency gains. Our preliminary studies also suggest that further performance enhancements for BDT-BT-Cn and BDT-FBT-Cn polymers are possible through morphology optimization (Table S2, Fig. S9, and Table S3, ESI†). In the past few years, the development of new materials in the PSC community has centered on the design/manipulation of conjugated backbones and energy level tuning. Generally, a limited variety of standard side-chain substituents or even more typically only one kind of side chain is introduced for a new polymer framework unless solubility/processability proves problematic. As a result, the potential of many polymer systems was not fully explored due to taking little account of side chain optimization. The observations in this paper are based on a relatively large number of closely related polymers and the results provide a consistent view on the effects of side chain length on device performance that will support the molecular design of new photovoltaic active semiconducting conjugated polymers in future.
4. Experimental section
4.1. Synthesis
All synthetic procedures were performed under an argon atmosphere. All reactants and reagents are commercially available and used as received, unless otherwise specified. Solvents were dried over 4 Å molecular sieves. [60]PCBM (purity of 99%) and [70]PCBM (purity of 99%) were purchased from Solenne BV. 2,6-Bis(trimethyltin)-4,8-di(2,3-didecylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (M1), 2,6-bis(trimethyltin)-4,8-di(2,3-dioctylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (M2), 2,6-bis(trimethyltin)-4,8-di(2,3-dihexylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (M3), and 5,6-difluoro-4,7-dibromobenzo[2,1,3]thiadiazole (M6) were purchased from SunaTech Inc. 2,6-Bis(trimethyltin)-4,8-di(2-(2-ethylhexyl)thiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (M4) was purchased from Solarmer Materials, Inc. 5,6-Dibromobenzo[2,1,3]thiadiazole (M5) was synthesized according to our previous method.:M4:M6 = 0.5:0.5:1 as a dark shiny solid (85 mg, yield 89%). Mn = 11.3 kg mol−1, and PDI = 1.7.
4.2. Measurements and characterization
Molecular weights and polydispersity index (PDI) were determined by gel permeation chromatography (GPC) at 140 °C on a PL-GPC 120 system using a PL-GEL 10 μm MIXED-B column and 1,2-dichlorobenzene (o-DCB) as the eluent against polystyrene standards. All the polymer samples were dissolved in o-DCB at 140 °C overnight and the solutions were filtered through PTFE filters (0.2 μm) prior to injection. UV-visible spectra were recorded on a Perkin Elmer Lambda 900 UV/vis/near IR spectrophotometer at room temperature unless otherwise noted. All solution UV-vis experiments were performed in o-DCB with a sample concentration of 0.05 mg mL−1. Films were prepared by spin coating o-DCB solutions on glass substrates. Cyclic voltammetry (CV) studies were performed with a scan rate of 0.1 V s−1 under an inert atmosphere with 1 M tetrabutylammonium hexafluorophosphate in acetonitrile as the electrolyte. The working electrode was an indium tin oxide (ITO) bar and the counter electrode was a silver electrode. A silver wire coated with silver chloride (Ag/AgCl) was used as the quasi-reference electrode in combination with Fc/Fc+ as an internal standard. The samples were spin coated on the top of the ITO working electrode to form ∼10 nm thick films. Atomic force microscopy (AFM) was performed on an extended Veeco MultiMode AFM connected to a Nanoscope III controller in tapping mode using PPP-NCH-50 probes (Nanosensors). Transmission electron microscopy (TEM) was performed on a Tecnai G2 Sphera transmission electron microscope (FEI) operating at 200 kV. Field-effect transistors (FETs) were fabricated in a bottom-contact top-gate configuration on pre-cleaned glass substrates. Source and drain contacts were defined by evaporating chromium (2 nm) and gold (40 nm) through a shadow mask. Subsequently, the polymer was applied by spin coating in a nitrogen-filled glove-box from a 3 mg mL−1 solution in o-DCB, at room temperature, for 60 s at 1500 rpm. The polymer film was annealed at 200 °C for 20 min and cooled down to room temperature slowly. Next, a layer of CYTOP (AGC Chemicals) was spin coated as a gate dielectric. This spin coating step was performed at 1500 rpm for 60 s in a nitrogen-filled glovebox. The CYTOP was annealed at 80 °C for 10 min and at 200 °C for 15 min. The top gate electrode was applied by thermal evaporation of gold (40 nm) through a shadow mask. The transistors were electrically characterized with a Keithley 2400 measuring unit. The length and the width of the transistor channel are 70 μm and 1000 μm, respectively.4.3. Fabrication and characterization of solar cells
Photovoltaic devices were made by spin coating poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) (Clevios P, VP AI 4083) onto pre-cleaned, patterned indium tin oxide (ITO) substrates in air (14 Ω per square) (Naranjo Substrates). The polymer–fullerene photoactive layers were deposited by spin coating in air from the solutions containing the corresponding polymers and [60]PCBM or [70]PCBM with different content ratios at room temperature. Unless indicated otherwise, no thermal annealing was applied to the blend films. The thickness of active layer films was controlled by the spin speed. Before the evaporation of the Al electrode, a thin PFN layer was spin-coated on the top of the active layer from its methanol solution (0.2 mg mL−1) at 3000 rpm. For BDT-FBT-C20:PCBM, LiF (1 nm) was evaporated to serve as the ECL due to the film cracking upon PFN solution deposition. Finally, Al (100 nm) was deposited by vacuum evaporation at ∼3 × 10−7 mbar as the back electrode. The active area of the cells was 0.09 or 0.16 cm2, which provided similar results. Current density–voltage (J–V) curves were measured under simulated solar light (100 mW cm−2) from a tungsten–halogen lamp filtered by using a Hoya LB100 daylight using a Keithley 2400 source meter. No mismatch correction was done. All measurements were conducted in a nitrogen-filled glovebox. The accurate short-circuit current density (Jsc) was determined from the EQE by convolution with the AM 1.5G solar spectrum. External quantum efficiency (EQE) measurements were performed in a homebuilt set-up, with the devices kept in a nitrogen filled box with a quartz window and illuminated through an aperture of 2 mm. Mechanically modulated (Stanford Research, SR 540) monochromatic (Oriel, Cornerstone 130) light from a 50 W tungsten halogen lamp (Osram 64610) was used as probe light, in combination with continuous bias light from a solid state laser (B&W Tek Inc. 532 nm, 30 mW). The intensity of the bias laser light was adjusted using a variable-neutral density filter. The response was recorded as the voltage over a 50 Ω resistance, using a lock-in amplifier (Stanford Research Systems SR 830). For all the single junction devices, the measurements were carried out under representative illumination intensities (AM1.5G equivalent, provided by the 532 nm laser).Acknowledgements
We thank Ralf Bovee for GPC analysis, Wijnand Dijkstra and Marco van der Sluis for AFM measurements. C. Duan thanks Fei Huang and Zhicheng Hu of the South China University of Technology for providing PFN. The work was performed in the framework of the Mujulima project that received funding from the European Commission's Seventh Framework Programme (Grant Agreement No. 604148). The work of JJVF forms part of the research programme of the Dutch Polymer Institute (DPI), project #734. The research leading to these results has also received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 339031. The research forms part of the Solliance OPV program and has received funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035).Notes and references
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035 CrossRef CAS PubMed.
- S. Zhang, L. Ye, W. Zhao, B. Yang, Q. Wang and J. Hou, Sci. China: Chem., 2015, 58, 248 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174 CrossRef CAS.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403 CrossRef CAS.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- L. Ye, S. Zhang, W. Zhao, H. Yao and J. Hou, Chem. Mater., 2014, 26, 3603 CrossRef CAS.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65 CrossRef CAS.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. W. Samuel, Nat. Commun., 2013, 4, 2867 Search PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, S. C. J. Meskers, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 1565 CrossRef CAS PubMed.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385 CrossRef CAS.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006 CrossRef CAS PubMed.
- S. R. Cowan, W. L. Leong, N. Banerji, G. Dennler and A. J. Heeger, Adv. Funct. Mater., 2011, 21, 3083 CrossRef CAS.
- E. T. Hoke, K. Vandewal, J. A. Bartelt, W. R. Mateker, J. D. Douglas, R. Noriega, K. R. Graham, J. M. J. Fréchet, A. Salleo and M. D. McGehee, Adv. Energy Mater., 2013, 3, 220 CrossRef CAS.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847 CrossRef CAS PubMed.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364 CrossRef CAS.
- T. Heumueller, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, J. A. Bartelt, T. M. Burke, T. Ameri, C. J. Brabec and M. D. McGehee, Energy Environ. Sci., 2014, 7, 2974 CAS.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS PubMed.
- Y. Y. Liang and L. P. Yu, Acc. Chem. Res., 2010, 43, 1227 CrossRef CAS PubMed.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed.
- C. H. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- H. X. Zhou, L. Q. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595 CrossRef CAS PubMed.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299 CrossRef CAS.
- A. Dhanabalan, J. K. J. van Duren, P. A. van Hal, J. L. J. van Dongen and R. A. J. Janssen, Adv. Funct. Mater., 2001, 11, 255 CrossRef CAS.
- J. Cao, Q. Liao, X. Du, J. Chen, Z. Xiao, Q. Zuo and L. Ding, Energy Environ. Sci., 2013, 6, 3224 CAS.
- Y. Dong, X. Hu, C. Duan, P. Liu, S. Liu, L. Lan, D. Chen, L. Ying, S. Su, X. Gong, F. Huang and Y. Cao, Adv. Mater., 2013, 25, 3683 CrossRef CAS PubMed.
- Z. Zhang, F. Lin, H.-C. Chen, H.-C. Wu, C.-L. Chung, C. Lu, S.-H. Liu, S.-H. Tung, W.-C. Chen, K.-T. Wong and P.-T. Chou, Energy Environ. Sci., 2015, 8, 552 CAS.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H.-Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702 CrossRef CAS PubMed.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS PubMed.
- C. Cui, W.-Y. Wong and Y. Li, Energy Environ. Sci., 2014, 7, 2276 CAS.
- J. Warnan, C. Cabanetos, R. Bude, A. El Labban, L. Li and P. M. Beaujuge, Chem. Mater., 2014, 26, 2829 CrossRef CAS.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2014, 26, 5880 CrossRef CAS PubMed.
- C. B. Nielsen, R. S. Ashraf, N. D. Treat, B. C. Schroeder, J. E. Donaghey, A. J. P. White, N. Stingelin and I. McCulloch, Adv. Mater., 2015, 27, 948 CrossRef CAS PubMed.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604 CrossRef CAS.
- Z.-G. Zhang and Y. Li, Sci. China: Chem., 2015, 58, 192 CrossRef CAS.
- E. G. Wang, M. Wang, L. Wang, C. H. Duan, J. Zhang, W. Z. Cai, C. He, H. B. Wu and Y. Cao, Macromolecules, 2009, 42, 4410 CrossRef CAS.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180 CrossRef CAS PubMed.
- C. Duan, W. Cai, B. B. Y. Hsu, C. Zhong, K. Zhang, C. Liu, Z. Hu, F. Huang, G. C. Bazan, A. J. Heeger and Y. Cao, Energy Environ. Sci., 2013, 6, 3022 CAS.
- Y. Li, J. Zou, H.-L. Yip, C.-Z. Li, Y. Zhang, C.-C. Chueh, J. Intemann, Y. Xu, P.-W. Liang, Y. Chen and A. K. Y. Jen, Macromolecules, 2013, 46, 5497 CrossRef CAS.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Fréchet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656 CrossRef CAS PubMed.
- W.-H. Chang, J. Gao, L. Dou, C.-C. Chen, Y. Liu and Y. Yang, Adv. Energy Mater., 2014, 4, 1300864 Search PubMed.
- J. Gao, L. Dou, W. Chen, C.-C. Chen, X. Guo, J. You, B. Bob, W.-H. Chang, J. Strzalka, C. Wang, G. Li and Y. Yang, Adv. Energy Mater., 2014, 4, 1300739 Search PubMed.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. El Labban, G. O. Ngongang Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608 CrossRef CAS PubMed.
- I. Osaka, M. Saito, T. Koganezawa and K. Takimiya, Adv. Mater., 2014, 26, 331 CrossRef CAS PubMed.
- D. Dang, W. Chen, S. Himmelberger, Q. Tao, A. Lundin, R. Yang, W. Zhu, A. Salleo, C. Müller and E. Wang, Adv. Energy Mater., 2014, 4, 1400680 Search PubMed.
- N. Cho, C. W. Schlenker, K. M. Knesting, P. Koelsch, H.-L. Yip, D. S. Ginger and A. K. Y. Jen, Adv. Energy Mater., 2014, 4, 1301857 Search PubMed.
- S. Zhang, M. A. Uddin, W. Zhao, L. Ye, H. Y. Woo, D. Liu, B. Yang, H. Yao, Y. Cui and J. Hou, Polym. Chem., 2015, 6, 2752 RSC.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357 CrossRef CAS.
- C. Duan, A. Furlan, J. J. van Franeker, R. E. M. Willems, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2015, 27, 4461 CrossRef CAS PubMed.
- J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F. H. Scholes, X. Chen, S. E. Watkins, G. J. Wilson, A. B. Holmes, W. W. H. Wong and D. J. Jones, Adv. Mater., 2015, 27, 702 CrossRef CAS PubMed.
- Z. Xiao, K. Sun, J. Subbiah, T. Qin, S. Lu, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2015, 6, 2312 RSC.
- W. Li, S. Albrecht, L. Yang, S. Roland, J. R. Tumbleston, T. McAfee, L. Yan, M. A. Kelly, H. Ade, D. Neher and W. You, J. Am. Chem. Soc., 2014, 136, 15566 CrossRef CAS PubMed.
- H. Wang, X. Yu, C. Yi, H. Ren, C. Liu, Y. Yang, S. Xiao, J. Zheng, A. Karim, S. Z. D. Cheng and X. Gong, J. Phys. Chem. C, 2013, 117, 4358 CAS.
- X. He, S. Mukherjee, S. Watkins, M. Chen, T. Qin, L. Thomsen, H. Ade and C. R. McNeill, J. Phys. Chem. C, 2014, 118, 9918 CAS.
- P. Liu, K. Zhang, F. Liu, Y. Jin, S. Liu, T. P. Russell, H.-L. Yip, F. Huang and Y. Cao, Chem. Mater., 2014, 26, 3009 CrossRef.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616 CrossRef CAS PubMed.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi and P. W. M. Blom, Appl. Phys. Lett., 2006, 88, 093511 CrossRef.
- C. H. Duan, C. D. Wang, S. J. Liu, F. Huang, C. H. W. Choy and Y. Cao, Sci. China: Chem., 2011, 54, 685 CrossRef CAS.
- J. Lee, M. Kim, B. Kang, S. B. Jo, H. G. Kim, J. Shin and K. Cho, Adv. Energy Mater., 2014, 4, 1400087 Search PubMed.
- Y. Yang, R. Wu, X. Wang, X. Xu, Z. Li, K. Li and Q. Peng, Chem. Commun., 2014, 50, 439 RSC.
- C. G. Shuttle, R. Hamilton, B. C. O'Regan, J. Nelson and J. R. Durrant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16448 CrossRef CAS PubMed.
- D. J. Wehenkel, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Org. Electron., 2012, 13, 3284 CrossRef CAS.
- L. J. A. Koster, M. Kemerink, M. M. Wienk, K. Maturová and R. A. J. Janssen, Adv. Mater., 2011, 23, 1670 CrossRef CAS PubMed.
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941 CrossRef CAS.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057 CrossRef CAS PubMed.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038 CrossRef CAS PubMed.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS PubMed.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456 CrossRef CAS PubMed.
- K. Maturová, S. S. van Bavel, M. M. Wienk, R. A. J. Janssen and M. Kemerink, Nano Lett., 2009, 9, 3032 CrossRef PubMed.
- N. E. Jackson, B. M. Savoie, T. J. Marks, L. X. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2015, 6, 77 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphs and tables. See DOI: 10.1039/c5ta09483f |
| This journal is © The Royal Society of Chemistry 2016 |