
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomically thin Pt shells on Au nanoparticle cores: facile synthesis and efficient synergetic catalysis†
C.
Engelbrekt
a,
N.
Šešelj
a,
R.
Poreddy
b,
A.
Riisager
b,
J.
Ulstrup
a and
J.
Zhang
*a
aNanoChemistry, Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800, Kgs. Lyngby, Denmark. E-mail: jz@kemi.dtu.dk
bCentre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800, Kgs. Lyngby, Denmark
First published on 26th January 2016
Abstract
We present a facile synthesis protocol for atomically thin platinum (Pt) shells on top of gold (Au) nanoparticles (NPs) (Au@PtNPs) in one pot under mild conditions. The Au@PtNPs exhibited remarkable stability (> 2 years) at room temperature. The synthesis, bimetallic nanostructures and catalytic properties were thoroughly characterized by ultraviolet-visible light spectrophotometry, transmission electron microscopy, nanoparticle tracking analysis and electrochemistry. The 8 ± 2 nm Au@PtNPs contained 24 ± 1 mol% Pt and 76 ± 1 mol% Au corresponding to an atomically thin Pt shell. Electrochemical data clearly show that the active surface is dominated by Pt with a specific surface area above 45 m2 per gram of Pt. Interactions with the Au core increase the activity of the Pt shell by up to 55% and improve catalytic selectivity compared to pure Pt. The Au@Pt NPs show exciting catalytic activity in electrooxidation of sustainable fuels (i.e. formic acid, methanol and ethanol), and selective hydrogenation of benzene derivatives. Especially high activity was achieved for formic acid oxidation, 549 mA (mgPt)−1 (at 0.6 V vs. SCE), which is 3.5 fold higher than a commercial < 5 nm PtNP catalyst. Excellent activity for the direct production of γ-valerolactone, an alternative biofuel/fuel additive, from levulinic acid and methyl levulinate was finally demonstrated.
Introduction
Platinum (Pt) catalysts are crucial for a range of chemical reactions in automotive, petroleum and energy conversion industries.1 Pt has excellent catalytic properties but low availability and high cost have shifted focus from pure Pt to advanced bimetallic Pt-based nanostructures.Nanomaterial properties depend critically on chemical composition, size, shape, crystallinity, and surface structure. Bimetallic nanostructures provide a strategy for design of advanced functional nanomaterials. A second element can, for example, reduce the concentration of surface sites for catalytic poisons – the so-called third-body effect.2 Metal monolayers also have dramatically altered properties by electronic interaction with the underlying core.2 Facile synthesis of thin metal shells on top of a metal core offers attractive approaches to tuning the catalytic activity as well as efficiently utilizing Pt.3 Gold (Au) nanostructures have been among the most studied systems due to unique optical and electronic properties, and application as catalysts for CO oxidation.4
Core–shell Au–Pt NPs show improved electrocatalysis, benefitting from electrochemical stability of Au, high catalytic activity of Pt, and a performance boost from core–shell synergy.5 Au–Pt core–shell NPs can be prepared by precursor co-reduction followed by thermal treatment,6 by underpotential deposition (UPD) of copper (Cu) on Au followed by galvanic displacement by Pt precursors,7 or reduction via seed-mediated growth.8 Thermally driven methods require high temperature, strong reducing agents, controlled atmosphere, or non-aqueous solvents.6 UPD is limited by the need for immobilization on conducting supports. UPD methods are superior for atomically thin Pt shells, while wet syntheses often provide thicknesses >1 nm.8 Seed-based wet chemical syntheses offer potential for large-scale one-pot, green preparation, but the complexity of the processes gives poor yield and control of product morphology. Shell precursor reduction, diffusion through seed coatings, and deposition on seed surfaces all have to be controlled.9
Non-toxic procedures and sustainability have emerged as criteria in nanomaterial synthesis and applications.10 In our previous work, a starch and glucose based recipe, the saccharide-based approach to metal nanostructure synthesis (SAMENS), for the synthesis of gold nanoparticles (AuNPs)11 and other metal nanostructures was developed. A dynamics study showed that glucose acts as reducing agent and starch as capping agent in phosphate buffer. The Good's buffer 2-(N-morpholino)ethanesulfonic acid (MES) participates in the reduction of HAuCl4 and adsorbs on the AuNP surface as well as controlling pH.12 Very small (1.6–1.8 nm) catalytically active starch coated platinum nanoparticles (PtNPs) have also been produced by the SAMENS recipe.13 In the present study we have explored SAMENS for the synthesis of stable and uniform Au–Pt bimetallic nanostructures with high catalytic activity. Uniform AuNPs serve as seeds for Au–Pt core–shell (Au@Pt) NPs with atomically thin Pt shells. Ultraviolet-visible light spectroscopy (UV-vis), NP tracking analysis (NTA), transmission electron microscopy (TEM) and electrochemical methods were employed to characterize the synthesis, structure, and properties. A catalyst of well dispersed Au@PtNPs with 12 wt% metal (3 wt% Pt, 9 wt% Au) on graphitized carbon black (G-CB) was prepared and comprehensively characterized. Starch capping was successfully removed thermally and the catalytic activity investigated by electrocatalytic oxidation of small organic fuel molecules, and by selective aromatic ring hydrogenation (ARH) processes and hydrocyclization of levulinic acid (LA) and methyl levulinate (MLA).
Small organic molecules such as formic acid (FA), methanol (MeOH), and ethanol (EtOH), have been widely used for fuel cell (FC) systems as these fuels can be produced from renewable sources without harmful waste. FA is attractive since it is non-toxic and can be used at high concentrations due to low rates of crossing commonly used membranes. This leads to good FC performance despite the low energy density compared to MeOH and EtOH. FA oxidation entails no C–C bond breaking which is a serious obstacle for EtOH oxidation. MeOH has received much attention as a C1 fuel with high energy density but faces challenges of toxicity and membrane crossing. EtOH exhibits low crossover and as a potential 12-electron oxidation process has a very high energy density.14 The Au@PtNP catalysts exhibit high specific electrochemical Pt surface area (ECSA) and excellent catalytic activity for all the FA (FAOR), MeOH (MOR) and EtOH (EOR) oxidation reactions. Currents reach 549, 219 and 229 mA (mgPt)−1 for FA, MeOH and EtOH, respectively, in 0.1 M fuel (scan rate 50 mV s−1), or 3.5, 2.0 and 3.1 times that of a < 5 nm commercial PtNP catalyst (Sigma). Apart from improved Pt utilization in the atomically thin shell, synergy between the Pt shell and the Au core improves the Pt surface activity by as much as 55% relative to pure Pt.
ARH to cyclohexane derivatives is important in petrochemical and pharmaceutical industry.15,16 Chemoselective ARH of benzoic acid (BA) to cyclohexane carboxylic acid (CCA) is crucial in the synthesis of ε-caprolactam,17 but ARH requires harsh conditions with a suitable catalyst to selectively hydrogenate the ring. Noble metal catalysts (Ru–Pt,18 Rh/C,19 Pd/C,17,20 Pd–Ru21) have been extensively studied. Pd/C is traditionally used though there are a number of limitations for ARH of BA to CCA, i.e. fast deactivation, and need for high temperature (423 K) and high H2 pressure (15 MPa).22–24 Recently, Pt was reported as an active ARH catalyst with activity/selectivity depending strongly on NP shape and size.25
Catalytic conversion of cellulosic biomass to various platform molecules, e.g. γ-valerolactone (GVL), is a key step in methyltetrahydrofuran production. GVL can be obtained by hydrocyclization of LA, an inexpensive starting material obtainable from biomass. Multiple catalytic strategies to produce LA via biomass transformation are available,26 but efficient conversion of LA to GVL using green technology is warranted. To date, numerous supported metal catalysts have been employed to hydrogenate LA to GVL with Ru-based catalysts being superior.27,28 Pt is active for LA hydrogenation, but activity comparable to that of Ru using green technologies is yet to be obtained.
The Au@Pt catalyst introduced in this work efficiently and selectively catalyzes the ARH of BA and benzamide (BM), with high activity (TOF = 62 h−1) for GVL synthesis from both LA and MLA. To the best of our knowledge, ARH of BA and its derivatives, and catalytic production of GVL from LA/MLA over Au–Pt catalysts has not been reported before.
Experimental section
Au–Pt core–shell nanoparticles (Au@Pt NPs) with atomically thin Pt shells
5–200 mL AuNP seed solution was prepared by pre-heating a MES buffered mixture of glucose and starch for 5–10 min in a 95 °C water or oil bath, adding gold precursor, and allowing 30 min for AuNP formation. The AuNP seed solution contained 2 mM HAuCl4, 10 mM glucose, 0.6 wt% starch and 10 mM MES (pH 7). Following AuNP formation, the seed solution was diluted 4 times with water and 20 mM H2PtCl6 was added to obtain a Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt mol ratio of 5:2 while heating was maintained for another 1–2 h. Complete reduction of the Pt precursor was confirmed with UV-vis. The original seed solution had a deep red colour while the final core–shell nanoparticle solution was dark brown.
:Pt mol ratio of 5:2 while heating was maintained for another 1–2 h. Complete reduction of the Pt precursor was confirmed with UV-vis. The original seed solution had a deep red colour while the final core–shell nanoparticle solution was dark brown.
Electrocatalytic oxidation
Details about electrochemical measurements are given in the supplementary information. After potential stabilization and CV in 0.1 M H2SO4, formic acid (FA), methanol (MeOH) or ethanol (EtOH) were admitted into the cell to obtain a concentration of 0.1 M. All the experiments were performed at room temperature (r.t.).Catalytic hydrogenation
A detailed description of hydrogenation experiments is given in the supplementary information. In brief, chemoselective aromatic hydrogenation of BA and BM, and cyclohydrogenation of LA and MLA was conducted in a 100 mL stainless steel autoclave reactor (Fig. S1†). Substrate, solvent, and catalyst were mixed in the autoclave, the reactor evacuated and pressurized with dihydrogen, heated to the target temperature, and maintained for 0.5 to 24 h. After cooling to r.t., aliquots of the reaction mixture were submitted to gas chromatography (GC), and the structure of the products identified using GC coupled with mass spectrometry (GC-MS).Results and discussion
Controlling the size of the Au core allows tuning of the core–shell nanostructure properties. AuNPs in the range of 8–80 nm were successfully prepared using a seeded growth approach inspired by Bastus et al.29 The synthesis procedure and AuNP characterization are provided in the supplementary information, Fig. S2–S6 and Tables S1–S2.† Fractions were obtained at various phases of the synthesis and characterized by UV-vis, TEM and NTA (Fig. 1a–c, respectively). A systematic increase in size was confirmed with stable NP dispersions in the range of 8–80 nm. With increasing particle size, the localized surface plasmon resonance (LSPR) band initially narrowed followed by red-shifting and broadening. Above 80 nm the dispersions became unstable leading to aggregation due to a thinning of the starch capping with increasing size as seen by comparing TEM and NTA data. | ||
| Fig. 1 Size control of AuNP cores. 3D plots of (a) extinction, (b) core diameter and (c) hydrodynamic diameter of the 10 partitions. Plots for individual partition are stacked along the y-axis with extinction (in a) and frequency (in b and c) represented with the colour scheme from 0 (black) to max (purple). The UV-vis spectra were normalized at the LSPRmax. The y-axis labels represent the size of the AuNPs derived from TEM images. UV-vis spectra and size distributions for the individual fractions are provided in Fig. S2 and S3.† | ||
Synthesis of Au@Pt core–shell NPs
The two-step, one-pot synthesis strategy for the preparation of Au@PtNPs from the SAMENS method is illustrated in Fig. 2a. The AuNP cores were first prepared by chemical reduction of HAuCl4 in buffered glucose–starch solution.12 Secondly, an atomically thin Pt shell was formed by reduction of H2PtCl6 to provide Au@Pt core–shell NPs. The precursor was reduced directly after the formation of AuNPs with the temperature maintained at 95 °C. The structures of the synthesis components are given in Scheme S1.† Formation of Au and Pt bimetallic NPs requires control of precursor reduction. Too fast reduction of the Pt precursor leads to formation of separate PtNPs. Characteristic UV-vis spectra of the SAMENS prepared Au@PtNPs, PtNPs and AuNPs are shown in Fig. 2b. The extinction from the Au@PtNPs (a continuum with a weak shoulder around 500 nm) resembles that of PtNPs with an indication of spectral features from AuNPs, indicating a drastic change in the AuNP surface structure. No unreacted metal precursors could be detected.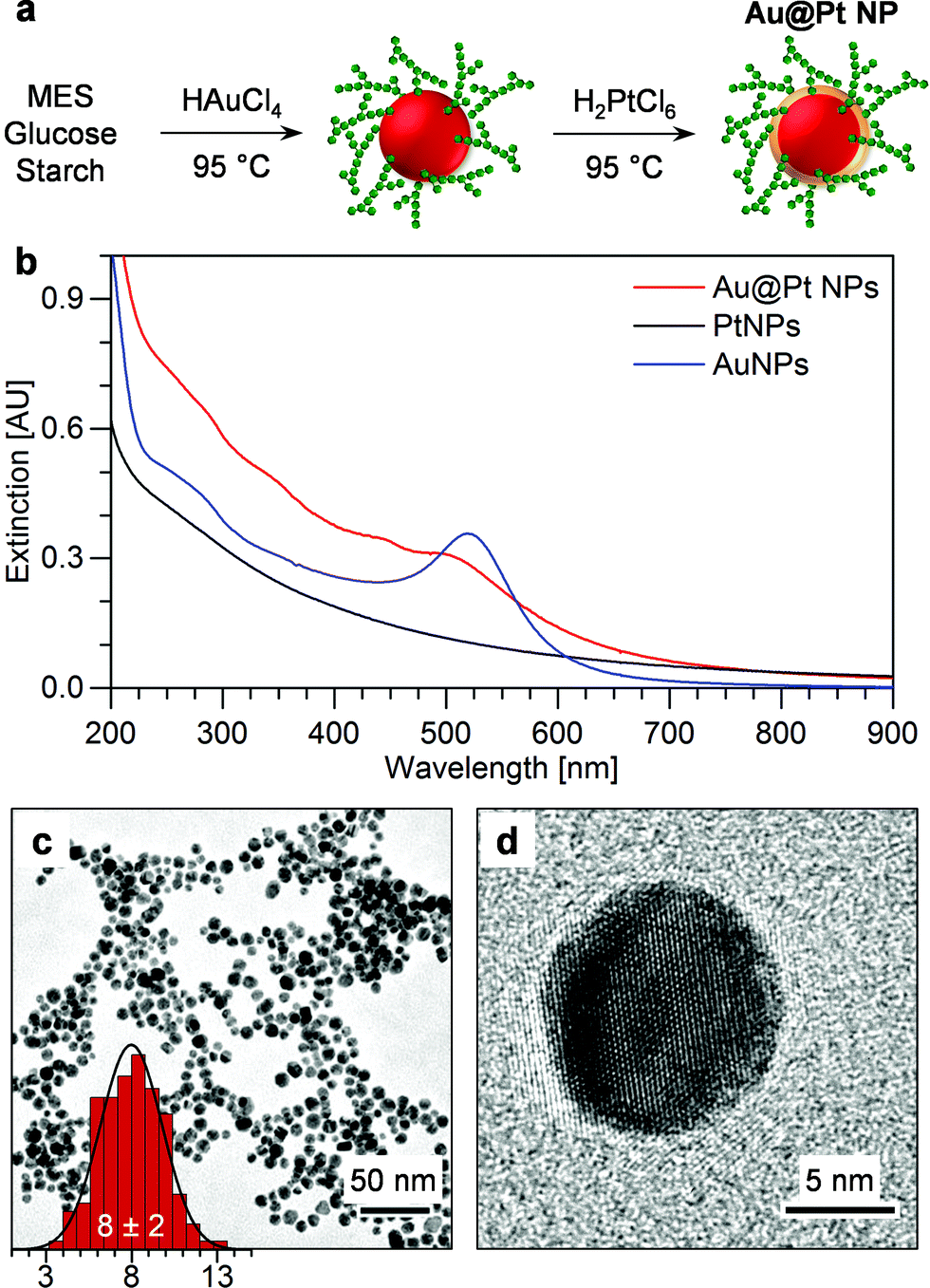 | ||
| Fig. 2 (a) Principle of synthesis of Au@Pt NPs. AuNP cores (red) coated by starch (green) are synthesized and the Pt shell (orange) of the Au@Pt NPs subsequently formed in the same pot. (b) Extinction spectra of as-synthesized Au@Pt NPs (red), PtNPs (black) and AuNPs (blue) from the SAMENS method showing almost complete LSPR quenching by the Pt shell. (c) TEM micrograph of as-synthesized Au@Pt NPs. No evidence of separate PtNPs was observed. Insert in (c) shows the size distribution in nm of the NPs measured from TEM images. (d) HR-TEM of as-synthesized Au@Pt nanostructures showing no indication of a Pt shell despite consistent Pt signal in EDX measurements. | ||
More details about the change in optical properties of the Au@PtNPs were obtained by varying the concentration of the Pt precursor. 0.2, 0.5 and 1.0 mM Pt precursor (AuPt-0.2, -0.5 and -1.0, respectively) was employed while keeping the concentration of Au fixed at 0.5 mM. Fig. S7† shows UV-vis spectra of the three samples after terminated heating (solid lines) and 2 years of storage at r.t. (dotted lines). The lowest Pt precursor concentration led to a dramatic broadening of the LSPR peak of the AuNP core strongly indicative of formation of a Pt shell around the AuNPs. This damping by deposited Pt was previously reported by Henglein when reducing Pt precursor with hydrogen gas.30 No significant LSPR damping was observed with higher Pt concentrations after terminated heating or even after complete Pt precursor reduction. Under these conditions, separate PtNPs rather than core–shell NPs were formed and the colloids retained the characteristic red colour of the AuNPs, Fig. S8.† All NP dispersions remain stable after more than 2 years at r.t. owing to the efficient steric stabilization by the starch coating. The LSPR peak of the AuNP core grew slightly over time due to rearrangement of the Pt shell, Fig. S9.†
TEM was used to evaluate the size and elemental composition of the Au@PtNPs, Fig. 2c and d. The size of the AuNP seeds was 8 ± 2 nm, which was also found for Au@PtNPs. The size of the freshly formed AuNPs and AuNPs which had undergone cooling and reheating (to simulate the Pt shell formation process) was measured to 8 ± 2 and 7 ± 2 nm, respectively (Fig. S10†). In all cases, no significant change in size was identified. Energy dispersive X-ray spectroscopy (EDX) confirmed that the Au@PtNPs contained both Au and Pt, Fig. 3a, at 76 ± 1 and 24 ± 1 mol%, respectively (based on 12 EDX measurements).
 | ||
| Fig. 3 Identification of the Pt shell. (a) EDX spectrum of as-synthesized Au@Pt NPs. (b) Estimated Pt shell thickness based on EDX and HR-TEM images of individual Au@Pt NPs. Red band shows mean ± 1 standard deviation and the green dashed line the diameter of metallic Pt. CVs of (c) as-synthesized Au@Pt NPs, single-crystal Pt and Au, (d) CVs of as-synthesized monometallic NPs and bare glassy carbon (0.1 M H2SO4, RE = RHE, CE = Pt, scan rate = 50 mV s−1). Pt and Au oxide reduction and hydrogen adsorption/desorption are highlighted. | ||
This ratio was also found for Au@PtNP samples in which the Pt shell was produced immediately after AuNP formation. The Pt shell thickness was estimated based on ensemble EDX measurements assuming perfectly spherical 8 nm particles (as measured from TEM) with ideal core–shell geometry to 3 Å. This was substantiated by individual NP EDX data providing a shell thickness of 2.9 ± 0.7 Å, Fig. 3b, which fits with a single Pt atom (2.77 Å). In fact, scanning transmission electron microscopy with EDX (STEM-EDX) of single NPs shows evenly distributed Pt and that Pt signals dominate around the NP perimeters, Fig. 4.
 | ||
| Fig. 4 Elemental mapping of individual Au@PtNPs by STEM-EDX. High-resolution STEM images are shown in the left column, elemental maps of Au and Pt in the middle columns, and a Au/Pt signal ratio map in the right column (white/yellow = high Pt/Au, red = low Pt/Au). Scale bars correspond to 3 nm. | ||
Further evidence of a Pt shell was found in the electrochemical response of the bimetallic NPs. Cyclic voltammetry (CV) of the Au@PtNPs drop cast on glassy carbon electrodes revealed broad anodic signals from 0.6 to 1.3 V from oxidation of the Pt shell and uncovered areas of the Au core, and clear cathodic peaks corresponding to the reduction of PtOx (0.36 V) and AuOx (0.86 V) showing that both metals are present at the surface, Fig. 3c. The oxide reductions in the bimetallic NPs accord with observations for pure SAMENS Au and Pt NPs tested under the same conditions (Fig. 3d) though the reduction potentials are shifted slightly, Table S3.† The Au oxide reduction is shifted 40 mV negatively suggesting that Au oxide is stabilized by the Pt shell whereas the Pt oxide reduction is shifted 30 mV positively. The hydrogen adsorption/desorption region at low potentials is characteristic of Pt and further supports the abundance of exposed metallic Pt.
Catalyst preparation and characterization
The Au@PtNPs were immobilized on a graphitized carbon black support (G-CB), which is robust against degradation due to a high content of sp2 carbon.31 Good wetting of the support was ensured by ultrasonication, prolonged stirring and heating in water prior to mixing with as-synthesized Au@PtNPs. The G-CB suspension was added to the Au@PtNP colloid shortly after Pt shell formation, and adsorption completed within 1 h, Fig. S11.† The starch coating used in this work was not covalently bound to the NP surface and could be removed via oxidation by heating in air at 300 °C for 1 h. Fig. S12a† shows thermogravimetric analysis (TGA) confirming that starch began decomposing/oxidizing around 200 °C, while the bare G-CB support was stable up to 700 °C. TGA of the Au@Pt/G-CB before and after thermal treatment are identical except for the low-temperature region corresponding to mass loss from starch and no indication of starch was found after thermal treatment. Oxidation of the carbon support was catalyzed by the Au@PtNPs. The onset of oxidation thus shifted from 730 to 570 °C in the presence of the bimetallic NPs. The oxidation of the starch capping was also catalyzed by the Au@PtNPs as indicated by shifts of the two exothermic peaks in the differential thermal analysis (DTA) of starch from 300/390 °C to 190/260 °C, Fig. S12b.† The masses finally obtained were assigned to the adsorbed Au@PtNPs and corresponded to 12–13 wt% of the catalyst (excluding starch).X-ray photoelectron spectroscopy (XPS) based on the Au (93–81 eV) and Pt (80–68 eV) 4f signals provided 67 ± 3 wt% of Au@Pt/G-CB, Fig. S13,† with Pt comprising 21.8 ± 0.7 wt% and Au 45 ± 3 wt%. More Pt was found with XPS, a surface specific technique, than EDX due to Pt-rich NP surface. The thermal treatment did not affect the Au and Pt 4f spectra, but the shoulder in the C 1s spectrum at 286.5 eV corresponding to C–O and the O 1s signal disappeared after treatment. X-ray powder diffraction (XRD) and TEM were used to verify that the thermal treatment did not compromise the bimetallic NP nanostructure as no change in the diffraction pattern was found, Fig. S14.† Evaluation of the crystallite size using the Scherrer equation on the metal peaks at 38 and 65° confirmed that no line narrowing and crystallite growth had occurred. TEM further supported that the overall catalyst structure was stable during the starch removal process, Fig. 5, though slight agglomeration of the NPs was observed.
 | ||
| Fig. 5 TEM micrographs of (a) as-synthesized Au@Pt/G-CB catalyst and (b) Au@Pt/G-CB catalyst after thermal treatment to remove the starch coating. | ||
Further electrochemical analysis of the Au@Pt/G-CB surface was undertaken, Fig. 6a. The CVs of bimetallic catalysts match that of immobilized, unsupported Au@Pt NPs but provide six times higher currents attributed to the optimized dispersion in three dimensions on the G-CB. The commercial catalyst exhibits characteristic Pt response throughout the applied potential range. The well-defined features of the supported catalysts allow for deeper evaluation of their electrochemical properties. The electrochemically active surface areas (ECSAs) of the Au@Pt/G-CB catalysts and a commercial Pt/G-CB catalyst were evaluated by integrating the charge in the hydrogen adsorption/desorption region (H-UPD) for Pt and the metal oxide reduction peaks for Pt and Au, Table 1 and Fig. S15.† The specific ECSA of Pt in the commercial catalyst was 21.2 and 21.8 m2 g−1 from H-UPD and PtOx, respectively, which is consistent with previous reports.32 The specific ECSA of Pt in Au@Pt/G-CP was as high as 45.3–52.5 m2 g−1, 2.1–2.4 times that of the commercial catalyst depending on whether hydrogen adsorption or PtOx reduction is considered, Table 1. The Pt ECSA decreased drastically with thermal treatment (39% from H-UPD and 33% from PtOx) which was unexpected from the XRD and TEM results. Indeed, the Au ECSA dropped by only 10% suggesting that agglomeration is not the only effect of heating. It is likely that the Pt shell rearranges forming thicker areas during heating. Considering the ECSAs from metal oxide reduction, the Pt coverage can be estimated by ECSAPt/ECSAPt+Au. The Pt coverage decreases from 73 to 66% during thermal treatment due to contraction of the Pt shell. Braidy et al. showed with detailed EDX mapping how Au–Pt core–shell nanoparticles separate into pure phases joined by a flat interface at 600 °C in inert atmosphere.33 They reported that the core–shell structure is stable at 300 °C. The smaller NP, thinner Pt shell, air atmosphere and catalysed starch oxidation in the current system could thus facilitate shell rearrangement at lower temperatures, e.g. 300 °C. The increase in the Au:Pt ratio after thermal treatment observed in XPS further supports that more Au is found at the particle surface. The greater reduction in Pt ECSA relative to Au ECSA could also be caused by partial dissolution of the Pt shell in the bulk of the NP. Both as-synthesized and thermally treated Au@Pt/G-CB catalysts were further tested in catalytic systems.
 | ||
| Fig. 6 Cyclic voltammograms of Au@Pt/G-CB (red), t-Au@Pt/G-CB where starch has been removed (blue) and commercial Pt/G-CB (black) in (a) pure electrolyte (0.1 M H2SO4), or electrolyte with (b) 0.1 M formic acid, (c) 0.1 M methanol or (d) 0.1 M ethanol. In all cases RE = RHE, CE = Pt and scan rate = 50 mV s−1. | ||
| Catalyst | ECSA [m2 g−1] | Coverage ΓPta | Mass activity [mA mgPt−1] | Surface area activityb [mA cm−2] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FAOR, 0.61 V | MOR, 0.54 V | EOR, 0.62 V | FAOR, 0.61 V | MOR, 0.54 V | EOR, 0.62 V | |||||
| H-UPD | PtOx | AuOx | ||||||||
| a Pt surface area relative to total metal surface area based on metal oxide reduction. b Data normalized to the average ECSA from H-UPD and PtOx. | ||||||||||
| Au@Pt/G-CB | 45.3 | 52.5 | 6.50 | 73% | 556 | 219 | 229 | 1.14 | 0.45 | 0.47 |
| t-Au@Pt/G-CB | 27.5 | 35.1 | 5.85 | 66% | 315 | 197 | 162 | 1.02 | 0.64 | 0.53 |
| Pt/G-CB | 21.2 | 21.8 | — | (100%) | 158 | 110 | 74 | 0.74 | 0.51 | 0.34 |
Electrocatalysis
The Au@PtNP performance in FC anode electrocatalysis was evaluated by studying electrooxidation of small organic molecules used in direct alcohol/carboxylic acid FCs. The three tested systems vary in complexity from simple 2-electron oxidation of FA to the oxidation of EtOH in a 12-electron process. Fig. 6b–d shows CVs recorded at 50 mV s−1 in pure 0.1 M H2SO4 electrolyte with 0.1 M FA, MeOH or EtOH, respectively, Table 1. As-synthesized Au@Pt/G-CB, thermally treated Au@Pt/G-CB without starch (t-Au@Pt/G-CB) and commercial PtNPs (Sigma-Aldrich) on G-CB (Pt/G-CB) were tested and are presented in Fig. 6 as red, blue and black lines, respectively. All data were recorded at identical Pt loadings and are shown as Pt mass specific activities.The activity of the three catalysts towards the EOR is illustrated by the CVs in Fig. 6d showing a main oxidation peak at 0.62 V and a second peak at 1.0 V which is due to further oxidation of intermediate products.47 The catalyst activities follow the same trend as for the FAOR with mass-specific peak currents of Au@Pt/G-CB (229 mA (mgPt)−1) and t-Au@Pt/G-CB (162 mA (mgPt)−1) being 3.1 and 2.2 times higher than for commercial Pt/G-CB (74 mA (mgPt)−1). As with the MOR, the Pt ECSA-specific activity was highest for the thermally treated catalyst, t-Au@Pt/G-CB followed by the as-synthesized Au@Pt/G-CB being 55% and 38% higher than Pt/G-CB, respectively (Table 1). Notably the ratio between the 0.62 and 1.0 V peaks for Au@Pt catalysts was opposite that of Pt/G-CB indicating modification of surface specificity for EtOH oxidation in the presence of the Au core. This is supported by a recent theoretical study in which EtOH and acetyl radical were found to exhibit closer binding interactions on Pt/Au(111) than on pure Pt(111).48
Catalytic hydrogenation of benzenes
The activity of Au@Pt NPs towards hydrogenation (reduction) reactions was probed using substituted benzenes and tested in the hydrocyclization of LA to GVL, Scheme S2.† ARH of BM to cyclohexane carboxamide (CCM) was chosen as a standard reaction to optimize the reaction parameters, such as solvent effects and thermal pre-treatment, before testing BA, and LA/MLA hydrogenation. The Au@Pt/G-CB was screened in its as-synthesized and thermally treated forms using both protic (water) and aprotic (toluene) solvents to study how the solvent environment affects the catalyst activity. Fig. 7 (and Table S4†) shows the catalysts activity for chemoselective hydrogenation of BM to CCM in both water and toluene using H2 as reducing agent. The Au@Pt catalyst was active and highly selective for BM hydrogenation under all reaction conditions. The thermally treated catalyst (Fig. 7III) showed no superior activity over the as-synthesized catalyst in water (I). Instead, a slight decrease (from 78 to 71% CCM yield) was noticed, most likely due to the nanostructural changes induced by the thermal treatment, as discussed previously. The activity of as-synthesized Au@Pt/G-CB dropped to 28% when going from water to toluene (II), but could be recovered to nearly that of water by removing starch (IV). This is due to the low solubility of starch in toluene, Fig. S16.† The non-hydrated starch molecules on the Pt surface in non-polar environment do not extend into the solution but rather collapse resulting in limited access to the active sites. The hydrogenation rate can be related to H-bond donation of the environment around the active site. A lower activation energy barrier for protic solvents, due to strong interaction between protic solvents and substrate by hydrogen bonding, leads to higher hydrogenation rates.49 This is of minor importance in the present system where the activity of thermally treated catalyst is similar in both solvents (III and IV). Finally, it is worth noting that no hydrogenation product of toluene was observed. | ||
| Fig. 7 (a) Conversion (green), yield (black) and TOF (red) for catalytic hydrogenation of 0.5 mmol BM (I to IV), 0.5 mol MLA (V to VII). Schemes of catalyst NP structures are shown on top of the columns. (b) Reaction schemes and general conditions for BM and MLA hydrogenation. Catalyst: (I, II, and V) as-synthesized Au@Pt/G-CB, (III, IV, and VI) thermally treated Au@Pt/G-CB, (VII) commercial Pt/G-CB. Catalyst loading: (I–IV) 0.32 mol% Pt, (V–VII) 1.5 mol% Pt. Solvent: (I, III, V–VII) water, (II and IV) toluene. Reaction time: (I–IV) 24 h, (V–VII) 30 min. | ||
The Au@Pt/G-CB catalyst showed also excellent catalytic activity for chemoselective hydrogenation of BA under mild conditions. Quantitative yields (100%) of CCA were achieved with > 99% selectivity in 24 h at 1 atm H2 pressure and 80 °C. N-doped carbon supported Pd nanoparticles was recently shown to bring a nine-fold increase in activity compared to commercial Pd on activated carbon for the hydrogenation of BA under similar conditions.49 The Au@Pt/G-CB introduced in this work provided as much as 15-fold increased turn-over frequency (TOF) at 0.3 mol% Pt with Au (1.0 mol%). No traces of BA hydrogenation to benzyl alcohol were observed under the given reaction conditions and CCA was the only product.
Hydrocyclization of LA to GVL
Carbon supported Pt catalysts have previously shown poor activity and over-hydrogenation of GVL resulting in low selectivity.27,50 Improved activity was reported by Kon et al. with Pt/C resulting in 14% yield of GVL in 1 h at relatively low pressure (8 bars) and high temperature (200 °C).51 Au@Pt/G-CB was screened for LA hydrogenation followed by cyclization to GVL. Quantitative yields of GVL were attained with excellent selectivity (> 99%) (Table S5, VIII†). The commercial Pt/G-CB catalyst was applied as a reference providing similar results. To determine the TOF, shorter reaction times were chosen. Reaction with commercial Pt/G-CB for 5 h gave quantitative yields of GVL at a TOF of 13 h−1 (Table S5, IX†). The activities of Au@Pt NP and commercial Pt catalysts (Fig. 7V–VII) were compared at 30 min reaction times. Commercial Pt/G-CB catalyst yielded 40% more GVL than as-synthesized Au@Pt/G-CB but the highest yields were obtained with thermally treated Au@Pt/G-CB. The removal of the starch capping thus resulted in a significantly higher catalytic activity than that of commercial Pt/G-CB (48% yield vs. 42%) at a TOF of 62 h−1. This observation highlights the need for starch removal for LA hydrocyclization. The larger molecular size of LA relative to other tested substrates (FA, MeOH, EtOH, BA, BM) may hinder diffusion through the starch layer and limit the activity. The surface structure changes induced by the heating might also increase the activity towards LA hydrocyclization. All the catalysts showed excellent selectivity to GVL with no detectable over-hydrogenation (formation of valeric acid, 2-Me-THF).Conclusions
The green synthesis protocol SAMENS was developed for synthesis of highly active starch protected 8 ± 2 nm core–shell NPs (Au@PtNPs) consisting of a spherical Au core and a 3 Å atomically thin Pt shell. The G-CB supported Au@Pt catalyst (3 wt% Pt, 9 wt% Au) was thoroughly characterized. Starch removal by heating changed the chemistry of the Pt shell as seen in the electrochemical characterization, and strongly affected the catalytic performance in electrochemical oxidation and hydrogenation, which possibly relate to structural changes of the Pt shell. Catalytic activities for several electrooxidation and hydrogenation reactions were investigated and compared to commercial Pt/G-CB. The high specific Pt surface area of the thin Pt shell led to superior activity of Au@Pt/G-CB over Pt/G-CB for electrochemical oxidation of important FC fuels (FA, MeOH and EtOH). Au@Pt/G-CB mass activities (in mA (mgPt)−1) of 556 for FAOR, 219 for MOR and 229 for EOR were 3.5, 2.0 and 3.1 times higher than for the commercial Pt/G-CB catalyst. The core–shell structure gave synergy between the Pt shell and the Au core, as well as limiting inactive Pt. The activity of the Pt surface increased with up to 55%.The presence of the starch coating had insignificant effects on the catalytic activity for ARH of BM in water. Removal of starch prompted a 2.5-fold increase in activity in toluene in which the hydrophilic starch chains collapse, passivating the surface. The Au@Pt/G-CB catalyst was active for GVL production from LA/MLA via hydrogenation/cyclization. For this system, the highest activity (TOF = 62 h−1) was exhibited by the thermally treated catalyst.
The facile, green and robust synthesis presented here produces highly active and well-defined bimetallic nanostructures and offers prospects of further tuning catalytic activity/selectivity by the Au core size, Pt shell thickness and thermal treatment.
Acknowledgements
Assistance with STEM-EDX experiments from Wilhelmus Huyzer (DTU CEN), TGA measurements from Larisa Seerup (DTU Energy Conversion and Storage) and the experimental hydrogenation setup from Eduardo J. García-Suárez (DTU Chemistry) are appreciated. Financial support from Danish Council for Independent Research Technology and Production Sciences (DFF-1335-00330) and the Lundbeck Foundation (R141-2013-13273) are acknowledged.Notes and references
- N. Tian, J. Xiao, Z. Zhou, H. Liu, Y. Deng, L. Huang, B. Xu and S. Sun, Faraday Discuss., 2013, 162, 77–89 RSC.
- K. Jiang, H. Zhang, S. Zou and W. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55–68 CAS.
- B. N. Wanjala, J. Luo, R. Loukrakpam, B. Fang, D. Mott, P. N. Njoki, M. Engelhard, H. R. Naslund, J. K. Wu, L. Wang, O. Malis and C. Zhong, Chem. Mater., 2010, 22, 4282–4294 CrossRef CAS.
- D. F. Yancey, E. V. Carino and R. M. Crooks, J. Am. Chem. Soc., 2010, 132, 10988–10989 CrossRef CAS PubMed.
- J. Luo, L. Wang, D. Mott, P. N. Njoki, Y. Lin, T. He, Z. Xu, B. N. Wanjana, I.-S. Lim and C. Zhong, Adv. Mater., 2008, 20, 4342–4347 CrossRef CAS.
- C. J. Serpell, J. Cookson, D. Ozkaya and P. D. Beer, Nat. Chem., 2011, 3, 478–483 CAS.
- C. J. Murphy, J. Mater. Chem., 2008, 18, 2173–2176 RSC.
- C. Engelbrekt, K. H. Sorensen, J. Zhang, A. C. Welinder, P. S. Jensen and J. Ulstrup, J. Mater. Chem., 2009, 19, 7839–7847 RSC.
- C. Engelbrekt, P. S. Jensen, K. H. Sorensen, J. Ulstrup and J. Zhang, J. Phys. Chem. C, 2013, 117, 11818–11828 CAS.
- C. Engelbrekt, K. H. Sorensen, T. Lubcke, J. Zhang, Q. Li, C. Pan, N. J. Bjerrum and J. Ulstrup, ChemPhysChem, 2010, 11, 2844–2853 CrossRef CAS PubMed.
- G. L. Soloveichik, Beilstein J. Nanotechnol., 2014, 5, 1399–1418 CrossRef PubMed.
- S. Patton, T. Cropp and K. Reynolds, Biochemistry, 2000, 39, 7595–7604 CrossRef CAS PubMed.
- B. Moore, H. Cho, R. Casati, E. Kennedy, K. Reynolds, U. Mocek, J. Beale and H. Floss, J. Am. Chem. Soc., 1993, 115, 5254 CrossRef CAS.
- H. Weissermel and H. J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 1997 Search PubMed.
- J. Thomas, B. Johnson, R. Raja, G. Sankar and P. Midgley, Acc. Chem. Res., 2003, 36, 20–30 CrossRef CAS PubMed.
- H. Wang and F. Zhao, Int. J. Mol. Sci., 2007, 8, 628–634 CrossRef CAS.
- B. Zong, X. Zhang and M. Qiao, AIChE J., 2009, 55, 192–197 CrossRef CAS.
- R. Raja, T. Khimyak, J. Thomas, S. Hermans and B. Johnson, Angew., Chem. Int. Ed., 2001, 40, 4638–4642 CrossRef CAS.
- SNIA VISCOSA, 1968, IE 1122162.
- SNIA VISCOSA, 1966, IE 1019795.
- SNIA VISCOSA, 1964, IE 967918.
- C. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816–5822 CrossRef CAS PubMed.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
- L. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- P. P. Upare, J. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS.
- N. G. Bastus, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef CAS PubMed.
- A. Henglein, J. Phys. Chem. B, 2000, 104, 2201–2203 CrossRef CAS.
- L. N. Cleemann, F. Buazar, Q. Li, J. O. Jensen, C. Pan, T. Steenberg, S. Dai and N. J. Bjerrum, Fuel Cells, 2013, 13, 822–831 CAS.
- M. Gong, F. Li, Z. Yao, S. Zhang, J. Dong, Y. Chen and Y. Tang, Nanoscale, 2015, 7, 4894–4899 RSC.
- N. Braidy, G. R. Purdy and G. A. Botton, Acta Mater., 2008, 56, 5972–5983 CrossRef CAS.
- M. Neurock, M. Janik and A. Wieckowski, Faraday Discuss., 2008, 140, 363–378 RSC.
- M. Rana, M. Chhetri, B. Loukya, P. K. Patil, R. Datta and U. K. Gautam, ACS Appl. Mater. Interfaces, 2015, 7, 4998–5005 CAS.
- S. Chen, H. Su, Y. Wang, W. Wu and J. Zeng, Angew., Chem. Int. Ed., 2015, 54, 108–113 CrossRef CAS PubMed.
- A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef CAS.
- A. Hamnett, Catal. Today, 1997, 38, 445–457 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- S. Wang, N. Kristian, S. Jiang and X. Wang, Nanotechnology, 2009, 20, 025605 CrossRef PubMed.
- S. Yan, S. Zhang, W. Zhang, J. Li, L. Gao, Y. Yang and Y. Gao, J. Phys. Chem. C, 2014, 118, 29845–29853 CAS.
- X. Xu, Y. Zhou, J. Lu, X. Tian, H. Zhu and J. Liu, Electrochim. Acta, 2014, 120, 439–451 CrossRef CAS.
- X. Zhao, J. Zhu, L. Liang, J. Liao, C. Liu and W. Xing, J. Mater. Chem., 2012, 22, 19718–19725 RSC.
- X. Wang, W. Wang, Z. Qi, C. Zhao, H. Ji and Z. Zhang, J. Power Sources, 2010, 195, 6740–6747 CrossRef CAS.
- J. de Souza, S. Queiroz, K. Bergamaski, E. Gonzalez and F. Nart, J. Phys. Chem. B, 2002, 106, 9825–9830 CrossRef CAS.
- S. Sen Gupta and J. Datta, J. Chem. Sci., 2005, 117, 337–344 CrossRef CAS.
- G. Camara and T. Iwasita, J. Electroanal. Chem., 2005, 578, 315–321 CrossRef CAS.
- A. O. Pereira and C. R. Miranda, Appl. Surf. Sci., 2014, 288, 564–571 CrossRef CAS.
- X. Xu, M. Tang, M. Li, H. Li and Y. Wang, ACS Catal., 2014, 4, 3132–3135 CrossRef CAS.
- A. M. Hengne, N. S. Biradar and C. V. Rode, Catal. Lett., 2012, 142, 779–787 CrossRef CAS.
- K. Kon, W. Onodera and K. Shimizu, Catal. Sci. Technol., 2014, 4, 3227–3234 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08922k |
| This journal is © The Royal Society of Chemistry 2016 |