
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved stability of reversible solid oxide cells with a nickelate-based oxygen electrode†
M. A.
Laguna-Bercero
 *,
H.
Monzón
,
A.
Larrea
and
V. M.
Orera
*,
H.
Monzón
,
A.
Larrea
and
V. M.
Orera
Instituto de Ciencia de Materiales de Aragón (ICMA), CSIC – Universidad de Zaragoza, C/ Pedro Cerbuna 12, E-50009, Zaragoza, Spain. E-mail: malaguna@unizar.es
First published on 15th December 2015
Abstract
The stability and performance of YSZ (yttria stabilized zirconia) based solid oxide cells with Ruddlesden–Popper phases as the oxygen electrode have significantly improved. Microtubular Solid Oxide Fuel Cells (mT-SOFCs) using Pr2NiO4+δ (PNO) as the oxygen electrode along with different electrolyte–electrode interlayers were fabricated and characterized in both fuel cell (FC) and electrolysis (SOEC) operation modes. The stability and performance of the cells strongly depend on the barrier layer used. In the FC mode, cells with the PNO–Ce0.9Gd0.1O2−δ (CGO) composite barrier layers showed power densities of ca. 0.63 W cm−2 at 800 °C and 0.7 V. In addition, they presented excellent stability as no degradation was observed after 100 hours under the operating conditions. Their performance in the electrolysis mode is also remarkable (−0.78 A cm−2 at 800 °C and 1.3 V). As anticipated, nickelates withstand the excess of oxygen at the electrode–electrolyte interface better than other oxygen electrode materials. Oscillatory current behaviour has been observed and ascribed to the partial decomposition reaction of the Pr2NiO4+δ phase into PrNiO3 and PrO2−y which, on the other hand, seems not to deteriorate the electrochemical properties of the cell. However, the PNO–CGO in situ reaction, forming mixed praseodymium, cerium and gadolinium oxides (PCGO) at the electrolyte–oxygen electrode interface, appears to be essential for the good stability and performance of the cells. In this study we demonstrate, for the first time, the excellent reversible SOFC/SOEC performance and stability under current load of a cell with nickelate based oxygen electrodes.
1. Introduction
One of the crucial aspects to achieving long lifetime and highly efficient SOFCs (solid oxide fuel cells) and SOECs (solid oxide electrolysis cells) is to enhance the durability and performance of the oxygen electrode, as they typically degrade over time under the operating conditions. In recent years, research has focused on finding complex oxides without alkaline earth metal elements, such as Sr or Ba, as they tend to segregate forming undesirable products, especially at the electrode–electrolyte interface.1 Cobalt-free electrodes are also desirable, due to the difficult material disposal associated with Co-containing oxides.2 Recently, lanthanide nickelates (Ln = La, Nd, Pr) have received considerable interest as an alternative to conventional manganite, cobaltite and ferrite oxygen electrodes for SOFCs and also as oxygen separation membranes.3,4 These materials present a K2NiF4-type structure consisting of alternate LnNiO3 perovskite layers and LnO rock-salt layers where excess oxygen atoms occupy the interstitial sites between the LnO layers. Ln2NiO4±δ materials belong to the Ruddlesden–Popper series and present flexible oxygen stoichiometry. As a consequence, they can accommodate excess interstitial oxygen under high pO2 atmospheres and withstand low pO2 atmospheres via loss of oxygen. The materials exhibit fast oxygen ion diffusion through bulk and rapid surface exchange kinetics. The most studied nickelate material for SOFC applications is the lanthanum nickelate (La2NiO4+δ, LNO).5,6In addition to their potential use in SOFCs, they also seem to be very attractive for electrolysis applications.7 In the electrolysis mode, there is an increase of pO2 at the oxygen electrode/electrolyte interface due to oxygen evolution. Electrochemically induced oxygen pressure increase at the electrolyte–oxygen electrode interface and subsequent membrane failure have been theoretically predicted and experimentally observed in the electrolysis mode.8,9 Electrode materials with oxygen hyperstoichiometry, such as the Ruddlesden–Popper phases, are believed to be favourable for effective oxygen evolution in the SOEC mode.10,11 Moreover, it has been reported that nickelates are less prone to Cr poisoning from the interconnectors than standard LSCF materials.12
The main drawback of the nickelate phases is their reactivity with the standard electrolytes: yttria stabilized zirconia (YSZ) and gadolinium doped ceria (CGO).13 As a consequence, it is necessary to use barrier layers between the electrolyte and the oxygen electrode. Montenegro-Hernandez et al.13 conducted a study about the chemical compatibility between Ln2NiO4±δ (Ln = La, Nd, Pr) and both YSZ and gadolinium doped ceria (CGO) electrolytes. Evidence of the chemical reaction between LNO and both CGO and YSZ was detected at temperatures as low as 700 °C, whereas Nd2NiO4+δ (NNO) was found to react at higher temperatures (1000 °C). Additionally, Sayers et al.14 observed that under an air atmosphere at 900 °C, LNO rapidly reacts with CGO electrolytes forming a higher order Ruddlesden–Popper phase (Lan+1NinO3n+1) as one of the reaction products. More recently, the same authors found that through in situ high-resolution synchrotron X-ray powder diffraction, there was no such reaction in an unsealed capillary tube.15 Pr2NiO4+δ (PNO) is the phase presenting the highest oxygen ion diffusion values and surface exchange kinetic parameters. This is possibly due to the fact that PNO exhibits a larger range of oxygen nonstoichiometry variation compared to LNO and NNO, because of the smaller size of the A-site cations. However, the reactivity of Pr2NiO4+δ (PNO) with both CGO and YSZ is still a matter of discussion in the literature. For example, Philippeau et al.16 found that after heating at 800 °C for 5 days, PNO is decomposed into a perovskite phase (PrNiO3 type) with traces of nickel oxide. They also found some reactivity with CGO. Montenegro-Hernandez et al.13 reported that the PNO phase is not thermodynamically stable below 900 °C as well.
In spite of the remarkable oxygen conductivity values, few studies about the performance of PNO as SOFC cathodes are found in the literature. This might be connected to the broadly assumed thermodynamic metastability of the PNO phase at temperatures in the range of 800–950 °C in oxidizing atmospheres.17 In fact, it was established that at these temperatures, PNO decomposes into another Ruddlesden–Popper phase, according to reaction (1).
 | (1) |
In addition, Odier et al.18 suggested that under pure oxygen, ambient pressure and temperature above 850 °C, PrO2−y is expelled out from the Pr2NiO4+δ structure.
In spite of this unfavourable evidence, Philippeau et al. studied the behaviour of PNO/CGO/PNO cells in air at 600 °C.16 They found low polarization resistance values (Rp = 0.28 Ω cm2) for these symmetrical cells. Recently, researchers from Jülich GmbH19 reported that the highly defective praseodymium PNO phase (Pr1.9NiO4+δ) is thermo-chemically stable under the typical cell operation conditions. Although the rest of the used cell materials were not specified in this communication, they reported rather high current densities of 1.2 A cm−2, similar values as in cells with LSCF (lanthanum strontium cobalt ferrite) cathodes.
To our knowledge, the only published report of a fuel cell test using PNO as the cathode and YSZ as the electrolyte is that of Ferchaud et al.20 They used a ∼1.3 μm thick Co-doped GCO cathode barrier layer and measured maximum power densities of ∼ 400 mW cm−2 at 600 °C. PNO–Ag composite cathodes have also been used in low temperature SOFCs with ceria/carbonate composite electrolytes,21 and also in intermediate temperature protonic fuel cells with BaCe0.9Y0.1O3−δ (BCY) electrolytes.22,23 Very recently, oxygen electrodes produced by PNO infiltration onto an LSGM (Lanthanum Strontium Gallium Magnesium Oxide) porous scaffold have been reported.24 In summary, although PNO is a very promising material for oxygen electrodes in SOFC/SOEC systems, its stability under real operating conditions still remains to be verified.
In our current work, we have fabricated and studied, for the first time, the electrochemical performance of microtubular Solid Oxide Fuel Cells (mT-SOFC) with YSZ electrolytes and PNO oxygen electrodes. mT-SOFCs are interesting from the point of view of its higher resistance to thermal cycling, and because they present shorter start-up/shut-down times as well-as higher volumetric power densities in comparison with the traditional planar geometry.25–27 The effect of different cathode configurations using several CGO-based barrier layers, as well as the PNO stability under SOFC operation, are reported.
2. Experimental
2.1. Cell fabrication
NiO–YSZ anode supported tubes were fabricated by Powder Extrusion Moulding (PEM) following the procedure described by Monzón et al.28 and Arias et al.29 The composition was adjusted to obtain a final structure after NiO to Ni reduction of 25% Ni – 25% YSZ – 50% pore (in vol%). Subsequently, YSZ electrolyte suspensions were prepared using TZ-8YS (Tosoh) powders in an isopropanol–ethanol azeotropic mixture, and using PVB (polyvinyl butyral) as the binder and Beycostat as the dispersant agent. Viscosities of all suspensions were characterized using a rheometer (Haake Mars, Thermo Scientific, Germany). YSZ suspensions, with typical viscosities in the range of 0.05 Pa s, were deposited by dip-coating (three immersions) utilizing an in-house built device. The half cells were then dried at RT and co-sintered at 1500 °C for 2 hours. Three types of cells (Table 1), all with a PNO (Marion Technologies) cathode, were fabricated applying three different electrolyte–cathode barrier layer configurations: a pure CGO (Ce0.9Gd0.1O2−δ, FuelCell Materials) barrier layer (one immersion, sintered at 1300 °C for 2 h), a PNO–CGO (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in wt%) composite barrier layer (one immersion, sintered at 1100 °C for 2 h) and another one without a barrier layer. Notice that due to the highly refractory nature of CGO, higher sintering temperatures had to be used for the pure CGO coating than for the PNO–CGO composite layer. PNO powders were finally used to produce the cathode suspensions in ethanol media, using PVB as the binder and Beycostat as the dispersant agent. Pure PNO suspensions, with typical viscosities in the range of 0.03 Pa s, were deposited by dip-coating (four times), then dried at RT and sintered at 1100 °C for 2 hours. Final dimensions of the microtubular cells were: 50 mm in length, outer diameter of 3.4 mm, NiO–YSZ wall thickness (T) of ca. 700 μm, ca. 20 μm YSZ electrolyte, ca. 2–3 μm barrier layer and ca. 55 μm PNO cathode.
:1 in wt%) composite barrier layer (one immersion, sintered at 1100 °C for 2 h) and another one without a barrier layer. Notice that due to the highly refractory nature of CGO, higher sintering temperatures had to be used for the pure CGO coating than for the PNO–CGO composite layer. PNO powders were finally used to produce the cathode suspensions in ethanol media, using PVB as the binder and Beycostat as the dispersant agent. Pure PNO suspensions, with typical viscosities in the range of 0.03 Pa s, were deposited by dip-coating (four times), then dried at RT and sintered at 1100 °C for 2 hours. Final dimensions of the microtubular cells were: 50 mm in length, outer diameter of 3.4 mm, NiO–YSZ wall thickness (T) of ca. 700 μm, ca. 20 μm YSZ electrolyte, ca. 2–3 μm barrier layer and ca. 55 μm PNO cathode.
| Cell name | Anode T = 700 μm | Electrolyte T = 20 μm | Interlayer T = 2–3 μm | Cathode T = 55 μm |
|---|---|---|---|---|
| PNO1 | NiO–YSZ | YSZ | — | PNO sintered at 1100 °C |
| PNO2 | NiO–YSZ | YSZ | CGO sintered at 1300 °C | PNO sintered at 1100 °C |
| PNO3 | NiO–YSZ | YSZ | CGO:PNO sintered at 1100 °C |
PNO sintered at 1100 °C |
2.2. Structural and microstructural characterization
Chemical compatibility of PNO and CGO at different temperatures was studied using uniaxially pressed pellets of PNO–CGO mixtures (4:1 and 2:1 in wt%). XRD (X-ray diffraction) of the milled powders were collected using CuKα radiation in a D-Max Rigaku instrument. In order to study the possible decomposition of the PNO phase after SOFC/SOEC operation, XRD patterns were also directly collected over the surface of the cells. Lattice parameters were determined by Rietveld refinements using the Fullprof software.30 Microstructural studies were analysed using a Merlin (Carl Zeiss, Germany) field emission scanning electron microscope (FE-SEM) equipped with an INCA-450 Energy Dispersive Spectroscopy system (Oxford Instruments, United Kingdom).
2.3. Electrochemical characterization
Electrochemical studies were performed at 600–800 °C in both fuel cell and electrolysis modes using the experimental setup described in ref. 31 and 32. A fuel composition of 3% H2O – 97% H2 was used for operation in the fuel cell mode and 50% H2O – 50% H2 in the electrolysis mode. Electrical contacts were made using silver and gold wires in a four-probe setup configuration. For the inner contact (hydrogen electrode), silver wires were mechanically attached into featured holes on both ends of the microtubes. For the outer contact (oxygen electrode), gold wire was coiled around the PNO cathode surface (1 cm2) adding Au or Pt paste to improve electrical contact and current collection. The cells were then sealed to alumina tubes using a ceramic sealant (Aremco, Ceramabond 503) and heated up to 800 °C under nitrogen, while the oxygen electrode side was exposed to ambient air. Subsequently, nitrogen gas was switched to pure humidified hydrogen, reducing NiO to metallic Ni at the anode. Steam was supplied by the use of a direct vapour humidifier controlling the relative humidity with a resolution of ±1.3%. All gas lines located downstream of the humidifier were externally heated in order to prevent steam condensation. j–V (current density–voltage) was recorded in the galvanodynamic mode using a scan rate of 2.5 mA cm−2 s−1. EIS (electrochemical impedance spectroscopy) measurements were performed under OCV (open circuit voltage) conditions using 20 mV of sinusoidal amplitude and a frequency range from 100 kHz to 100 mHz. Those experiments were performed using a VSP potentiostat/galvanostat (Princeton Applied Research, Oak Ridge, US).3. Results and discussion
3.1. Stability of PNO as a novel oxygen electrode
We have studied the reactivity of PNO and CGO as a means of broadening current knowledge of their chemical compatibility. We have used two different PNO:CGO compositions (4:1 and 2:1 in wt%) that were heated for 2 hours at 1100 °C and 1200 °C in air. XRD patterns of the reacted samples and of the PNO and CGO as received powders are shown in Fig. 1. It is clearly observed that after the thermal treatment, the PNO intensity of the major peak at 31.8° (113) is decreasing (Fig. 1b). PNO decomposition increases on increasing the temperature and also on increasing the CGO content in the CGO:PNO mixture. In fact, after the thermal treatment, the PNO phase is almost no longer present in the 2:1 sample. However, for the 4:1 sample, the PNO phase is still present after annealing. This experiment clearly indicates that some reaction between PNO and CGO takes place in this temperature range.
 | ||
| Fig. 1 (a) XRD patterns of different mixtures of PNO and CGO powders, collected after annealing at 1100 and 1200 °C for 2 hours. XRD patterns of the as-received PNO and CGO powders are shown as the reference. • Pr4Si3O12; * NiO. (b) Magnification of the region at 2θ = 30–35°. | ||
The chemical reaction between PNO and CGO results in the production of mixed Pr and Ce oxides. In fact, as observed in Table 2, the lattice parameters for the ceria phase decrease on increasing the PNO content in the composite, and also on increasing the annealing temperature. Furthermore, two fluorite-type phases were identified for the 4:1 mixture, which corresponds to the PCGO and praseodymium oxide, respectively. In relation to this, Chiba et al.33 studied Ce–Pr mixed oxides as functional layer materials for SOFC cathodes. They found that on increasing the Pr content in the Ce1−xPrxO2−δ mixed oxide, the oxygen vacancy concentration and the total conductivity of the material increase. They concluded that a mixed oxide with a composition near x = 1 is the most favourable composition for an active layer in a SOFC cathode. In a similar trend, Sadykov et al. studied composites of praseodymium based perovskites (PrNi1−xCoxO3+δ) and doped ceria (Ce0.9Y0.1O2−δ) as cathodic materials.34 They also observed an increase of oxygen mobility in comparison with the pure phases due to redistribution of cations between perovskite (PrNi1−xCoxO3) and fluorite (Ce1−xPrxO2−δ) domains.
:PNO composites. All phases correspond to the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The chemical formula corresponds to estimations according to the obtained lattice parameters
m space group. The chemical formula corresponds to estimations according to the obtained lattice parameters
| Sample | Phase | Lattice parameters a = b = c (Å) |
|---|---|---|
| CGO | Ce0.9Gd0.1O2−δ | 5.4136(3) |
| PNO:CGO 2:1, 1100 °C |
Ce0.332Gd0.037Pr0.631O2−δ | 5.4044(2) |
| PNO:CGO 2:1, 1200 °C |
Ce0.332Gd0.037Pr0.631O2−δ | 5.4033(2) |
| PNO:CGO 4:1, 1100 °C |
Ce0.203Gd0.023Pr0.774O2−δ | 5.3985(3) |
| PrO2−y | 5.4563(14) | |
| PNO:CGO 4:1, 1200 °C |
Ce0.203Gd0.023Pr0.774O2−δ | 5.3979(3) |
| PrO2−y | 5.4585(8) | |
In our experiments, the observed decrease in the lattice parameters of the ceria phase is a clear indication that Pr is entering the fluorite structure and substituting for the Ce ions, hence forming a Gd- and Pr-doped mixed cerium oxide. The final composition of these mixed oxides could not be fully determined from our XRD experiments. Although, by comparing our data with the experiments of Chiba et al.,33 it can be established that the compositions should be close to the theoretical stoichiometric formula (Ce0.332Gd0.037Pr0.631O2−δ and Ce0.203Gd0.023Pr0.774O2−δ for the 2:1 and 4:1 studied samples, respectively) calculated assuming full equilibrium substitution of Pr into the fluorite structure. As a consequence of the PNO decomposition, NiO (*) is also formed as observed in the X-ray diffractograms. In addition, all the studied samples also showed the presence of small amounts of Pr4Si3O12 impurities (•). The presence of this impurity phase has also been previously observed in the analogue NNO.35,36 Schuler et al.,35 reported the formation of Nd-silicate as a thermally induced reaction phase with amorphous Si impurity during the processing of the NNO raw powder.
In summary, these experiments confirm the reaction between PNO and CGO at temperatures as low as 1100 °C. Pr from the PNO phase diffuses into the CGO fluorite structure forming the mixed oxide, PCGO. However, according to previous studies reported in the literature, it is very interesting to point out here that the presence of these mixed Pr–Ce oxides, especially at the electrolyte/electrode interface, could be beneficial rather than detrimental in terms of oxygen electrode performance in SOFC and SOEC.37–39
3.2. Microstructure of novel oxygen electrodes and interface characterization
In order to study the performance of the PNO cathodes in a real device under the operating conditions, three different types of microtubular cells were fabricated. The anode and electrolyte of the cells are Ni/YSZ microtubes coated with a dense and thin YSZ electrolyte.28 As summarized in Table 1 we have fabricated cells with three different PNO-based cathode configurations only differing between them in the nature of the electrolyte–electrode barrier. Typical microstructures of the interlayer between the electrolyte and the barrier layer (or cathode for the PNO1 cell) are shown in Fig. 2. | ||
| Fig. 2 SEM images showing the interface between the electrolyte and the oxygen electrode for the different cells. | ||
Notice that higher sintering temperatures are required for pure CGO layers (1300 °C) than for the PNO–CGO composite layer (1100 °C). As a consequence, the microstructure of the interlayer of the PNO2 cell (Fig. 2b) is coarser in comparison with the PNO3 cell (Fig. 2c). The thicknesses of the barrier layers are about 2 μm and 3 μm for the CGO (PNO2) and CGO:PNO (PNO3) cells, respectively. In Fig. 3, the microstructure of the fabricated PNO3 cell is shown. A good adhesion between layers is observed at all the different interfaces: fuel electrode/electrolyte, electrolyte/barrier layer and barrier layer/oxygen electrode.
 | ||
| Fig. 3 SEM image showing the typical microstructure of the PNO3 cell. | ||
3.3. Electrochemical SOFC experiments
A chronoamperometric study in the fuel cell mode was performed for each type of cell using pure humidified hydrogen (3% H2O – 97% H2) as the fuel and static air as the oxidant. The experiments were performed at 800 °C at a voltage of 0.7 V and the results are given in Fig. 4a. | ||
| Fig. 4 (left) Chronoamperometric studies for each cell at 800 °C and at a voltage of 0.7 V; (right) j–V curves performed before and after the chronoamperometry experiments. | ||
The best initial performance (580 mA cm−2) was obtained by the PNO1 cell (with no interlayer), although the current density rapidly decreases as a function of operating time. The degradation in this cell is very severe, about 50% after only two hours under the current load. The initial performance of the cells with the barrier layer is in the range of 450 mA cm−2. It is slightly reduced because of the increase in ohmic resistance caused by the extra layer. The PNO2 cell (pure CGO interlayer) also degrades with operation time, although the degradation rate was decreased (ca. 4% per hour) in comparison with the PNO1 cell (ca. 25% per hour). However, in spite of the improvement achieved, this degradation rate is still high to meet the typical SOFC requirements, in the range of less than 1% per 1000 hours. We believe that the layer fabrication method we have used was not efficient enough to produce the dense CGO barrier layer, required to suppress the YSZ–PNO reactions. In fact, as observed in the micrograph in Fig. 2, the CGO barrier layer is still relatively porous, and as a consequence, reactivity between PNO and YSZ is still expected to happen. Alternatively to the dip coating method, dense CGO barrier layers can be developed through electrophoresis or sputtering methods.
However, the PNO3 cell showed remarkable stability with operation time. In fact, as observed in Fig. 4a, the current density increased from 461 to 567 mA cm−2 after ∼70 hours under current load, confirming the excellent stability of the oxygen electrode in the PNO3 cell. The observed enhancement is about 0.3% per hour, although this enhancement is not linear. The following studies will be focussed on these improved cells.
Another significant fact observed in Fig. 4 left is the relatively large fluctuation in the recorded data during the experiment. This is in contrast to the smooth and low scattering sets of data usually found in these experiments in other cells using standard LSM (La0.8Sr0.2MnO3−δ) or LSCF (La0.6Sr0.4Co0.2Fe0.8O3−δ) oxygen electrodes.40,41 Even for other Ruddlesden–Popper based cathodes such as LNO42,43 or NNO,44 chronoamperometric studies also show low dispersion in the measured values. We believe that these fluctuations are not due to any type of electronic instrumental noise or drift. A possible explanation could be associated with the presence of some chemical reactions. Low frequency oscillatory behaviours in electrochemical experiments have been rarely observed and are related to reversible redox reactions.45 This particular effect may be explained considering that, at the operation temperature (800 °C), and probably enhanced by the current load, Pr2NiO4+δ could partially decompose into PrNiO3 (or Pr4Ni3O10−δ) and PrO2−δ, as expressed in eqn (1). As reported by other authors, this reaction seems to be reversible, which is a necessary ingredient in oscillatory behaviour.46 Regarding the stability of the PNO phase, in situ powder neutron diffraction studies have recently reported this decomposition under oxygen-rich atmospheres.47
Fig. 4b shows the j–V curves for the three studied cells before and after the chronoamperometry experiment. Initial performances are rather similar for the three analysed cells. PNO1 and PNO2 cells showed a significant decrease in performance after the experiment under a constant current load, possibly associated with YSZ/PNO reactivity. However, the performance of the PNO3 cell even increased after operation. EIS experiments recorded before and after the chronoamperometric study, also confirmed this result (see ESI†). The enhancement for the PNO3 cell is not fully understood. It could be related to microstructural reorganization induced by the current flux (electrode activation) perhaps involving the aforementioned formation of Pr, Ce and Gd mixed oxide (PCGO) at the interface.
3.4. Reversible operation studies
Experiments under both fuel cell and electrolysis modes were performed for the PNO3 cell using 50% H2/50% H2O as the fuel. Those experiments were done after the chronoamperometric studies in the FC mode described in the previous section; that is to say after 100 hours under the operating conditions (∼70 hours under the current load). As observed in Fig. 5, the cell is completely reversible in the range of temperatures studied (600–800 °C). For example, in the electrolysis mode (at 800 °C and 1.3 V), current densities of ca. 800 mA cm−2 and ASR (area specific resistance) of ∼0.41 Ω cm2 were measured. ASR values were obtained from the j–V slopes. These reported current densities are much higher than those obtained for identical mT-SOFC using LSM–YSZ standard oxygen electrodes at the same temperature (ca. 500 mA cm−2).48 In order to reach the same performance with standard cells, it is necessary to increase the operation temperature up to 900 °C.48 From our knowledge, this SOEC performance is the highest reported for microtubular electrolysis cells. In addition, the measured ASR in the fuel cell mode is slightly higher (∼0.45 Ω cm2) than the one in the SOEC mode. Similar behaviour was observed at lower temperatures under the same fuel conditions (at 700 °C: ASRfuel cell ∼ 1.16 Ω cm2vs. ASRelectrolysis ∼ 1.01 Ω cm2). As indicated in the introduction, under the SOEC operation mode and at high current values, a high concentration of oxygen is produced and it usually accumulates at the electrode–electrolyte interface when conventional cathode materials are used. Oxygen accumulation creates a barrier to the transport of gas and can even lead to rupture of the cells.49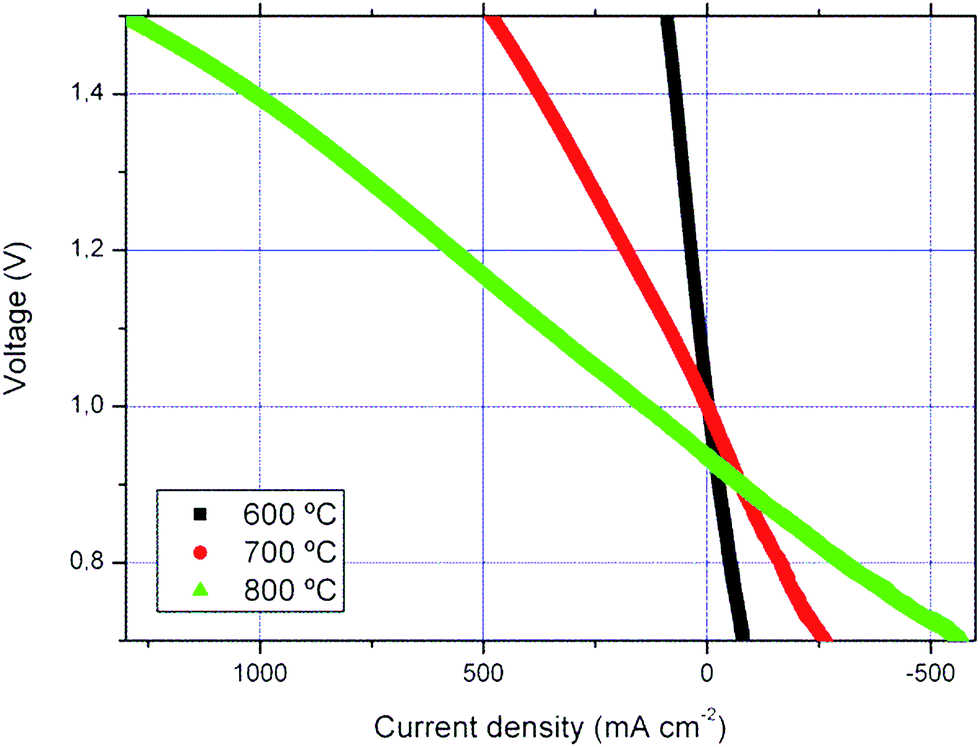 | ||
| Fig. 5 j–V curves (fuel cell and electrolysis modes) performed for the PNO3 cell at different temperatures. | ||
However, this oxygen excess can accommodate itself into the PNO phase as previously reported in other Ruddlesden–Popper compounds.10,11,44 In our opinion, this is probably the reason for the outstanding enhancement in the performance of these novel cells in the electrolysis mode. In conclusion, we have been able to take advantage of the hyperstoichiometry of some Ruddlesden–Popper phases, making them extraordinarily appealing for SOFC cathodes and particularly for electrolysis applications.
3.5. Post-operation phase analysis
To gain insight about the processes taking place during the operation of these cells, we have performed post operation studies. Fig. 6 shows the XRD collected on the surface of the oxygen electrodes after the electrochemical studies. For comparison, the XRD of a blank cell (PNOblank, fabricated under the same conditions but not operated) is also shown. For the blank cell, the XRD pattern resulted in two major phases: Pr2NiO4+δ (orthorhombic Fmmm, a = 5.4591(8) Å; b = 5.3769(9) Å; c = 12.3842(18) Å) and YSZ (cubic Fmm, a = 5.1470(9) Å), and no apparent decomposition of the Pr2NiO4+δ phase was observed. However, some Pr4Si3O12 and Pr2Zr2O7 small XRD peaks about the detection limit may indicate the presence of these minority phases. The presence of Pr4Si3O12 was previously discussed in Section 3.1,35,36 and the presence of Pr2Zr2O7 is a consequence of the expected reactivity between YSZ and Pr2NiO4+δ (ref. 13 and 14) during the sintering process, as no protection barrier interlayer was used in this case. Nevertheless, the XRD pattern collected for the PNO1 sample after operation shows, in addition to YSZ (cubic Fmm, a = 5.1470(8) Å), the complete decomposition of Pr2NiO4+δ into PrNiO3 (orthorhombic Pbnm, a = 5.3788(27) Å; b = 5.4114(70) Å; c = 7.6320(66) Å) and PrO2−y (cubic Fmm, a = 5.4713(17) Å) together with small amounts of NiO. Decomposition under the operation of this cathode has been previously described by several authors.13,17,18,50 Furthermore, the observed Au peaks correspond to the current collector used for the electrochemical measurements.
 | ||
| Fig. 6 XRD patterns collected from the oxygen electrode surface after the electrochemical studies for the PNO1 and PNO3 cell. A blank cell (nonoperated) is also shown for comparison. The detected phases are: ■ YSZ; □ PrNiO3; ○ Pr4Si3O12; ● Pr2NiO4+δ; ▲PrO2−y; * NiO; x Ce9xGdxPryO2−z; & Pr2Zr2O7; Δ Au (or Pt). | ||
Finally, the XRD analysis of the cathode in the improved PNO3 sample (after a long operation period) appears as major phases YSZ (cubic Fmm, a = 5.1425(4) Å), PrNiO3 (orthorhombic Pbnm, a = 5.3454(82) Å; b = 5.4364(39) Å; c = 7.6338(66) Å), Ce9xGdxPryO2−z (cubic Fmm, a = 5.4038(4) Å) and Pr2NiO4+δ (orthorhombic Fmmm, a = 5.4168(24) Å; b = 5.4542(16) Å; c = 12.4740(27) Å). The most striking feature in the case of the successful PNO3 cell is the formation of mixed Pr and Gd doped cerium oxides. In this case, the partial decomposition of PNO takes place via the reaction with CGO, forming the composite oxygen electrode according to reaction (2). Note that this reaction is a modification of reaction (1) in order to include the formation of PCGO due to the incorporation of Pr into the Ce–fluorite structure. As previously discussed, it is proposed that this reaction is continuously taking place, causing the jumps observed during the chronoamperometry test.
| Pr2NiO4+δ + Ce0.9Gd0.1O2−δ → Pr2NiO4+δ + PrNiO3 + Ce9xGdxPryO2−z | (2) |
We cannot assure the influence of the reported reactivity for this cell over longer times (thousands of hours), as this is out of the scope of the present study. Our findings clearly show that despite these phase changes, these cells presented excellent stability as no degradation was observed after 100 hours under the operating conditions. It is also remarkable that by using CGO/PNO composite barrier layers, the formation of the insulating zirconate phase is also avoided.
Finally, SEM experiments were performed after the operation. In Fig. 7, two polished transverse-cross sections of cells PNO1 (left) and PNO3 (right) are shown. Both images were taken at low kV (0.5 kV) using an InLens detector in order to enhance the contrast between conductive and non-conductive phases. Both electrodes of the two samples show a homogenous distribution of particles and there are no signs of delamination. This experiment confirms that the composite oxygen electrode of the cell PNO3 (mainly PrNiO3 and Ce9xGdxPryO2−z mixture), also presents a homogeneous distribution of conductive particles in spite of the phase change, assuring a good current collection by the PrNiO3 phase, whereas the PCGO phase is responsible for the oxygen diffusion at the electrolyte interface. It is worth noting that PrNiO3, as well as the higher order Ruddlesden–Popper phase Pr4Ni3O10−δ, are excellent electronic conductors.17 Moreover, PrO2−y presents decent oxygen diffusion parameters to be contemplated as a component for SOFC cathodes.51 In particular, the closely related lanthanum derivate (La4Ni3O10−δ) has been proposed as a SOFC cathode because of its excellent conductivity.52 EDS analysis and low-energy backscattering electron SEM observations in the electrolyte with the oxygen electrode interface confirmed that there is no evident reaction between the oxygen electrode and the YSZ electrolyte for PNO3 cells (see ESI†). However, some nanometric Pr-rich grains at the electrode–electrolyte interface were observed in the EDS-SEM experiments which agrees with the observation of small amounts of Pr2Zr2O7 by XRD in the PNO1 cell. The PNO3 cell showed a better adherence between the electrolyte and the oxygen electrode than the PNO1 cell, even after operation conditions. The PNO–CGO interlayer seems also to be acting as an adhesion layer between PNO and YSZ. Based on these results, we can allow ourselves to think that the newly formed PCGO oxide at the interface could also be enhancing the electrochemical performance of the oxygen electrode. PrO2−y and CeO2 mixtures are presented as a novel alternative for SOFC oxygen electrodes, and are worth further study.
 | ||
| Fig. 7 SEM images showing the microstructure of the PNO1 and PNO3 cell after the electrochemical studies. The images were obtained using 0.5 kV electrons and the InLens detector to produce high contrast between the conductive and isolating particles. | ||
4. Conclusions
Pr2NiO4+δ oxygen electrodes were successfully integrated into anode supported mT-SOFC using Ni–YSZ as the anode support, YSZ as the electrolyte and a CGO:PNO composite intermediate layer. Although Pr2NiO4+δ decomposes into PrNiO3 and PrO2−y, the deterioration in electrochemical performance of the cell is completely eliminated. In contrast, we observe a slight enhancement after 70 hours under current load. According to the post operation analysis, the formation of the mixed PCGO oxide at the interface between the electrolyte and the oxygen electrode plays a crucial role in the stability and performance of the cells. In addition, the Pr2NiO4+δ electrode is confirmed as a reliable oxygen electrode under real SOFC and SOEC conditions. This is the first evidence of a successful reversible SOFC/SOEC using Ruddlesden–Popper phases as the oxygen electrode, though there is still room for further studies to improve the durability of the cell.
Acknowledgements
The authors would like to acknowledge grant MAT2012-30763 financed by the Spanish Government (Ministerio de Economía y Competitividad) and Feder program of the European Community for funding the project. The use of Servicio General de Apoyo a la Investigación (SAI, University of Zaragoza) is also acknowledged. We finally acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- Y. Chen, W. Zhou, D. Ding, M. Liu, F. Ciucci, M. Tade and Z. Shao, Adv. Energy Mater., 2015, 1500537 Search PubMed
.
- J. Q. Xu, H. R. Thomas, R. W. Francis, K. R. Lum, J. W. Wang and B. Liang, J. Power Sources, 2008, 177, 512 CrossRef CAS
- S. J. Skinner and J. A. Kilner, Solid State Ionics, 2000, 135, 709 CrossRef CAS
- J. M. Bassat, P. Odier, A. Villesuzanne, C. Marin and M. Pouchard, Solid State Ionics, 2004, 167, 341 CrossRef CAS
- N. Hildenbrand, P. Nammensma, D. H. A. Blank, H. J. M. Bouwmeester and B. A. Boukamp, J. Power Sources, 2013, 238, 442 CrossRef CAS
- M. Rieu, R. Sayers, M. A. Laguna-Bercero, S. J. Skinner, P. Lenormand and F. Ansart, J. Electrochem. Soc., 2010, 157, B477 CrossRef CAS
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4 CrossRef CAS
- A. V. Virkar, Int. J. Hydrogen Energy, 2010, 35, 9527 CrossRef CAS
- M. A. Laguna-Bercero and V. M. Orera, Int. J. Hydrogen Energy, 2011, 36, 13051 CrossRef CAS
- F. Chauveau, J. Mougin, J. M. Bassat, F. Mauvy and J. C. Grenier, J. Power Sources, 2010, 195, 744 CrossRef CAS
- M. A. Laguna-Bercero, N. Kinadjan, R. Sayers, H. El Shinawi, C. Greaves and S. J. Skinner, Fuel Cells, 2011, 11, 102 CrossRef
- S.-N. Lee, A. Atkinson and J. A. Kilner, ECS Trans., 2013, 57, 605 CrossRef
- A. Montenegro-Hernandez, J. Vega-Castillo, L. Mogni and A. Caneiro, Int. J. Hydrogen Energy, 2011, 36, 15704 CrossRef
- R. Sayers, J. Liu, B. Rustumji and S. J. Skinner, Fuel Cells, 2008, 8, 338 CrossRef
- R. Sayers, J. E. Parker, C. C. Tang and S. J. Skinner, J. Mater. Chem., 2012, 22, 3536 RSC
- B. Philippeau, F. Mauvy, C. Mazataud, S. Fourcade and J.-C. Grenier, Solid State Ionics, 2013, 249–250, 17 CrossRef CAS
- A. V. Kovalevsky, V. V. Kharton, A. A. Yaremchenko, Y. V. Pivak, E. V. Tsipis, S. O. Yakovlev, A. A. Markov, E. N. Naumovich and J. R. Frade, J. Electroceram., 2007, 18, 205 CrossRef CAS
- P. Odier, C. Allanion and J. M. Bassat, J. Solid State Chem., 2000, 153, 381 CrossRef
-
I. C. Vinke, Materials for SOFC, decreasing the operating temperature, Materials Workshop H2FC-Infrastructure, San Sebastian, Spain, March 18 2014, available online at: http://h2fc.eu/files/downloads/workshops/Mat_Workshop/H2FC-Materials-WS_Materials_4_SOFC_Vinke.pdf Search PubMed
- C. Ferchaud, J.-C. Grenier, Y. Zhang-Steenwinkel, M. M. A. van Tuel, F. P. F. van Berkel and J.-M. Bassat, J. Power Sources, 2011, 196, 1872 CrossRef CAS
- L. Fan, M. Chen, C. Wang and B. Zhu, Int. J. Hydrogen Energy, 2012, 37, 19388 CrossRef
- P. Batocchi, F. Mauvy, S. Fourcade and M. Parco, Electrochim. Acta, 2014, 145, 1 CrossRef CAS
- G. Taillades, J. Dailly, M. Taillades-Jacquin, F. Mauvy, A. Essouhmi, M. Marrony, C. Lalanne, S. Fourcade, D. J. Jones, J.-C. Grenier and J. Rozière, Fuel Cells, 2010, 10, 166 Search PubMed
- J. G. Railsback, Z. Gao and S. A. Barnett, Solid State Ionics, 2015, 274, 134 CrossRef CAS
- V. Lawlor, S. Griesser, G. Buchinger, A. G. Olabi, S. Cordiner and D. Meissner, J. Power Sources, 2009, 193, 387 CrossRef CAS
- V. Lawlor, J. Power Sources, 2013, 240, 421 CrossRef CAS
- V. M. Orera, A. Larrea and M. A. Laguna-Bercero, Frontiers in Energy Research, 2014, 2, 22, DOI:10.3389/fenrg.2014.00022
- H. Monzón, M. A. Laguna-Bercero, A. Larrea, B. I. Arias, A. Várez and B. Levenfeld, Int. J. Hydrogen Energy, 2014, 39, 5470 CrossRef
- B. I. Arias, M. E. Sotomayor, A. Várez, B. Levenfeld, H. Monzón, M. A. Laguna-Bercero and A. Larrea, RSC Adv., 2015 Search PubMed
- J. Rodriguez-Carvajal, Physica B, 1993, 192, 55 CrossRef CAS
- M. A. Laguna-Bercero, A. Férriz, A. Larrea, L. Correas and V. M. Orera, Fuel Cells, 2013, 13, 1116 CrossRef
- H. Monzón and M. A. Laguna-Bercero, Int. J. Hydrogen Energy, 2012, 37, 7262 CrossRef
- R. Chiba, H. Taguchi, T. Komatsu, H. Orui, K. Nowaza and H. Arai, Solid State Ionics, 2011, 197, 42 CrossRef CAS
- V. A. Sadykov, N. F. Eremeev, E. M. Sadovskaya, A. S. Bobin, Y. E. Fedorova, V. S. Muzykantov, N. V. Mezentseva, G. M. Alikina, T. A. Kriger, V. D. Belyaev, V. A. Rogov, A. S. Ulikhin, Y. S. Okhlupin, N. F. Uvarov, O. F. Bobrenok, N. McDonald, J. Watton, A. Dhir, R. Steinberger-Wilckens, J. Mertens and I. C. Vinke, Russ. J. Electrochem., 2014, 50, 669 CrossRef CAS
- J. A. Schuler, H. Luebbe, A. Hessler-Wysser and J. van herle, J. Power Sources, 2012, 213, 223 CrossRef
- M. A. Laguna-Bercero, H. Luebbe, J. Silva and J. van herle, Fuel Cells, 2015, 15, 98 CrossRef CAS
- R. Chiba, T. Komatsu, H. Orui, H. Taguchi, K. Nozawa and H. Arai, Electrochem. Solid-State Lett., 2009, 12, B69 CrossRef CAS
- H. Tagguchi, R. Chiba, T. Komatsu, H. Orui, K. Watanabe and K. Hayashi, J. Power Sources, 2013, 241, 768 CrossRef
- S. R. Bishop, T. S. Stefanik and H. L. Tuller, Phys. Chem. Chem. Phys., 2011, 13, 10165 RSC
- Y. Du, C. Finnerty and J. Jiang, J. Electrochem. Soc., 2008, 155, B972 CrossRef CAS
- G. DiGiuseppe and L. Sun, J. Fuel Cell Sci. Technol., 2013, 11, 021007 CrossRef
- X. Zhang, H. Zhang and X. Liu, J. Power Sources, 2014, 269, 412 CrossRef CAS
- Y.-S. Yoo, M. Choi, J.-H. Hwang, H.-N. Im, B. Singh and S.-J. Song, Ceram. Int., 2015, 41, 6448 CrossRef CAS
- M. A. Laguna-Bercero, A. R. Hanifi, H. Monzón, J. Cunningham, T. H. Etsell and P. Sarkar, J. Mater. Chem. A, 2014, 2, 9764 CAS
- J. L. Hudson and T. T. Tsotsis, Chem. Eng. Sci., 1994, 49, 1493 CrossRef CAS
- A. V. Kovalevsky, V. V. Kharton, A. A. Yaremchenko, Y. V. Pivak, E. N. Naumovich and J. R. Frade, J. Eur. Ceram. Soc., 2007, 27, 4269 CrossRef CAS
- M. A. Laguna-Bercero, J. Peña-Martínez, J. Hodges, A. Huq and S. J. Skinner, High temperature phase evolution of Pr2NiO4+δ determined by in-situ neutron diffraction experiments, in preparation.
- M. A. Laguna-Bercero, R. Campana, A. Larrea, J. A. Kilner and V. M. Orera, Fuel Cells, 2011, 11, 116 CrossRef CAS
- A. V. Virkar, Int. J. Hydrogen Energy, 2012, 37, 12609 CrossRef CAS
- Z. Zhang and M. Greenblatt, J. Solid State Chem., 1995, 117, 236 CrossRef CAS
- T. Ishihara, J. Xie, T. Ho Shin, Y.-W. Ju, S. Ida and J. A. Kilner, J. Power Sources, 2015, 275, 167 CrossRef CAS
- R. J. Woolley and S. J. Skinner, J. Power Sources, 2013, 243, 790 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08531d |
| This journal is © The Royal Society of Chemistry 2016 |