
A non-noble metal MoS2–Cd0.5Zn0.5S photocatalyst with efficient activity for high H2 evolution under visible light irradiation†
Shaojian
Zhao
,
Junjian
Huang
,
Qiuyue
Huo
,
Xiaozhou
Zhou
and
Weixia
Tu
*
State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: tuwx@mail.buct.edu.cn
First published on 11th November 2015
Abstract
Here we describe the synthesis and characteristics of MoS2–Cd0.5Zn0.5S, an efficient non-noble metal photocatalyst of H2 evolution from water under visible light irradiation. This new quaternary composite was successfully synthesized by using a two-step hydrothermal method. The formation of a uniform Cd0.5Zn0.5S solid solution through hydrothermal coprecipitation and the subsequent loading of a suitable amount of MoS2 were essential to enhance the catalytic activity of the MoS2–Cd0.5Zn0.5S photocatalyst, which exhibited a synergistic effect for H2 evolution derived from splitting water. This photocatalyst exhibited good photostability and high photocatalytic activity, which was maintained even after being re-used three times. An H2 evolution rate of 12.30 mmol h−1 g−1 was achieved under visible light irradiation from an aqueous solution of lactic acid, indicative of superior water-splitting activity for this noble metal-free photocatalyst.
1. Introduction
The gradual depletion of fossil fuels and serious environmental pollution have in recent times become issues of considerable concern. Photocatalytic technology offers an effective way to transform solar radiant energy into chemical energy,1 and has been considered as an ideal candidate for the replacement of fossil fuels. Since the first report on photocatalytic water splitting was published in 1972,2 hydrogen evolution via photocatalysis has received much attention due to its recycling potential and environmental friendliness.3 Because visible light makes up 40% of the radiant energy in solar light, much effort has been devoted to developing visible-light-responsive photocatalysts (such as CdSe,4 CdS,5 C3N4,6 In2S3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and other composites like CdSe–MoS2,8 (AgIn)xZn2(1−x)S2 solid solution,9 MoS2–CdS,10 CdS/ZnS/In2S311 and nickel-based cocatalysts).12 CdS, with a relatively narrow band gap (2.4 eV), has in particular shown high performance in photocatalytic H2 production under visible light irradiation.13 However, most of the reported photocatalysts have shortcomings such as photocorrosion14 and a reliance on noble metals as co-catalysts,15 which limit their wide application. Therefore, the development of inexpensive photocorrosion-resistant photocatalysts is still needed for H2 evolution.
7 and other composites like CdSe–MoS2,8 (AgIn)xZn2(1−x)S2 solid solution,9 MoS2–CdS,10 CdS/ZnS/In2S311 and nickel-based cocatalysts).12 CdS, with a relatively narrow band gap (2.4 eV), has in particular shown high performance in photocatalytic H2 production under visible light irradiation.13 However, most of the reported photocatalysts have shortcomings such as photocorrosion14 and a reliance on noble metals as co-catalysts,15 which limit their wide application. Therefore, the development of inexpensive photocorrosion-resistant photocatalysts is still needed for H2 evolution.
Some CdS catalysts loaded with non-noble metal compounds, such as MoS2,10c Ni(OH)216 and NiS,17 have been reported to exhibit enhanced H2 evolution rates of 1.47 mmol h−1 g−1, 5.09 mmol h−1 g−1 and 2.18 mmol h−1 g−1, respectively. But CdS loaded with these metal compounds is still not superior to CdS loaded with noble metals, such as the Pt–PbS/CdS photocatalyst, with a high H2 evolution rate of 8.77 mmol h−1 g−1,15b (Zn0.95Cu0.05)0.5Cd0.5S loaded with Pt, exhibiting an H2 evolution rate of 3.63 mmol h−1 g−1,18 and Pt-loaded ZnIn2S4, showing an H2 evolution rate of 8.42 mmol h−1 g−1.19 Therefore, there is a great urgency to develop an ideal visible-light-responsive photocatalyst, i.e., one that is inexpensive as well as highly stable and active. Such ideal properties may be provided by the newly discovered photocatalyst consisting of a solid solution of ZnxCd1−xS. When formed as a solid solution, ZnxCd1−xS is more stable and active than are CdS and ZnS. In an investigation by Li et al.,20 the Cd0.5Zn0.5S solid solution exhibited a high H2 evolution rate of 7.42 mmol h−1 g−1 under visible light irradiation. Many methods have been used to fabricate ZnxCd1−xS solid solutions, such as coprecipitation,21 thermal sulfuration,22 thermolysis,20,23 chemical bath deposition24 and so on. The Zn0.62Cd0.16S obtained through coprecipitation method showed an H2 evolution rate of 2.01 mmol h−1 g−1 under visible light irradiation.21 When prepared by the thermal sulfuration method, the Cd0.2Zn0.8S photocatalyst exhibited an H2 evolution rate of about 0.83 mmol h−1 g−1,22 and the titania nanotube–Cd0.65Zn0.35S nanocomposite also showed a high evolution rate.25 Many doping methods with different metal ions have been considered for improving the photocatalytic activity of the solid solution.26 A ZnxCd1−xS photocatalyst modified with a co-catalyst such as NiOH exhibited an H2 evolution rate of 7.16 mmol h−1 g−1, higher than that other than ZnxCd1−xS photocatalysts.27 In addition, controlling the molar ratio of ZnS to CdS has proven to be an effective way to enhance the photocatalytic activity of ZnxCd1−xS solid solution.20,28 Most of the ZnxCd1−xS photocatalysts synthesized through the above methods show irregular shapes with large and nonuniform sizes. Small and uniform-sized ZnxCd1−xS photocatalysts would be expected to improve the catalytic activities of the solid solutions and of their composited photocatalysts.
Herein, we aimed to achieve a ZnxCd1−xS solid solution with a uniform size and shape as well as high photocatalytic activity via hydrothermal coprecipitation. We also aimed to produce a new quaternary composite MoS2–CdxZn1−xS photocatalyst with superior photocatalytic activity through the subsequent loading of MoS2 as a co-catalyst on the ZnxCd1−xS solid solution. In our work, the addition of potassium hydroxide and a two-step hydrothermal process were employed for the synthesis of uniform MoS2–CdxZn1−xS nanoparticles. The MoS2–Cd0.5Zn0.5S photocatalyst without loaded noble metals was observed to be stable and to exhibit a high H2 production activity of 12.30 mmol h−1 g−1 under visible light irradiation from an aqueous solution of lactic acid. Our new photocatalysis technique efficiently provided a non-noble metal photocatalyst for the H2 evolution reaction.
2. Experimental
2.1. Synthesis of ZnxCd1−xS solid solution
ZnxCd1−xS solid solutions were prepared through a hydrothermal coprecipitation method with different molar ratios of Cd to Zn precursors. One millimole of cetyltrimethyl ammonium bromide (CTAB) was dissolved completely in 80 mL of distilled water at 50 °C, and was cooled to room temperature. Then x mmol cadmium acetate (Cd(CH3COO)2·2H2O), 1 − x mmol zinc acetate (Zn(CH3COO)2·2H2O), 2 mmol thiourea (CN2H4S) and 10 mmol KOH were added in that order into the CTAB solution prepared above with constant stirring (x = 0–1). The mixtures were stirred for 24 h, and then transferred into a Teflon vessel (100 mL), which was placed in a vacuum drying oven and heated at 200 °C for 24 h. The formed turbid liquid was filtered and washed with ethanol several times to remove CTAB and other ions, and the product was dried at 60 °C for 12 h. Finally, the ZnxCd1−xS powder was obtained. CdS and ZnS were also synthesized, using a similar method, for comparing the photocatalytic activities of different photocatalysts.2.2. Synthesis of the MoS2–CdxZn1−xS nanomaterial
The MoS2–Cd0.5Zn0.5S sample was synthesized by using a two-step hydrothermal method. Ten micromoles of ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and 2 mmol of thiourea were (completely) dissolved in 60 mL of distilled water, and then 1 mmol of Cd0.5Zn0.5S obtained from the above hydrothermal coprecipitation method was mixed in. After being ultrasonicated for 30 min, the mixture was transferred into a Teflon vessel (100 mL) and heated in a vacuum drying oven at 200 °C for 23 h. The obtained turbid liquid was filtered and washed with distilled water several times, and then dried under vacuum at 60 °C for 12 h. By varying the amount of the Mo precursor, different MoS2–Cd0.5Zn0.5S samples were prepared. The obtained CdS and ZnS were also loaded with MoS2 by the same hydrothermal method for comparison.2.3. Characterization
X-ray diffraction (XRD), using a Bruker D8 Advance diffractometer (Germany Bruker AXS Ltd.), was employed for phase identification of the powders. The morphology and size of the obtained samples were visualized with a scanning electron microscope (SEM, S-4700F, Japan JEOL Ltd.). Transmission electron microscopy images were taken using a high-resolution transmission electron microscope (HR-TEM, J-3010, Japan JEOL Ltd.). Ultraviolet-visible (UV-Vis) diffuse reflectance spectra were recorded with a UV-Vis spectrophotometer (Tu-1901, Beijing Persee General Instrument Co. Ltd.). X-ray photoelectron spectroscopy (XPS) measurements were taken using an ultrahigh vacuum VG ESCALAB 250 electron spectrometer equipped with a multichannel detector. Element analysis was carried out on an inductively coupled plasma spectrometry-atomic emission spectrometer (ICP-AES, ULTIMA, JY Inc.). A Micromeritics ASAP 2020HD88 nitrogen adsorption apparatus was employed to determine the Brunauer–Emmett–Teller (BET) specific surface areas (SBET) of the samples.2.4. Photocatalytic reaction and quantum efficiency (QE)
A photocatalytic H2 evolution reaction was carried out in a 500 mL photochemical reactor equipped with a magnetic stirrer and cooling system in a dark box. A 500 W Xe lamp was used as a light source for the photocatalytic H2 evolution. For a typical photocatalytic reaction, 60 mg of MoS2–ZnxCd1−xS photocatalyst was dispersed in a 400 mL aqueous solution containing 40 mL of lactic acid as a sacrificial reagent under ultrasonic treatment for 10 min. Then, the mixture was transferred into the photochemical reactor. During the reaction under light irradiation, the reaction solutions were maintained at room temperature with cooling water. The amount of hydrogen evolved during the reaction was analyzed and determined by using gas chromatography (TDX-01 molecular sieve column, TCD, N2 carrier).For comparison, another experiment for H2 production was carried out in a 50 mL Pyrex flask sealed with stopper-rubber. A 300 W Xe lamp with a UV-cutoff filter (λ ≥ 420 nm) was used as the visible light source. In contrast to the above experiment, 3 mg of MoS2–ZnxCd1−xS photocatalyst was dispersed in a 20 mL aqueous solution containing 2 mL of lactic acid in this experiment. The quantum efficiency (QE) was measured under the same photocatalytic reaction conditions, except that a monotone filter (λ = 420 nm) was used instead of the UV-cutoff filter. The average intensity of irradiation and the irradiation area were determined to be 31.69 mW cm−2 and 16.25 cm2, respectively. The QE was calculated according to the following equation:
| QE = (number of reacted electrons)/(number of incident photons) × 100% |
| = (number of evolved H2 molecules × 2)/(number of incident photons) × 100% |
3. Results and discussion
Using the hydrothermal coprecipitation method with the aid of potassium hydroxide, ZnxCd1−xS solid solutions were prepared and used for the subsequent hydrothermal syntheses of MoS2–ZnxCd1−xS photocatalyst. MoS2–Cd0.5Zn0.5S samples were obtained with the starting reactant molar ratios of 1:1 for Cd/Zn, 10:1 for KOH/(Cd + Zn), and 7.5:100 for Mo/(Cd + Zn) and were examined first. Their morphologies and crystal structures were characterized with electron microscopy and X-ray diffraction. Fig. 1a and b show SEM images of Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S, respectively. The obtained Cd0.5Zn0.5S particles were observed to be spherical with a uniform size of about 14 nm, without any evidence of adhesiveness and agglomerations. Thus, the present hydrothermal coprecipitation method provides a way to synthesize uniform Cd0.5Zn0.5S nanoparticles that are superior to the product obtained by other methods reported. Compared with the Cd0.5Zn0.5S nanoparticles, the obtained MoS2–Cd0.5Zn0.5S particles were observed to be a little bit larger, with some of them showing a distorted petal shape. These different observed sizes and shapes indicate that MoS2 may be generated on the surface of Cd0.5Zn0.5S particles.
 | ||
| Fig. 1 SEM images of Cd0.5Zn0.5S (a) and MoS2–Cd0.5Zn0.5S (b); HRTEM image of MoS2–Cd0.5Zn0.5S (c); X-ray diffraction patterns of Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S (d). | ||
An HRTEM image of the MoS2–Cd0.5Zn0.5S particles is shown in Fig. 1c. Petal-shaped MoS2 lamellas were observed to be generated on Cd0.5Zn0.5S nanoparticles, which formed separate and distorted lamellar shells without overlap. A repeat distance of 2.40 Å was observed (Fig. 1c), which is close to the distance between the (102) crystallographic planes in the Cd0.5Zn0.5S solid solution (Fig. 1d). The distance between lamellas on the surface of Cd0.5Zn0.5S nanoparticles was observed to be about 2.72 Å, corresponding to the (100) plane of the MoS2 nanosheets. The above SEM and HRTEM results indicate that MoS2 formed and combined with the Cd0.5Zn0.5S solid solution, i.e., quaternary MoS2–Cd0.5Zn0.5S nanoparticles formed. Energy dispersive X-ray spectrometry (EDS) mapping analysis of MoS2–Cd0.5Zn0.5S confirmed a homogenous distribution of the Cd, Zn, Mo, and S elements. Similarly, the EDS mapping analysis of Cd0.5Zn0.5S also confirmed a homogenous distribution of the Cd, Zn, and S elements (ESI, Fig. S1†).
Fig. 1d shows the XRD patterns of Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S nanoparticles. The XRD patterns of the two samples are in good agreement with JCPDS file no. 89-2943, indicating a spinel phase Cd0.5Zn0.5S solid solution. Well-ordered diffraction peaks corresponding to the (100) (002) (101) (102) (110) (103) (112) (203) (211) (300) planes of Cd0.5Zn0.5S were observed, with no other peaks present. The positions of these peaks were for each case at slightly lower two-theta values than those in the JCPDS file, indicative of a slightly enlarged crystal lattice constant, which is in agreement with the difference between the HRTEM image of MoS2–Cd0.5Zn0.5S and that of Cd0.5Zn0.5S. There is no great difference between the XRD pattern of MoS2–Cd0.5Zn0.5S and that of Cd0.5Zn0.5S particles. The former (as well as of course the latter) only showed the diffraction peaks of Cd0.5Zn0.5S particles, with no MoS2 diffraction peak observed. This observation can be attributed to the single-layer structure of MoS2.29
To gain deeper insight into the bulk compositions of the MoS2–Cd0.5Zn0.5S photocatalyst, a quantitative elemental characterization on MoS2–Cd0.5Zn0.5S was conducted by using ICP. The metal elements Cd, Zn, and Mo were detected in MoS2–Cd0.5Zn0.5S using this method. And these metals made up, respectively, 45.67 mol%, 46.17 mol%, and 8.16 mol% of the total metal content, consistent with an approximately 1:1 molar ratio of Cd to Zn and metal molar ratios similar to those of the starting materials.
X-ray photoelectron spectroscopy (XPS) can be used to help characterize the chemical states of nanomaterials. Fig. 2 shows the XPS spectra of MoS2–Cd0.5Zn0.5S and Cd0.5Zn0.5S. The binding energy (BE) scales were referenced by setting the C 1s BE to 285.0 eV. The BEs of Cd 3d5/2 and Cd 3d3/2 for Cd0.5Zn0.5S were found to be 405.1 and 412.0 eV, respectively, slightly higher than those of bulk. For MoS2–Cd0.5Zn0.5S, the BEs of Cd 3d shifted to values 0.1 eV higher than those of Cd0.5Zn0.5S. In contrast, the BEs of Zn 2p for MoS2–Cd0.5Zn0.5S shifted to values 0.4 eV lower than those of Cd0.5Zn0.5S, for which the BEs of Zn 2p were observed to be 1022.6 and 1045.7 eV. These differences may be due to the differences in the chemical states of the Zn and Cd atoms in the MoS2–Cd0.5Zn0.5S sample relative to those of the Cd0.5Zn0.5S sample. The BE peaks of S 2p for MoS2–Cd0.5Zn0.5S were observed at 162.9 and 161.9 eV, lower than that of plain sulfur, and an S 2p1/2 peak emerged. The altered S peaks probably resulted from the presence of Mo. The BEs of Mo 3d5/2 and Mo 3d3/2 centred at 228.5 and 232.4 eV, respectively. All of the XPS results indicated the involvement of positive-valence Cd, Zn and Mo metallic states and negative-valence sulfur, providing evidence for the formation of MoS2–Cd0.5Zn0.5S and Cd0.5Zn0.5S. Moreover, the changed chemical states of Zn and Cd in the MoS2–Cd0.5Zn0.5S atoms described above indicates an obvious interaction between MoS2 and Cd0.5Zn0.5S.
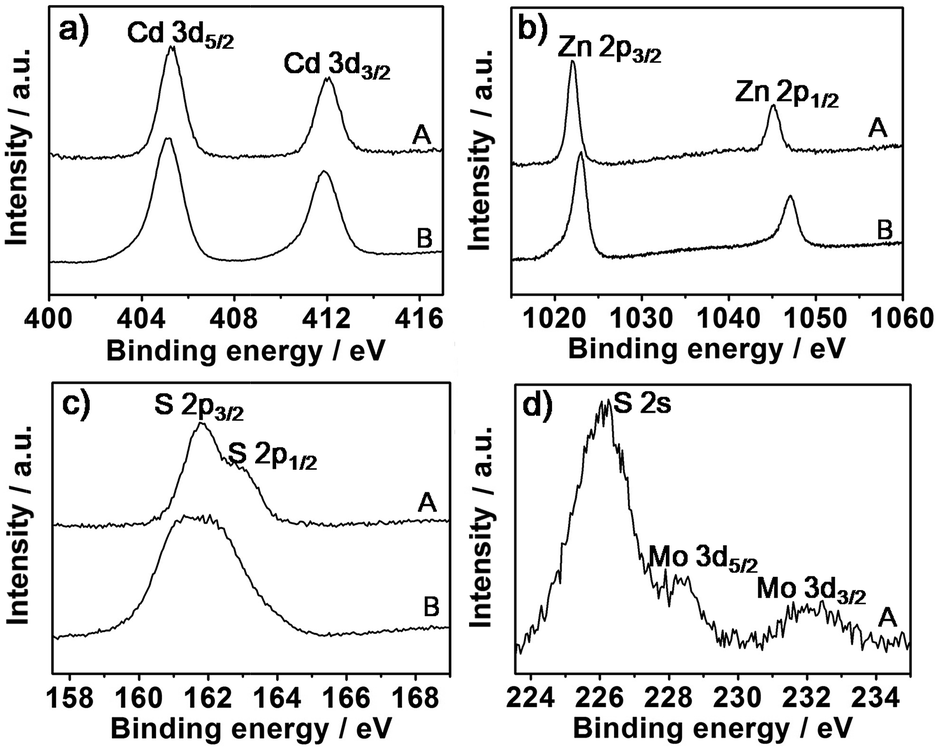 | ||
| Fig. 2 XPS spectra of MoS2–Cd0.5Zn0.5S (A) and Cd0.5Zn0.5S (B) with Cd 3d (a), Zn 2p (b), S 2p (c) and Mo 3d (d) binding energies. | ||
Diffuse reflectance spectroscopy is a useful tool to characterize the optical properties of photocatalysts. Fig. 3 shows the diffuse reflectance UV-Vis spectra of Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S nanoparticles. The inset was obtained by using the Kubelka–Munk function.30 The band gap Eg of each sample was obtained by extrapolating the linear relation to [Ahν]0.5 = 0. The optical absorption edge of the Cd0.5Zn0.5S nanoparticles was observed to be about 540 nm, corresponding to a band gap of 2.30 eV (Fig. 3). Furthermore, the MoS2–Cd0.5Zn0.5S nanoparticles yielded a band gap of 2.25 eV with an optical absorption edge of about 551 nm. Taken together, our results reveal the uniform Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S nanoparticles to be responsive to visible light, and can be applied in visible light catalytic reactions.
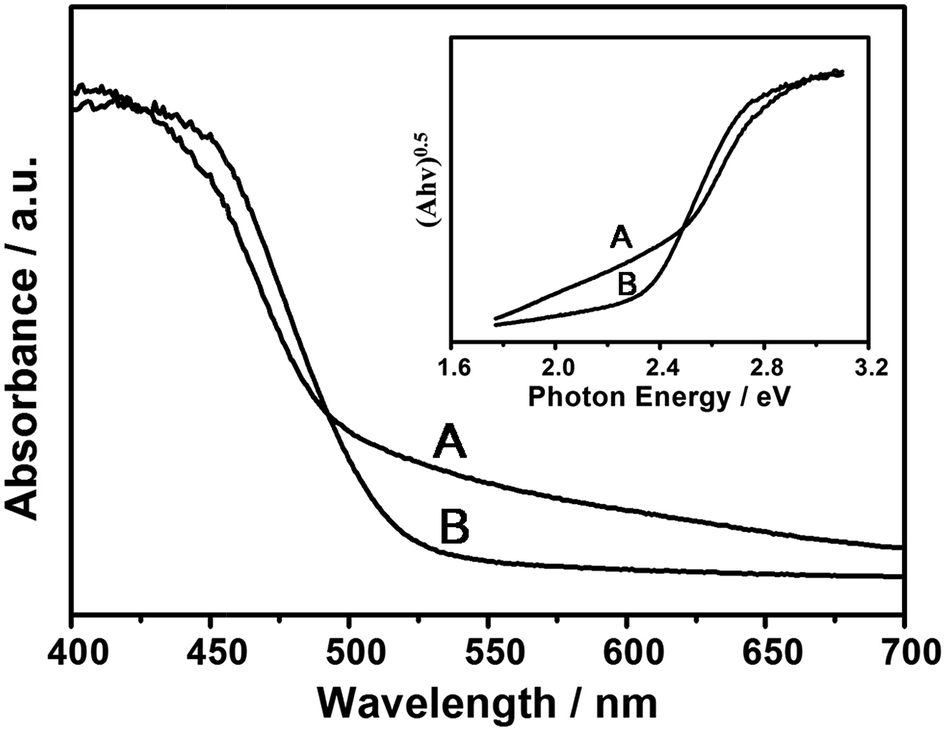 | ||
| Fig. 3 Diffuse-reflectance UV-Vis spectroscopy and the resulting band gap (inset) of MoS2–Cd0.5Zn0.5S (A) and Cd0.5Zn0.5S (B). | ||
Fig. 4 shows the nitrogen adsorption–desorption isotherms and pore-size distribution curves (inset) for the Cd0.5Zn0.5S solid solution and the MoS2–Cd0.5Zn0.5S composite. Both Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S were observed here to have type IV isotherms. According to the Brunauer–Deming–Deming–Teller (BDDT) classification, the hysteresis loops of type H2 at a relative pressure range of 0.7–1 and 0.8–1 for the Cd0.5Zn0.5S solid solution and the MoS2–Cd0.5Zn0.5S composite indicate the formation of mesopores of Cd0.5Zn0.5S and mesopores as well as macropores of MoS2–Cd0.5Zn0.5S. The Cd0.5Zn0.5S solid solution exhibited a narrow distribution of pore sizes, and peaks in the range of 5–30 nm with a peak pore diameter of 14 nm (inset in Fig. 4). The MoS2–Cd0.5Zn0.5S composite exhibited a broad distribution of pore sizes, and peaks in the range of 5–120 nm with a peak pore diameter of 50 nm. The specific surface areas of Cd0.5Zn0.5S solid solution and MoS2–Cd0.5Zn0.5S composite were determined to be 26.7 m2 g−1 and 38.4 m2 g−1, respectively. Both the pore sizes and specific surface areas of Cd0.5Zn0.5S increased when it was combined with MoS2.
 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms and the corresponding pore-size distribution curves (inset) of Cd0.5Zn0.5S and MoS2–Cd0.5Zn0.5S. | ||
In contrast to many other methods for synthesizing Cd0.5Zn0.5S solid solution reported in the literature,20-22 we introduced KOH into the synthesis reaction system. KOH was expected to promote the formation of uniform ZnxCd1−xS nanoparticles with high photocatalytic activity, and this expectation was based on the following considerations. First, adding KOH would increase the alkalinity of the reaction system and thus speed up the formation of the metal hydroxide and its colloidal solution.31 Secondly, sulphions would be easily generated from the hydrolysis reaction of thiourea in alkaline solution. The sulphions would react quickly with the hydroxides to form metal sulfides for the subsequent transformation into a solid solution with the assistance of a hydrothermal process. These two expected effects were observed in our experiments. Adding KOH was found to favor the formation of cadmium and zinc hydroxide colloidal solutions and accelerate the formation of their sulfides (ESI, Fig. S2†). That is, as indicated in Fig. 1, KOH actively effects the formation of a homogeneous ZnxCd1−xS solid solution and MoS2–ZnxCd1−xS nanoparticles.
In the preparation of the Cd0.5Zn0.5S nanoparticles, several different MoS2–Cd0.5Zn0.5S samples were synthesized with varying amounts of KOH in the preparation process based on the presence of Cd0.5Zn0.5S solid solution. The synthesized MoS2–Cd0.5Zn0.5S quaternary composites made with these different amounts of KOH were used as photocatalysts in the water splitting reaction for H2 evolution. The molar ratio of KOH to metal precursors (the total amount of Cd and Zn) varied between 2.1 and 20. Fig. 5a shows the rate of H2 evolution from water over the MoS2–Cd0.5Zn0.5S photocatalyst synthesized with the aid of KOH. The amount of KOH had a significant effect on the photocatalytic activity of MoS2–Cd0.5Zn0.5S. An optimum amount of KOH was necessary for improving photocatalytic activity of the MoS2–Cd0.5Zn0.5S photocatalyst. When the value of the molar ratio of KOH to Cd and Zn metal precursors was 10, the synthesized MoS2–Cd0.5Zn0.5S exhibited superior photocatalytic activity with a high H2 evolution rate of 12.30 mmol h−1 g−1 for a duration of five hours.
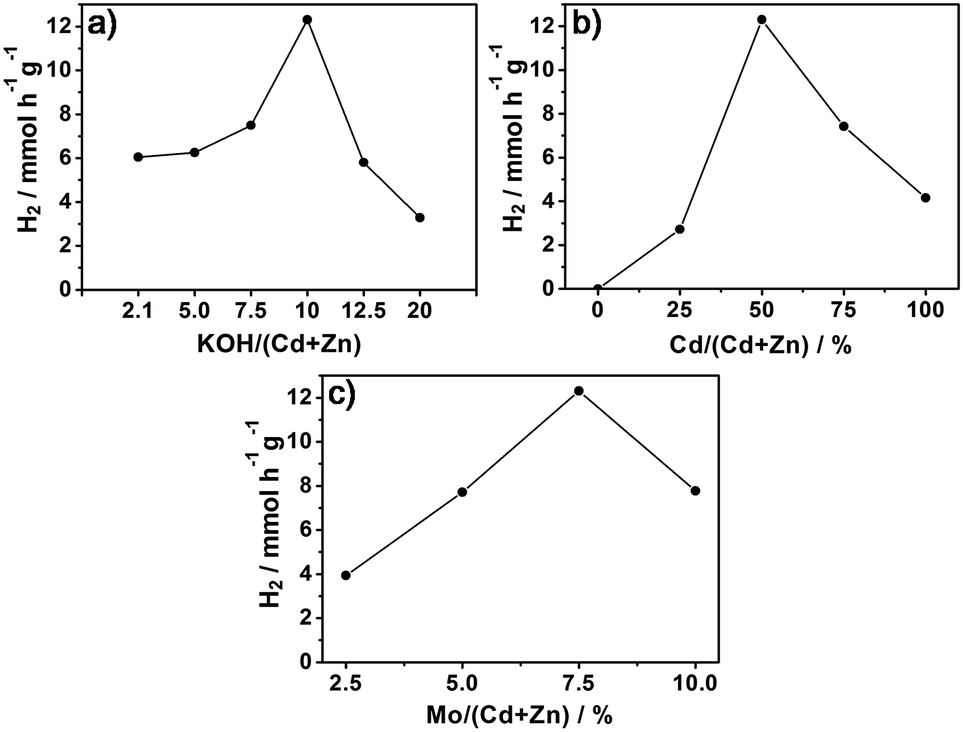 | ||
| Fig. 5 The dependence of the MoS2–Cd0.5Zn0.5S-catalyzed rate of H2 evolution on the MoS2–Cd0.5Zn0.5S synthesis conditions, including the molar ratios of KOH/(Cd + Zn) (a), Cd/(Cd + Zn) (b), and Mo/(Cd + Zn) (c). | ||
Different molar ratios of Zn to Cd precursors were also tested for their effects on the formation of the ZnxCd1−xS solid solution. The catalytic activities of the corresponding synthesized MoS2–ZnxCd1−xS samples are given in Fig. 5b. High H2 evolution over the MoS2–Cd0.5Zn0.5S photocatalyst was observed here when the molar ratio of Cd to Zn was 1:1. In addition, the Mo precursor was reacted with thiourea in the presence of Cd0.5Zn0.5S solid solution during the second hydrothermal process. The molar amount of Mo precursor was varied from 2.5% to 10%. Fig. 5c shows the H2 evolution results for the case of xMoS2–Cd0.5Zn0.5S photocatalysts prepared from different amounts of Mo precursor, ranging from 2.5% to 10%. The optimum molar percentage of Mo was found to be 7.5%. Similarly, an appropriate amount of the used metal precursors was preferred. In summary, an efficient MoS2–Cd0.5Zn0.5S photocatalyst can be synthesized through a two-step hydrothermal method when using optimum reaction conditions, i.e., with the molar ratios of Cd/Zn, KOH/(Cd + Zn) and Mo/(Cd + Zn) being set to 1:1, 10:1 and 7.5:100, respectively. The highest rate of H2 evolution observed over a duration of five hours was 12.30 mmol h−1 g−1. For comparison with experimental conditions used in other published investigations, the catalytic activity of the MoS2–Cd0.5Zn0.5S photocatalyst was also examined when irradiated under a 300 W Xe lamp. A high H2 evolution rate of 11.49 mmol h−1 g−1 was determined from the lactic acid solution. The corresponding apparent quantum efficiency (AQE) at 420 nm reached 1.34% for the MoS2–Cd0.5Zn0.5S photocatalyst.
In order to demonstrate the superior photocatalytic activity of MoS2–Cd0.5Zn0.5S for H2 production, pure CdS, ZnS, MoS2, as well as the MoS2–ZnS and MoS2–CdS composites were synthesized using the same method for comparison. Photocatalytic reactions for H2 evolution over the obtained different photocatalysts were carried out. ZnS, MoS2 and MoS2–ZnS hardly exhibited any photocatalytic activity for H2 production, and CdS had weak photocatalytic activity for H2 production, which was barely detected quantitatively. In contrast, as shown in Fig. 6a, MoS2–CdS and Cd0.5Zn0.5S exhibited relatively high photocatalytic H2 production activities of 4.15 mmol h−1 g−1 and 4.30 mmol h−1 g−1, respectively. Notably, MoS2–Cd0.5Zn0.5S delivered the highest photocatalytic activity with an H2 evolution rate of 12.30 mmol h−1 g−1, which was almost three times higher than those of MoS2–CdS or Cd0.5Zn0.5S. The results revealed that the MoS2–Cd0.5Zn0.5S quaternary photocatalyst exhibited a synergistic effect, which can improve H2 evolution in water splitting reactions. In fact, it was the combination of MoS2 and the Cd0.5Zn0.5S solid solution that provided the synergy rather than the combination of MoS2, CdS and ZnS (see ESI Fig. S3†).
 | ||
| Fig. 6 Photocatalytic activities of different photocatalysts (a); and H2 evolution for different recycling times of MoS2–Cd0.5Zn0.5S photocatalysts (b). | ||
Therefore, MoS2 was determined to play an important role in the high H2 evolution for the MoS2–Cd0.5Zn0.5S photocatalyst as well as in improving the photocatalytic activity of Cd0.5Zn0.5S. Both the average pore diameter and the specific surface area of MoS2–Cd0.5Zn0.5S were determined by using BET to be larger than those of Cd0.5Zn0.5S, indicating the role of MoS2 in changing the structure of the Cd0.5Zn0.5S solid solution. The improved pore size and surface area of the MoS2–Cd0.5Zn0.5S composites yielded improved adsorption and consequently the enhancement of the photocatalytic activity. As Low and co-works reported,32 MoS2 nanosheets could be used as a promising material for photocatalytic reactions. Their theoretical calculations and experimental results showed MoS2 to be able to activate H2, and showed the conduction band (CB) potential of nanoscale MoS2 to be about −0.20 eV versus the normal hydrogen electrode potential (NHE) because of the quantum confinement effect.33 An empirical equation (ECB = χ − EC − 0.5Eg) could be employed to determine the CB edge position of the prepared Cd0.5Zn0.5S solid solution. In the equation, ECB is the CB edge potential and χ is the electronegativity of the semiconductor, while EC is the energy of free electrons on the hydrogen scale (∼4.5 eV), and Eg is the direct band gap of the semiconductor. So the ECB of the Cd0.5Zn0.5S solid solution was calculated to be −0.294 eV, which is more negative than that of MoS2. Therefore, the photogenerated electrons may transfer from the CB of Cd0.5Zn0.5S to the CB of MoS2. Fig. 7 shows the CB and VB edge potentials of the Cd0.5Zn0.5S solid solution and MoS2. A possible mechanism of the MoS2–Cd0.5Zn0.5S photocatalyst for the high H2 evolution may be proposed. When the MoS2–Cd0.5Zn0.5S photocatalyst was irradiated under visible light, forming electron–hole pairs, photogenerated electrons were transferred from the Cd0.5Zn0.5S solid solution to MoS2 being used to evolve H2, and the holes oxidized the sacrificial agents.10a As a result, the co-catalyst MoS2 accelerated the separation of electron–hole pairs, which is a significant factor for photocatalytic reactions, and restrained the recombination of electron–hole pairs, contributing a high H2 evolution for the MoS2–Cd0.5Zn0.5S photocatalyst. However, when excess MoS2 was loaded, most of the surface of the Cd0.5Zn0.5S solid solution may have been covered by MoS2 nanosheets, decreasing the number of the electron–hole pairs that were formed, or the excess MoS2 nanosheets may have become recombination centres for photogenerated electrons–holes pairs, leading to a low photoactivity,34 which was observed, as shown in Fig. 5c.
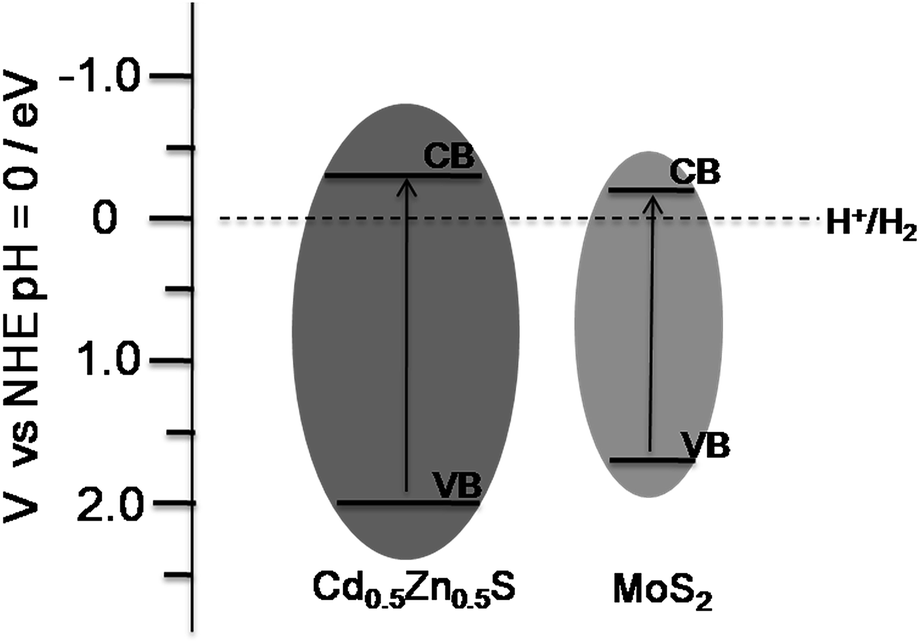 | ||
| Fig. 7 Schematic illustration of the potential and band positions of the Cd0.5Zn0.5S solid solution and MoS2. | ||
Also, the MoS2–Cd0.5Zn0.5S photocatalyst was reused in the catalytic reaction to evaluate its catalytic stability. Photocatalytic results of recycling the catalyst three times are shown in Fig. 6b. MoS2–Cd0.5Zn0.5S displayed very stable photocatalytic behaviour with high H2 evolution even when re-used three times.
Taken together, we achieved a visible-light-responsive non-noble metal MoS2–Cd0.5Zn0.5S photocatalyst with good photochemical stability and a five-hour duration of photocatalytic activity for H2 evolution of up to 12.30 mmol h−1 g−1.
4. Conclusions
In summary, Cd0.5Zn0.5S solid solution nanoparticles with uniform shape and small average sizes of 14 nm were synthesized by using the hydrothermal coprecipitation method. The obtained Cd0.5Zn0.5S nanoparticles showed a higher activity than those prepared by other methods reported. Based on the Cd0.5Zn0.5S solid solution, a new quaternary composite, MoS2–Cd0.5Zn0.5S, was formed successfully via the growth of MoS2 by using this hydrothermal method. The molar ratios of three metal precursors were optimized. The addition of a suitable amount of KOH and the formation of the Cd0.5Zn0.5S solid solution were found to be essential for achieving the small-sized and highly active MoS2–Cd0.5Zn0.5S photocatalyst. Over the MoS2–Cd0.5Zn0.5S photocatalyst, a high rate of hydrogen evolution from water of 12.30 mmol h−1 g−1 was achieved under visible light irradiation. The MoS2–Cd0.5Zn0.5S photocatalyst exhibited superior catalytic activity and photochemical stability, and hence provides an efficient non-noble metal photocatalyst for many promising applications in the field of clean energy.Acknowledgements
Financial support from the National Natural Science Foundation of China (21106006) is acknowledged.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(43), 15729–15735 CrossRef CAS PubMed.
- A. Fujishima, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- (a) Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414(6864), 625–627 CrossRef CAS PubMed; (b) X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2014, 3, 2485–2534 RSC.
- F. A. Frame, E. C. Carroll, D. S. Larsen, M. Sarahan, N. D. Browning and F. E. Osterloh, Chem. Commun., 2008, 19, 2206–2208 RSC.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133(28), 10878–10884 CrossRef CAS PubMed.
- (a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8(1), 76–80 CrossRef CAS PubMed; (b) X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117(19), 9952–9961 CrossRef CAS; (c) S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5(12), 2101–2107 CrossRef CAS PubMed.
- (a) W. Qiu, M. Xu, X. Yang, F. Chen, Y. Nan, J. Zhang, H. Iwai and H. Chen, J. Mater. Chem., 2011, 21(35), 13327–13333 RSC; (b) X. An, J. C. Yu, F. Wang, C. Li and Y. Li, Appl. Catal., B, 2013, 129, 80–88 CrossRef CAS.
- F. A. Frame and F. E. Osterloh, J. Phys. Chem. C, 2010, 114(23), 10628–10633 CAS.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126(41), 13406–13413 CrossRef CAS PubMed.
- (a) X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130(23), 7176–7177 CrossRef CAS PubMed; (b) J. Zhang, Z. Zhu and X. Feng, Chem.–Eur. J., 2014, 20(34), 10632–10635 CrossRef CAS PubMed; (c) J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., 2015, 127(4), 1226–1230 CrossRef; (d) J. Z. Chen, X. J. Wu, L. S. Yin, B. Li, X. Hong, Z. X. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 54(4), 1210–1214 CrossRef CAS PubMed; (e) J. Xiong, Y. Liu, D. Wang, S. Liang, W. Wu and L. Wu, J. Mater. Chem. A, 2015, 3(24), 12631–12635 RSC.
- Z. Shen, G. Chen, Q. Wang, Y. Yu, C. Zhou and Y. Wang, Nanoscale, 2012, 4(6), 2010–2017 RSC.
- Y. Xu and R. Xu, Appl. Surf. Sci., 2015, 351, 779–793 CrossRef CAS.
- (a) M. Matsumura, S. Furukawa, Y. Saho and H. Tsubomura, J. Phys. Chem. C, 1985, 89(8), 1327–1329 CrossRef CAS; (b) J. F. Reber and M. Rusek, J. Phys. Chem. C, 1986, 90(5), 824–834 CrossRef CAS.
- (a) R. Williams, J. Chem. Phys., 1960, 32(5), 1505–1514 CrossRef CAS; (b) A. J. Frank and K. Honda, J. Phys. Chem., 1982, 86(11), 1933–1935 CrossRef CAS.
- (a) H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266(2), 165–168 CrossRef CAS; (b) C. Huang and Q. Xu, J. Mater. Chem. A, 2015, 3(26), 13884–13891 RSC.
- J. Ran, J. Yu and M. Jaroniec, Green Chem., 2011, 13(10), 2708–2713 RSC.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46(40), 7631–7633 RSC.
- W. Zhang, Z. Zhong, Y. Wang and R. Xu, J. Phys. Chem. C, 2008, 112(45), 17635–17642 CAS.
- B. Chai, T. Peng, P. Zeng, X. Zhang and X. Liu, J. Phys. Chem. C, 2011, 115(13), 6149–6155 CAS.
- Q. Li, H. Meng, P. Zhou, Y. Zheng, J. Wang, J. Yu and J. Gong, ACS Catal., 2013, 3(5), 882–889 CrossRef CAS.
- C. Xing, Y. Zhang, W. Yan and L. Guo, Int. J. Hydrogen Energy, 2006, 31(14), 2018–2024 CrossRef CAS.
- K. Zhang, D. Jing, C. Xing and L. Guo, Int. J. Hydrogen Energy, 2007, 32(18), 4685–4691 CrossRef CAS.
- Y. M. Sung, Y. J. Lee and K. S. Park, J. Am. Chem. Soc., 2006, 128(28), 9002–9003 CrossRef CAS PubMed.
- S. Al Kuhaimi and Z. Tulbah, J. Electrochem. Soc., 2000, 147(1), 214–218 CrossRef.
- J. Li, L. Wu, L. Long, M. Xi and X. Li, Appl. Surf. Sci., 2014, 322, 265–271 CrossRef CAS.
- K. Zhang, D. Jing, Q. Chen and L. Guo, Int. J. Hydrogen Energy, 2010, 35(5), 2048–2057 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu and S. Z. Qiao, ChemSusChem, 2014, 7(12), 3426–3434 CrossRef CAS PubMed.
- D. Han, J. Cao, S. Yang, J. Yang, B. Wang, L. Fan, Q. Liu, T. Wang and H. Niu, Dalton Trans., 2014, 43(28), 11019–11026 RSC.
- (a) H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., 2010, 122(24), 4153–4156 CrossRef; (b) H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49(24), 4059–4062 CrossRef PubMed; (c) K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105(13), 136805 CrossRef PubMed.
- M. Nowak, B. Kauch and P. Szperlich, Rev. Sci. Instrum., 2009, 80(4), 046107 CrossRef CAS PubMed.
- (a) Y. Wang, J. Ren, K. Deng, L. Gui and Y. Tang, Chem. Mater., 2000, 12(6), 1622–1627 CrossRef CAS; (b) J. Zhang, Y. Wang, H. Ji, Y. Wei, N. Wu, B. Zuo and Q. Wang, J. Catal., 2005, 229(1), 114–118 CrossRef CAS.
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50(74), 10768–10777 RSC.
- (a) B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127(15), 5308–5309 CrossRef CAS PubMed; (b) Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134(15), 6575–6578 CrossRef CAS PubMed.
- Y. Min, G. He, Q. Xu and Y. Chen, J. Mater. Chem. A, 2014, 2(8), 2578–2584 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08365f |
| This journal is © The Royal Society of Chemistry 2016 |