
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid materials from organic electronic conductors and synthetic-lignin models for charge storage applications†
Tomasz
Rębiś‡
ab,
Ting Yang
Nilsson‡
a and
Olle
Inganäs
*a
aBiomolecular and Organic Electronics, IFM, Linköping University, S-581 83 Linköping, Sweden. E-mail: oling@ifm.liu.se
bInstitute of Chemistry and Technical Electrochemistry, Faculty of Chemical Technology, Poznań University of Technology, Berdychowo 4, 60-965 Poznań, Poland
First published on 4th January 2016
Abstract
Homopolymers and copolymers of the monolignols syringol (S) and guaiacol (G) were prepared as well-defined lignin model compounds. Polymerisation was performed by phenol–formaldehyde condensation, also including the monomer hydroquinone (HQ) to extend the range of redox processes in these synthetic lignins (SLig). The chemical structures of the SLig samples were characterized by 13C and quantitative 31P NMR, and the molecular weight was monitored by size exclusion chromatography (SEC). Subsequently, SLig were incorporated into two different electron-conducting matrix – single-wall carbon nanotubes (SWNT) and polypyrrole (PPy), respectively. As a result, the hybrid materials, with a controlled amount of SWNT or with an unknown amount of PPy, were assembled and compared. The charge storage properties in the investigated materials are attributed to contributions from both the double-layer capacitance of the conducting matrix, and the faradaic reactions provided by quinone groups immobilized in the electrodes. The results indicate a considerable improvement of charge capacity, with the synthetic lignins incorporated in the hybrid materials. With a PPy carrying S, G and HQ, better performance is obtained than has previously been obtained with lignin derivatives, showing a maximum capacity of 94 mA h g−1. Moreover, a low amount of electronic conductor (20% wt of SWNT) is adequate to perform efficient electron communication between redox active quinones and the electrode surface, providing 72 mA h g−1.
Introduction
A growing demand exists for the efficient and high charge density storage devices. Hence, the development of inexpensive, high-performance batteries or supercapacitors for large scale applications is a challenge. This is essential for sustainable energy delivery for intermittent solar or wind energy sources.1,2 Electroactive organic compounds or polymers involving reversible redox reactions are considered promising candidates as electrode materials for charge storage devices. Among large diversity of suitable organics, those containing quinones have received the greatest attention, since they have high theoretical capacity, high electron transfer kinetics, excellent redox reversibility and are of low cost.3–5 Much work has been done on the synthesis and application of more effective quinone or carbonyl based materials for lithium-ion batteries as well as supercapacitors.6–9 In the case of supercaps, the introduction of redox active moieties into the electrode matrix is a simple strategy to enhance the capacitance through the reversible faradaic reactions.10,11Interestingly, an unlimited quantity of quinone precursors occurs naturally in plants, for example in the form of biopolymers consisting of aromatic hydroxyphenolic or methoxyphenolic groups.12 Thus, one of the most attractive sources of quinones seems to be lignin which is the second most abundant natural polymer, binding cellulose fibers together in plants.13 The lignin derivatives from the pulping industry are delivered as complex polyphenols14 with significantly varying chemical and physical properties,15 depending on biological sources and chemical process history. The aromatic phenylpropane moieties of lignin can be converted into quinones by electrochemical redox, and thereby form an electroactive group introducing charge storage capacity in lignin.16 The two-electron redox reaction of the quinones makes lignin a material of choice to prepare energy storage systems with increased specific charge storage density. Hence, lignin derivatives have met with increasing research interest for energy storage applications in recent years.17 In order to increase the loading of quinones and retain facile electron transfer between reversible redox couples of non-conducting phenols and electrode surface, there is considerable interest in the incorporation of lignin derivatives into conducting polymer films17,18 or different carbon materials such as multi-walled carbon nanotubes19 or reduced graphene oxide.20
Our earlier research showed the charge capacity of polypyrrole to increase, from ca. 29 mA h g−1 to ca. 79 mA h g−1 at 1 A g−1 discharge rate, after introducing lignosulfonate in the system.17 With other choices of lignin derivatives, lower values were obtained, as the (sub) tropical alkaline lignin/polypyrrole (Lig/PPy) system with more phenolic amount than lignosulfonate showed a lower charge capacity <45 mA h g−1 at 1 A g−1 current.21 Recent work on alkaline lignins report enhanced capacity with polypyrrole.22 Adding extra quinones during the formation of the hybrid material can enhance charge capacity.23 This can also be done by adding different non-quinone redox species.24 The complexity of polypyrrole chain configuration and conformation,25 together with the ill-defined lignin derivatives with undefined structure,26 reactivity27 and purity,28,29 brought many uncertain parameters that may affect the charge capacity of the Lig/PPy system.
Herein, a series of well-defined lignin models were polymerized via phenol–formaldehyde condensation. Two of the main phenols from lignin, syringol and guaiacol, were used to mimic the natural hardwood and softwood lignin; hydroquinone was introduced to the system to extend the redox potential window and to add a quinone. The chemical structure, phenol reactivity and purity of the synthetic-lignin (SLig) derivatives were obtained by NMR and SEC characterization. Furthermore, we demonstrate the assembly and charge storage properties of two hybrid systems: SLig/single-wall carbon nanotubes (SLig/SWNT) with a controlled amount of SWNT and SLig/polypyrrole (SLig/PPy) with an unknown amount of PPy in the final material. PPy and SWNT both store charge and enable a fast electronic communication between the quinone sites of SLig and the electrode surface. In order to optimize the electrode performance of the SLig/SWNT, the mass ratio was varied in the range between 50 and 83 (wt%). In addition, the relationship between the chemical structure of SLig and the electrochemical properties of SLig/PPy and SLig/SWNT is also revealed. The amount of phenolic content, bonding between phenolic groups and types of phenolic groups were correlated to the electrochemical properties of the SLig/PPy and SLig/SWNT hybrid electrodes in a comparative analysis. With the best of these SLig polymers, we attain charge density values superior to the hybrids of polypyrrole and lignosulfonate.
Results and discussion
Characterization of the synthetic lignin derivatives by NMR and SEC
Syringol (S), guaiacol (G) and hydroquinone (HQ) monomers were chosen to prepare synthetic lignin derivatives via condensation reaction with formaldehyde (Scheme 1). Two types of homopolymers, PolyS and PolyG, were synthesized by syringol (PolyS) and guaiacol (PolyG), respectively. Two types of copolymers were synthesized by copolymerizing syringol and guaiacol (PolySG); and copolymerizing syringol, guaiacol and hydroquinone (PolySGHQ). The chemical structure of the synthetic lignin derivatives were characterized by 1H, 31P and 13C NMR, and the molecular weights were monitored by SEC.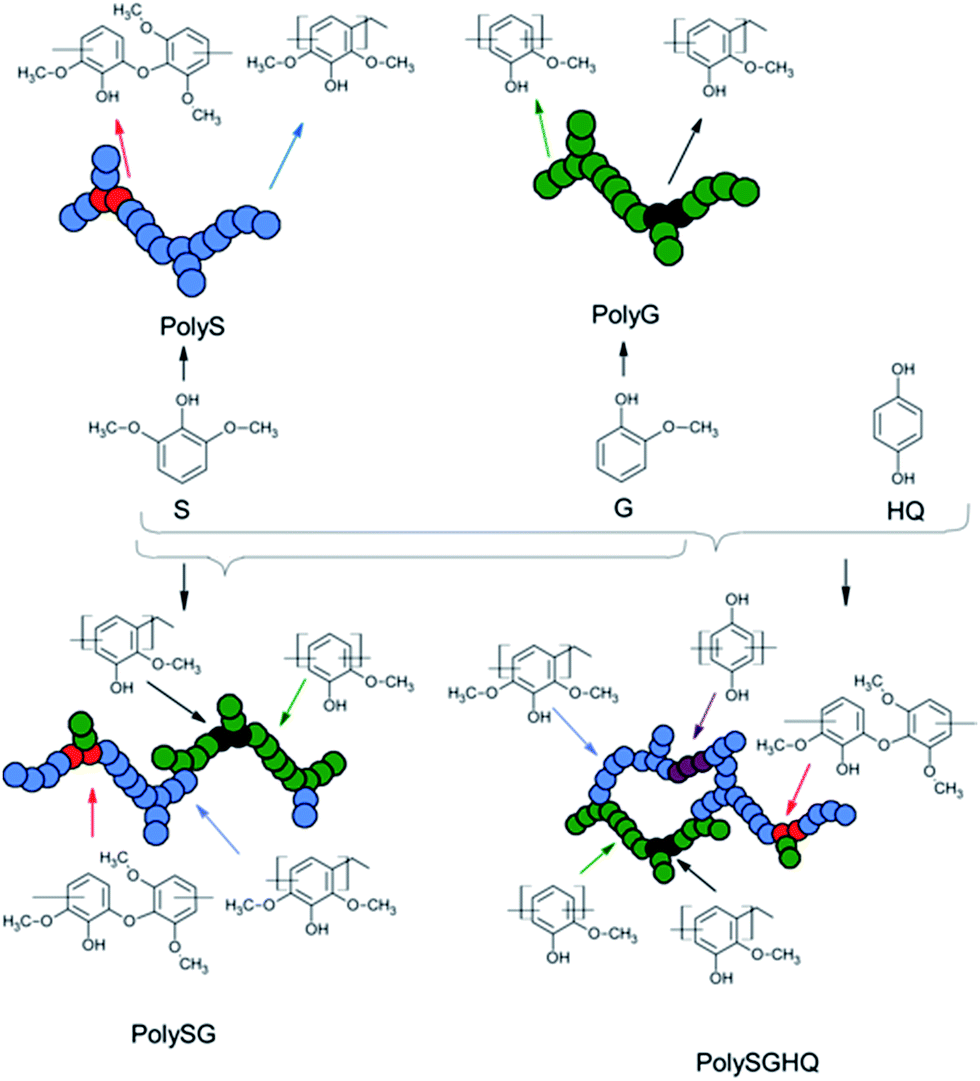 | ||
| Scheme 1 Schematic representation for synthesis of synthetic lignin derivatives via phenol–formaldehyde condensation. | ||
The hydroxyl groups in the synthetic lignin derivatives were coupled with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (phosphitylating reagent) and an internal standard to reveal the type and amount of phenolic hydroxyl groups. The 31P NMR spectrum in Fig. 1a for the homopolymer PolyS shows a significant peak at 142.7–143.2 ppm and a relatively small peak at 141.2–141.7 ppm, which are the spectral range of the diphenylmethane condensed and biphenyl ethers (4-O-5′) condensed phenolic unit.31 This indicates that the syringyl (S) units in PolyS were mostly condensed during the polymerization. In Fig. 1b, the spectrum of the other homopolymer PolyG showed relatively small peaks at 142.7–143.2 ppm and a prominent peak at 140.0–139.0 ppm, which correlate to diphenylmethane condensed phenolic and guaiacyl (G) unit. PolyG showed much less diphenylmethane phenolic condensation and no 4-O-5′ condensed compared to PolyS. In the spectrum of the copolymer PolySG (Fig. 1c), peaks are found at 142.7–143.2 ppm, and 140.0–139.0 ppm, which indicate the combination of the diphenylmethane condensed and biphenyl ethers (4-O-5′) condensed S unit and G unit in the copolymer. In the spectrum of the copolymer PolySGHQ (Fig. 1d), peaks are seen at 142.7–143.2 ppm, 140.0–139.0 ppm, and 138.8–137.2 ppm, which indicate the diphenylmethane condensed, biphenyl ethers (4-O-5′) condensed S unit, G unit and hydroquinone unit in the copolymer PolySGHQ.
 | ||
| Fig. 1 31P NMR spectra of synthetic lignin derivatives. Homopolymers are (a) PolyS, (b) PolyG; and copolymers (c) PolySG and (d) PolySGHQ. The internal standard is denoted as IS at 144.4 ppm. | ||
The molecular weights of the homopolymers PolyS, PolyG and the copolymers PolySG, PolySGHQ were monitored by SEC. In Fig. 2, the SEC trace showed that the PolyS, PolyG, PolySG and PolySGHQ were mostly high molecular weight polymers (compared to the standard polymer passing time). This indicated the successful polymerization of the SLig via polycondensation with formaldehyde. The SEC trace of PolyS and PolyG were almost identical, the main peaks found at 22.0 to 23.8 minutes and other less significant peaks at 23.8–24.6 minutes, 24.6–25.0 minutes, 25.2–25.8 minutes and 26.5–26.8 minutes. These indicated that the peak molecular weight of the PolyS at 15–19 kDa and for PolyG at 15–18.6 kDa, as well as that the distribution of the smaller molecular weight were similar. The copolymer PolySG showed main peaks at 22.0 to 23.8 minutes and a peak at 24.6–25.0 minutes and the other small peak at 26.5–26.8 minutes. This indicates that PolySG has a similar peak molecular weight of 15–19 kDa and less fractions of small molecular weights, compared to the homopolymers. The copolymer PolySGHQ showed the main peak at 20.9–23.8 minutes and the other peak at 24.6–25.0 minutes, which indicated the highest peak molecular weight as 14.7–25.3 kDa, and a much less fraction of small molecular weight, compared to the other polymers.
 | ||
| Fig. 2 SEC traces of the synthetic lignins PolyS, PolyG, PolySG and PolySGHQ at UV-vis 280 nm. | ||
Probing the electrochemical properties of monolignol monomers and SLig via SWNT
The electrochemical properties of SLig were first investigated by cyclic voltammetry. In order to get more insight into the redox behavior of SLig, the monomers (guaiacol and syringol) as well as ferulic acid, sinapic acid and hydroquinone, were studied for comparison as reference markers of quinone redox reaction of SLig on single-wall carbon nanotubes. SWNT are excellent electrode materials due to their good electrical conductivity, high porosity, as well as relatively good chemical stability.32 These properties of SWNT can be utilized to promote the electron-transfer reaction when applied as electrode materials in energy storage devices or electroanalysis. The surface of SWNT can be easily modified by noncovalent strong π–π stacking interaction between SWNT and hydrophobic electroactive molecules.10,19 In addition, SWNT being good electronic conductors, they can act as a network transporting electrons from the electrode surface to redox active moieties. As seen in Fig. 3, the cyclic voltammograms of the phenolics (guaiacol, syringol, ferulic acid, sinapic acid and hydroquinone) exhibit a well-defined reversible redox couple, typically observed for adsorbed functionalities with small peak-to-peak separations. Moreover, the large surface area of SWNT allows to significantly increase the surface coverage of electroactive species, thus enhancing the voltammetric signals that can be observed. On the basis of previous literature reports on molecules containing similar methoxyphenolic groups, such as capsaicine,33 hesperidin,34 curcumin35,36 or eugenol,37 it is possible to predict that the electrochemical oxidation of studied compounds leads to the development of a reversible redox couple involving o-benzoquinone/catechol moieties.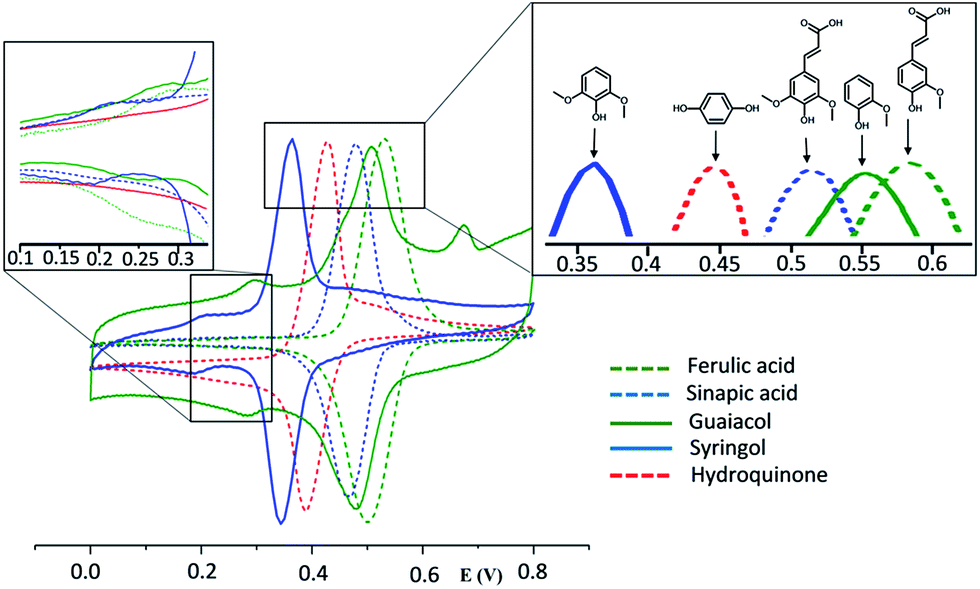 | ||
| Fig. 3 Cyclic voltammograms of ferulic acid/SWNT, sinapic acid/SWNT, guaiacol/SWNT, syringol/SWNT and hydroquinone/SWNT in 0.1 M HClO4, scan rate 10 mV s−1. | ||
The oxidative demethoxylation and formation of the reversible redox couple was observed during the first CV scan recorded in 0.1 M HClO4 for all studied systems, with the exception of hydroquinone. Upon first scan an irreversible oxidation wave is observed at ca. 0.7 V followed by the development of a redox couple positioned at lower potentials in subsequent scans (the representative example is ferulic acid on SWCNT which is presented in Fig. S1†). This process involves the chemical step (hydrolysis) resulting in removal of the methanol molecule.33,37 Similar behavior was also observed for lignosulfonates adsorbed on a gold electrode.16 Different numbers of oxygen containing groups substituted on the aromatic ring result in different values of E0′ of the recorded redox signals (Fig. 3). This phenomenon can be explained by the existence of electron donating effect arising from electron-rich oxygen atoms. Thus, the groups with a higher substitution degree produce redox couples with lower E0′ values. This is consistent with data obtained for the studied phenolic compounds. As seen, the predominant redox couple for molecules possessing two methoxyl groups, such as syringol (1,3-dimethoxy-2-hydroxybenzene) and sinapic acid, appear at 0.35 and 0.45 V, respectively. For comparison, peaks assigned to guaiacol (2-methoxyphenol) and ferulic acid are shifted toward more positive values (0.5 V for guaiacol and 0.55 V for ferulic acid). The existence of predominant couples can be ascribed to o-benzoquinone/catechol transitions originating from o-methoxyphenol oxidation.17,19 For syringol, guaiacol and ferulic acid, a second reversible, minor redox couple can be seen at lower potential values (see the inset in Fig. 3). Low values of E0′ suggest that the peaks can be derived from the additional substitution of aromatic ring by oxygen containing groups. The existence of such couples can probably be associated with the chemical reaction occurring when o-quinones are oxidized. Since electrogenerated o-quinone moieties are quite reactive and can lead to Michael adducts by nucleophilic attack, the potential assisted hydroxylation can occur in aqueous solution.37 The E0′ for hydroquinone is in good agreement with the data reported in the literature.38
The synthetic lignin model with redox functions has been created by polymerization of guaiacol and syringol as well as hydroquinone. Further, the SLig/SWNT hybrid materials were assembled to create the electronic communication between redox active polymer sites and electrode surface through the highly conducting carbon nanotube support. In order to investigate the electrochemical performance of the SLig/SWNT hybrid materials, CV was performed in 0.1 M HClO4. As seen in Fig. 4(a) the CV profile for the SWNT electrode shows a typical double-layer capacitive response with no distinctive redox peaks. In contrast, the CV of PolyG/SWNT exhibits the well-defined reversible quinone peaks at 0.48 V. The formal potential of the redox pair of PolyG/SWNT is very consistent with the position of guaiacol/SWNT peaks (Fig. 4(b)). On the other hand, the redox pair of PolyS/SWNT at 0.43 V is shifted to about 80 mV towards more positive values in comparison to syringol/SWNT studied as the reference (Fig. 4(a)). The observed difference may appear due partially to the existence of mixed products generated during the condensation of syringol. Hence, the present electrochemical behavior of PolyS/SWNT is rather similar to guaiacol/SWNT. As seen in Fig. 4(c), the peaks derived from PolySG/SWNT show electroactivity at a potential between the values corresponding to that obtained for syringol/SWNT and guaiacol/SWNT, suggesting the influence of both components to electrochemical response. As is demonstrated in Fig. 4(d), the PolySGHQ/SWNT hybrid material exhibits two slightly overlapping redox couples. Taking into account the electrochemical features mentioned above, the peaks recorded at 0.26 V and 0.45 V should be ascribed to hydroquinone and guaiacol/syringol electroactivity, respectively.
 | ||
| Fig. 4 Cyclic voltammograms of SLig/SWNT hybrid materials and corresponding various phenol derivatives in 0.1 M HClO4 at 10 mV s−1. | ||
Electrochemical characterization of SLig/PPy hybrid electrodes
The SLig/PPy hybrid films were electrochemically synthesized from 0.1 M pyrrole and tetraethylammonium tosylate in ethylene glycol (EG) solution with the synthetic-lignin derivatives PolyS, PolyG, PolySG and PolySGHQ respectively. Cyclic voltammetry was initially performed to characterize the electrochemical properties of SLig/PPy hybrid films. Upon first few cycles in an oxidative direction an activation process (demethoxylation) and formation of quinones can be observed (Fig. S2†).The cyclic voltammograms of the different SLig/PPy hybrid films in Fig. 5A show various contributions of redox activity. A pair of pronounced redox peaks similar to what is found for SLig/SWNT and lignosulfonate doped PPy17 show up at ca. 0.5 V for the PolyS/PPy, PolyG/PPy and PolySG/PPy hybrid films. The formal potential of PolyG/PPy peaks at 0.49 V was the highest among the hybrid films. This value is in good agreement with that obtained for PolyG/SWNT (0.48 V). The peak-to-peak potential separation (ΔEp) about 100 mV was measured for SLig/PPy hybrids. Therefore, when compared with SLig/SWNT, they exhibit larger ΔEp, suggesting that charge propagation between SLig and PPy is slower than that between SLig and SWNT. This may partially be due to slow diffusion of ions through the bulk of PPy electrode, which is a common drawback of conducting polymer based electrodes.17,39 Additionally, in SLig/SWNT, π–π stacking interaction can lead to a short distance between the conjugated carbon skeleton and the redox active sites. Consequently, the conversion of quinone to hydroquinone occurs at low charge-transfer resistance and rapidly responds to the applied potential (Fig. 4). The PolyS/PPy film with mostly condensed unit and PolySG/PPy with partly condensed units show a similar redox couple at 0.44 V, which is in good correlation with PolyS/SWNT (0.43 V). For the PolySGHQ/PPy hybrid film, there were two well-pronounced, partially overlapping features; one at ca. 0.25 V could be associated to HQ units and the other at 0.44 V could be associated to S, G and weakly bonded HQ units.40 The mass changes during the electropolymerization of SLig/PPy films were estimated by using EQCM. As seen from Fig. 5B, linear mass–time profiles were recorded. All of SLig/PPy materials present a very similar mass (192–197 μg).
 | ||
| Fig. 5 Cyclic voltammograms of the PolyS, PolyG, PolySG and PolySGHQ hybrid PPy films recorded at 1 mV s−1 in 0.1 M HClO4. The film thickness was ca. 1 μm (A). Mass changes obtained for the electrochemical polymerization of SLig/PPy hybrid films on the gold electrode (B). | ||
Charge storage properties of SLig/SWNT and SLig/PPy hybrid electrodes
The formation of the SLig/SWNT hybrid is a valuable strategy to obtain synergetic effects between the redox active polymer sites and a highly conducting carbon support. Moreover, it is possible to control the SWNT fraction in the final hybrid material by tailoring the mass ratio between SLig and the conductive matrix. This is in contrast to galvanostatic synthesis where the content of PPy in the final electrode material cannot really be controlled by optimization of the synthesis solution.17,18 Even so, the electrosynthesis can provide an effective and convenient one-step approach to the incorporation of SLig into the PPy film, though the mass ratio of both components in this case is unknown.The charge–discharge curves of the pristine SWNT and PolySGHQ/SWNT hybrid electrodes prepared with different mass ratios is shown in Fig. 6(a). SWNT store charge only in an electrochemical double-layer, which has been proved by its linear charge–discharge profile. The specific charge stored in pristine SWNT is 9.4 mA h g−1 (56 F g−1) in the studied potential range. The PolySGHQ/SWNT hybrid exhibits two plateaus in the ranges of 0.52–0.47 V and 0.32–0.25 V which implies a change in the mechanism towards pseudocapacitive storage. These faradaic contributions of PolySGHQ/SWNT are attributed to the fast electrochemical redox reaction of quinones due to the immobilized polymer.
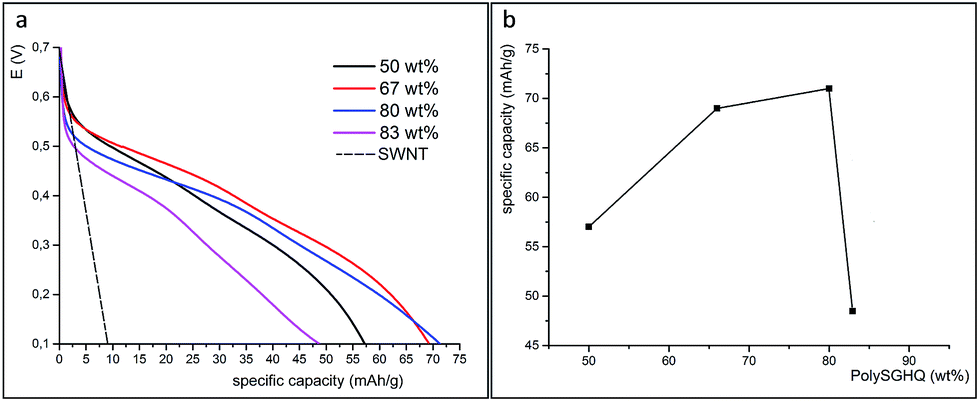 | ||
| Fig. 6 Charge–discharge curves for SWNT and PolySGHQ/SWNT hybrid material with different mass ratios in 0.1 HClO4 at 1 A g−1 (a). The relationship between charge storage capacity and mass ratio between PolySGHQ and SWNT (b). | ||
The galvanostatic charge–discharge measurements show that the faradaic contribution delivered by synthetic lignin activity dominates in the total capacity of the hybrid material. The results show that the hybrid material composed of 20 wt% of SWNTS and 80 wt% of PolySGHQ possesses the highest specific charge capacity of 72 mA h g−1 that corresponds to 432 F g−1 (Fig. 6(a)). Further increase of the redox polymer content above 80 wt% gives rise to a significant drop of specific charge capacity to 47 mA h g−1 (Fig. 6(b)). As can be seen in Fig. 6(b), a significant influence of the PolySGHQ content in the PolySGHQ/SWNT material on the charge storage capacity was observed. However, this contribution cannot be analyzed within a linear model based on the SWCNT fraction. The present values of charge storage capacity for PolySGHQ/SWCNT are comparable to that previously obtained for lignin polypyrrole (75 mA h g−1) composite.17
The charge–discharge tests of SLig/PPy hybrid films were performed at 1 A g−1, (Fig. 7A). Both PPy and SLig contributed to charge capacity in the SLig/PPy films. The phenolic amounts of PolyS, PolyG, PolySG, and PolySGHQ are collected and correlated with the specific capacity of the SLig/PPy hybrid films in Table 1. The mass ratio of SLig and pyrrole in the electrolyte used for electropolymerization was set at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, the mass ratio in SLig/PPy is unknown.
:1. However, the mass ratio in SLig/PPy is unknown.
 | ||
| Fig. 7 Specific charge capacity at 1 A g−1 (A) and specific capacity as a function of discharge current density (B) of the synthetic lignin derivative PolyS/PPy, PolyG/PPy, PolySG/PPy and PolySGHQ/PPy hybrid films. | ||
| Sample code | Condensed unit (mmol g−1) 143.5–141.4 ppm | G unit (mmol g−1) 139.6–139.0 ppm | HQ unit (mmol g−1) 139.0–136.0 ppm | Redox functional groups (mmol g−1) | Peak molecular weighta (kDa) | Specific capacity of the SLig/PPy film (PPy) (mA h g−1) |
|---|---|---|---|---|---|---|
| a Peak molecular weight from RI-SEC at 22.4–23.4 minutes. | ||||||
| PPy | — | — | — | — | — | 30 |
| PolyS | 15.7 | — | — | 15.7 | 14.9–19.0 | 49 |
| PolyG | 6.2 | 11.7 | — | 17.9 | 14.6–18.6 | 62 |
| PolySG | 11.9 | 4.7 | — | 16.6 | 14.7–19.0 | 57 |
| PolySGHQ | 8.0 | 3.4 | 5.0 | 16.4 | 14.7–25.3 | 94 |
At the 1 A g−1 discharge rate, within the potential range of 0.1–0.7 V, the SLig/PPy films yielded higher specific capacity than the PPy film, which gave 30 mA h g−1.18 Similar to the SLig/SWNT hybrids, the SLig/PPy materials exhibit a pronounced plateau region in the range of 0.55–0.40 V corresponding to the reduction of quinone species. The specific charge capacity increased to 49 mA h g−1 (for PolyS/PPy) and 62 mA h g−1 (for PolyG/PPy) with increasing amount of the total phenolic groups from 15.7 mmol g−1 (PolyS/PPy) to 17.9 mmol g−1 (PolyG/PPy). The specific capacity further increased to 94 mA h g−1 (specific capacitance 563 F g−1) for the PolySGHQ/PPy film with lower total phenolic amount in the PolySGHQ, 16.4 mmol g−1. This demonstrates that the charge storage capability increases with increasing amount of redox species of the S and G unit based SLig/PPy films; PolySGHQ/PPy film with HQ unit showed higher specific capacity than the S and G unit based SLig/PPy, regardless of the phenolic amount. Due to the unknown mass ratio between PPy and PolySGHQ, the causes of such improvement of charge storage in PolySGHQ are not clear. The plausible explanation may be an enhanced access to quinone moieties in PolySGHQ/PPy in comparison to PolyS/PPy and PolyG/PPy. Compared with the discharge plot in Fig. 7A of S and G unit based SLig/PPy films, a rapid decline is seen at the potential range ca. 0.35–0.10 V, whereas the discharge plot of the PolySGHQ/PPy film showed a consistent decline rate in this range. This indicated that introduction of a low redox potential species (<0.50 V) such as HQ to the model synthetic-lignin was more efficient than the increase of the phenolic amount of redox species higher than 0.50 V. The specific capacity of the SLig/PPy hybrid films at various current densities can be seen in Fig. 7B. The specific capacity decreases at higher currents. The fastest drop of specific capacity versus current density can be observed for PolySGHQ/PPy. However, the charge storage capacity of 57 mA h g−1 (62%) is still obtained at the high current density of 16 A g−1. Furthermore, at 16 A g−1, PolyG/PPy, PolyS/PPy and PolySG/PPy hold 80%, 90% and 87%, specific capacity of that at 1 A g−1, respectively, suggesting a good rate capability.
Different capacitance values are obtained within the studied potential range. From the inverse of the slopes of galvanostatic discharge curves, the separated capacitances contributions from PPy and SLig can be distinguished (Fig. 8).
 | ||
| Fig. 8 Respective specific capacitance versus specific current of contributing capacitances due to PPy and quinones (SLig, HQ) of PolySGHQ/PPy (A), PolyG/PPy (B), PolyS/PPy (C) and PolySG/PPy (D) electrodes extracted from the slopes of the galvanostatic discharge curve. | ||
HQ and SG fractions in PolySGHQ/PPy contributed to ca. 800 F g−1, while PPy provided ca. 280 F g−1. PolyG, PolyS and PolySG exhibited lower values of capacitances at 1 A g−1 (measured between 0.55 and 0.40 V) in comparison to PolySGHQ which is in good agreement with the CV data (Fig. 5A). It is worth mentioning that the PPy capacitance is higher than that estimated from the CV in Fig. 5A at 1 mV s−1 (210 F g−1). The influence of discharge rate on individual capacitances in the range of 0.55–0.40 V (quinones) is important, especially in the case of PolyG, PolyS and PolySG. Such behavior was typically observed for pseudocapacitive materials involving quinones.17,20
Conclusions
The characteristic monolignols syringol (S) and guaiacol were used to polymerize well-defined synthetic-lignins PolyS, PolyG and PolySG via polycondensation with formaldehyde. To extend the redox potential window, hydroquinone (HQ) was introduced to the synthetic-lignin PolySGHQ as a low oxidation potential phenolic unit. The methylene bridges in the chemical structure, which were revealed by 13C NMR, and the molecular weight peak over 14 kDa shown by the SEC trace, confirmed the success of the SLig derivative polymerization. The phenolic content in the SLig was measured by 31P NMR. By combination with different amounts of single-wall carbon nanotubes, the correlation between the loading of the electronic conductor and the charge storage capacity of the assembled SLig/SWNT hybrid material, was investigated. The charge performance of the PolySGHQ/SWNT is highest when the content of SWNT is 20 wt%. Thus, a low amount of electronic conductor (20% wt) is adequate to provide good access to the quinones and efficient electron communication between redox active phenolics and the electrode surface. The specific capacity was enhanced by ca. 7 times from 9.4 mA h g−1 (pure SWNT) to 72 mA h g−1 (PolySGHQ/SWNT), at the current density of 1 A g−1. The PolySGHQ/PPy film showed the highest specific capacity of 94 mA h g−1 (563 F g−1) after introducing the HQ unit with low oxidation potential (<0.5 V, pH = 1), whereas PolyG with the higher phenolic amount showed a lower specific capacity of 62 mA h g−1. According to the results, the introduction of HQ, possessing a redox potential <0.5 V (at pH = 1) enables a larger improvement of charge storage capacity for the lignin derivative/polypyrrole system. The chemical procedure highlighted here is a promising way to develop advanced electrodes for energy-storage applications from the cheap and easily accessible organic materials.Experimental
Materials
Single-wall carbon nanotube (SWNT) conductive aqueous ink (1 mg mL−1), dimethylformamide (DMF), pyridine, cyclohexanol, chromium(III) acetylacetonate, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (phosphitylating reagent), deuterated chloroform (CDCl3), diethyl ether, heptane, acetone, formaldehyde water solution (37%), 2,6-dimethoxyphenol (syringol, S), 2-methoxyphenol (guaiacol, G), 4-hydroxy-3-methoxycinnamic acid (ferulic acid), 3,5-dimethoxy-4-hydroxycinnamic acid (sinapic acid) hydroquinone (HQ), pyrrole, tetraethylammonium tosylate, ethylene glycol (EG), perchloric acid (HClO4) were purchased from Sigma-Aldrich. Pyrrole was distilled at 131 °C, the other chemicals were used as received.:0.9 (phenolic monomer:formaldehyde). Oxalic acid (0.01 g, 1 wt%) was added as a catalyst. The reactions were allowed to proceed at 110 °C for 12 hours. The finished products were dissolved in EtOH and precipitated in distilled water to remove residues, and freeze-dried. The dried products were precipitated in 500 mL diethyl ether to remove the unreacted monomers followed by vacuum drying. The products were isolated as light brown powders. The interpretation of 13C NMR (300 Hz, CDCl3) spectra were based on the literature:30 PolyS δ (ppm) = 25.13–32.31 (methylene bridges: orthol ortho, paral ortho and paral para), 56.19–56.30 (–O–CH3), 60.47 (–C–OH), 91 (hemiacetal carbons CH3O–CH2–OH), 118.4–120.2 (unsubstituted para carbons), 124.63–130.98 (meta carbons and substituted ortho carbons), 136.93–138.66 (substituted para carbons), 145.45–146.30 (phenoxy carbons). PolyG δ (ppm) = 31.95–41.17 (methylene bridges: orthol ortho, paral ortho and paral para), 56.00 (–O–CH3), 111.23–121.39 (unsubstituted ortho and para carbons), 131.2–134.5 (substituted para carbons), 143.7–146.4 (phenoxy carbons). The phenolic content of the products were measured by 31P NMR.
PolySG. Syringol (1 g, 6.5 mmol) and guaiacol (0.814 g, 6.5 mmol) at a molar ratio of 1
:1 were added to formaldehyde (0.957 g, 11.8 mmol) 37% water solution in test tubes, respectively, phenolic monomer:formaldehyde molar ratio was set to 1:0.9. Oxalic acid (0.027 g, 0.1 mmol) was added as the catalyst. The reactions were allowed to proceed at 110 °C for 12 hours. The finished product was dissolved in EtOH and precipitated in distilled water to remove residues and freeze-dried. The dried products were precipitated in 500 mL heptane to remove the unreacted monomers followed by vacuum drying. The products were isolated as light brown powder. The phenolic content of the product was measured by 31P NMR.
PolySGHQ. Syringol (0.349 g, 2.2 mmol), guaiacol (0.273 g, 2.2 mmol) and hydroquinone (0.242 g, 2.2 mmol) at a molar ratio of 1
:1:1 were added to formaldehyde (0.47 g, 5.9 mmol) 37% water solution in test tubes, respectively, and the phenolic monomer:formaldehyde molar ratio was set to 1:0.9. Oxalic acid (0.027 g, 0.1 mmol) was added as the catalyst. The reactions were allowed to proceed at 110 °C for 12 hours. The finished product was dissolved in EtOH and precipitated in distilled water to remove residues and freeze-dried. The dried products were precipitated in 500 mL diethyl ether to remove the unreacted monomers followed by vacuum drying. The products were isolated as light brown powder. The phenolic content of the product was measured by 31P NMR.
Methods
NMR experiments were performed on a BrukerAvance 300 MHz instrument. 1H NMR and 13C NMR spectra were acquired at 45° pulse angle by inverse gated proton decoupling and delay time was 1 second. 31P NMR spectra were obtained using cyclohexanol (δ = 144.5 ppm) as an internal standard, with 90° pulse angle by inverse gated proton decoupling and delay time was 2 seconds. The remaining phosphitylating reagent and its water adduct were identified at δ = 174.7 ppm and δ = 131.8 ppm, respectively. The polymer (20 mg) sample was dried at 40 °C in an oven overnight to remove water and then dissolved totally in 100 μL DMF. 100 μL DMF/pyridine (1:1, v/v) solution containing cyclohexanol (1.1 mg, 1.1 × 10−2 mmol) as the internal standard and chromium(III) acetylacetonate (0.5 mg, 1.4 × 10−3 mmol) as the relaxation agent was added subsequently. Phosphitylating reagent 100 μL was added to the lignin mixture and CDCl3 500 μL was then added. The final mixture was vortexed and transferred to a 5 mm NMR tube for 31P NMR analysis.
Size exclusion chromatography (SEC) was used for measuring the molecular weights of the lignin samples, using a 10 mM NaOH aqueous solution as the mobile phase on 3 TSK-gel columns (3000 PW, 4000 PW, 3000 PW) coupled in series. The flow rate was 1 mL min−1, and detection was achieved by UV at 280 nm. The system was calibrated with polystyrene standards with specific molecular weights ranging from 0.342 to 805 kDa. The Millennium 2 software supplied by Waters was used to process data.
Galvanostatic polymerization of synthetic-lignin/polypyrrole (SLig/PPy) film and cyclic voltammetry (CV) of the SLig/PPy films were performed on an Autolab PGStat 10 (EchoChemie, the Netherlands) in a three-electrode system. Lignin (16.7 mg) was totally dissolved by ultrasonication in 2.5 mL ethylene glycol with tetraethylammonium tosylate (75.4 mg, 0.1 M) as the dopant and distilled. Pyrrole (16.7 mg, 0.1 M) was added to prepare 1:1 mass ratio solution. This solution was used for galvanostatic polymerization of SLig/PPy films on a glassy carbon working electrode (A = 0.07 cm−2) by application of constant current density (0.4 mA cm−2) for 19 minutes. All the results reported in this work refer to Ag/AgCl (3 M KCl) reference electrode. The counter electrode was a platinum wire. The SLig/PPy film rinsed by using distilled water, and CV was subsequently performed in 0.1 M HClO4 aqueous solution in the range of −0.2 to 0.7 V. For galvanostatic charge–discharge tests, a constant current was applied between 0.1 and 0.7 V.
An electrochemical quartz crystal microbalance (QCM Q-Sense E4) was used to measure the mass deposition on a gold quartz crystal (A = 1 cm2) of the SLig/PPy film during the galvanostatic polymerization in a three-electrode system, with Ag/AgCl as the ref. electrode and platinum as the counter electrode. The frequency change was monitored during the deposition at 0.4 mA cm−2 for 19 minutes and analyzed by the Sauerbrey equation to deduce the mass. The deposited mass of the different SLig/PPy composites on gold (A = 1 cm2) were 197 μg (PolyS/PPy), 196 μg (PolyG/PPy), 197 μg (PolySG/PPy) and 192 μg (PolySGHQ/PPy).
The SLig/SWNT hybrid materials were prepared as follows. The single-wall carbon nanotube solution (1 mg mL−1) was added to the SLig ethanol solution (1 mg mL−1) in four volume ratios: 1:1, 2:1, 4:1 and 5:1 respectively to prepare the solution with 50 wt%, 67 wt%, 80 wt% and 83 wt% of SLig. All the mixed solutions were sonicated for 10 minutes for homogeneous dispersion. Working electrodes with SLig/SWNT were prepared by drop-casting (1 μL, 1 μg) the mixture solution on a glassy carbon electrode surface and dried properly at 60 °C in an oven. The SLig/SWNT electrodes were charged and discharged respectively at 1 A g−1 current density and at 0.1 V to 0.7 V in three electrode system containing 0.1 M HClO4 of the supporting electrolyte. Electrochemical characterization by cyclic voltammetry was performed for the materials prepared by drop-casting (1 μL, 1 μg) the mixed solutions (50 wt%) of SWNT and each studied compound (guaiacol, syringol, hydroquinone, ferulic acid, sinapic acid, PolyG, PolyS, PolySG, PolySGHQ).
Acknowledgements
This work was supported by the Knut and Alice Wallenberg Foundation through the project Power Papers. Dr Michal Wagner is acknowledged by conducting some of the EQCM measurements, and discussions with colleagues in BIORGEL, Chemistry at the Linköping University and KTH.References
- J. Liu, J.-G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. Li, Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- D. Vonlanthen, P. Lazarev, K. See, F. Wudl and A. J. Heeger, Adv. Mater., 2014, 26, 5095–5100 CrossRef CAS PubMed.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 CrossRef CAS.
- L. Zhao, W. Wang, A. Wang, K. Yuan, S. Chen and Y. Yang, J. Power Sources, 2013, 233, 23–27 CrossRef.
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS PubMed.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 4, 448–450 RSC.
- X. Chen, H. Wang, H. Yi and X. Wang, J. Phys. Chem. C, 2014, 118, 8262–8270 CAS.
- A. le Comte, D. Chhin, A. Gagnon, R. Retoux, T. Brousse and D. Bélanger, J. Mater. Chem. A, 2015, 3, 6146–6156 CAS.
- C. Heitner, D. R. Dimmel and J. A. Schmidt, Lignin and Lignans: Advances in Chemistry, CRC Press, Boca Raton, FL, 2010 Search PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2013, 39, 1266–1290 CrossRef.
- V. L. Chiang, Environ. Chem. Lett., 2006, 4, 143–146 CrossRef CAS.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- G. Milczarek, Langmuir, 2009, 25, 10345–10353 CrossRef CAS PubMed.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- F. N. Ajjan, M. J. Jafari, T. Rębiś, T. Ederth and O. Inganäs, J. Mater. Chem. A, 2015, 3, 12927–12937 CAS.
- G. Milczarek and M. Nowicki, Mater. Res. Bull., 2013, 48, 4032–4038 CrossRef CAS.
- S.-K. Kim, Y. K. Kim, H. Lee, S. B. Lee and H. S. Park, ChemSusChem, 2014, 7, 1094–1101 CrossRef CAS PubMed.
- S. Admassie, T. Y. Nilsson and O. Inganas, Phys. Chem. Chem. Phys., 2014, 16, 24681–24684 RSC.
- S. Leguizamon, K. P. Díaz-Orellana, J. Velez, M. C. Thies and M. E. Roberts, J. Mater. Chem. A, 2015, 3, 11330–11339 CAS.
- D. H. Nagaraju, T. Rębiś, R. Gabrielsson, A. Elfwing, G. Milczarek and O. Inganäs, Adv. Energy Mater., 2013, 4, 1–7 Search PubMed.
- S. Admassie, A. Elfwing, E. W. H. Jager, Q. Bao and O. Inganäs, J. Mater. Chem. A, 2014, 2, 1974–1979 CAS.
- T. V. Vernitskaya and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443–457 CrossRef.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- S. Shimizu, T. Yokoyama, T. Akiyama and Y. Matsumoto, J. Agric. Food Chem., 2012, 60, 6471–6476 CrossRef CAS PubMed.
- K. Minu, K. K. Jiby and V. V. N. Kishore, Biomass Bioenergy, 2012, 39, 210–217 CrossRef CAS.
- A.-S. Jönsson, A.-K. Nordin and O. Wallberg, Chem. Eng. Res. Des., 2008, 86, 1271–1280 CrossRef.
- R. Rego, P. J. Adriaensens, R. A. Carleer and J. M. Gelan, Polymer, 2004, 45, 33–38 CrossRef CAS.
- D. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 45–63 CrossRef CAS.
- G. G. Wildgoose, C. E. Banks, H. C. Leventis and R. G. Compton, Microchim. Acta, 2005, 152, 187–214 CrossRef.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Electroanalysis, 2008, 20, 2495–2500 CrossRef CAS.
- M. J. Sims, Q. Li, R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Electrochim. Acta, 2009, 54, 5030–5034 CrossRef CAS.
- L. Zheng and J. Song, Sens. Actuators, B, 2009, 135, 650–655 CrossRef CAS.
- M. A. N. Manaia, V. C. Diculescu, E. D. S. Gil and A. M. Oliveira-Brett, J. Electroanal. Chem., 2012, 682, 83–89 CrossRef CAS.
- A. Ciszewski and G. Milczarek, Electroanalysis, 2001, 13, 860–867 CrossRef CAS.
- H. Hong and W. Park, Langmuir, 2001, 17, 2485–2492 CrossRef CAS.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- M. R. Arcila-Velez and M. E. Roberts, Chem. Mater., 2014, 26, 1601–1607 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06821e |
| ‡ Tomasz Rębiś and Ting Yang Nilsson contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |