
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titanium oxynitride microspheres with the rock-salt structure for use as visible-light photocatalysts†
Jung Bo
Yoo
a,
Hyo Jin
Yoo
a,
Hyuk Joon
Jung
a,
Han Sol
Kim
a,
Sora
Bang
a,
Jongmyung
Choi
a,
Hoyoung
Suh
b,
Ji-Hyun
Lee
b,
Jin-Gyu
Kim
b and
Nam Hwi
Hur
*a
aDepartment of Chemistry, Sogang University, Seoul 121-742, Korea. E-mail: nhhur@sogang.ac.kr
bNano-Bio Electron Microscopy Research Group, Korea Basic Science Institute, Daejeon 34133, Korea
First published on 30th November 2015
Abstract
Novel photocatalysts (TiO2@TiO1−xNx) with the core–shell geometry were synthesized by controlled nitridation of TiO2 microspheres using ammonia gas. The oxynitride phases (TiO1−xNx) with a cubic rock-salt structure are exclusively formed on the surface of the TiO2 microspheres while the cores of the microspheres retain the TiO2 phase without nitrogen doping. Various spectroscopic data confirm the formation of the core–shell structure, denoted as TiO2@TiO1−xNx. The TiO2@TiO1−xNx materials showed superior photocatalytic activities for the decomposition of methylene blue as well as the generation of photocurrent under visible-light. It is evident that the TiO1−xNx phase is the key element to induce the photocatalytic activity. Specifically, the partial doping of nitrogen into the TiO matrix is crucial for visible-light absorption.
Introduction
Titanium dioxide (TiO2) has been extensively studied because of its wide applications in solar-driven environmental and energy technologies since Honda and Fujishima discovered water splitting on a TiO2 electrode under UV light.1 TiO2 has several merits as a photocatalyst including large abundance, low toxicity, and excellent stability.2,3 However, a major drawback is that it absorbs only UV light due to its large band gap energy (3.0 and 3.2 eV for the rutile and anatase phases, respectively).4,5 A wide range of approaches have been explored to narrow the band gap of TiO2. Chen et al. reported that disordered TiO2 nanoparticles (NPs) prepared by the hydrogenation method exhibit significantly enhanced activity in the production of hydrogen (H2) from water under visible-light.6 The chemical composition of TiO2 was also modified by doping nonmetal atoms such as carbon (C), hydrogen (H), nitrogen (N), fluorine (F), and sulfur (S) into the oxygen (O) site.7–13 The dopants typically generate impurity states above the valence band of TiO2, which results in the upshift of the valence band edge and the improvement in the solar absorption efficiencies of TiO2. In particular, N-doped TiO2 (nominally TiO2−xNx) materials with low N content (∼1 wt%) have been thoroughly studied.10,14 They are typically prepared by annealing TiO2 at temperatures below 600 °C under an ammonia (NH3) atmosphere, which exhibit excellent catalytic activities in the visible light range. But their absorption is not fully covered in the solar spectrum. Moreover, TiO2−xNx is unstable after going through the photocatalytic reaction and easily returns to parent TiO2.Despite the extensive interest in N-doping in the TiO2 matrix, there are few studies on N-doped titanium monoxide (TiO) materials with a cubic rock-salt structure due to the difficulty to control the doping level and the metallic character of TiO.15,16 Simon et al. reported the synthesis of N-doped TiO NPs using laser pyrolysis, which shows a large shift of the absorption threshold in the visible region.17 Similar to TiO, titanium nitride (TiN) also adopts a cubic rock-salt structure and is considered to be metallic. In contrast to bulk TiN materials, TiN films prepared by using a cathodic arc technique have a band gap of about 2.0 eV, indicating that TiN can be semiconducting if the particle size reduces to the nanometer scale.18 Very recently, Zheng et al. prepared TiOxNy@TiN composites through the flash oxidation of commercial TiN particles, demonstrating a high photocatalytic activity for H2 production under visible light irradiation.19 However, the non-equilibrium quick oxidation of TiN particles in the presence of 2,4,6-trinitrophenol hampers the control of the optimal doping level.
Here we report a convenient and reproducible method to make a new visible-light active composite photocatalyst. Controlled nitridation of monodisperse TiO2 microspheres, accomplished by flowing ammonia gas, converted the shell into the TiO1−xNx phase and transformed the core into the amorphous TiO2 phase. As a result, the nitrided product has the core–shell type structure, which is composed of two phases: a crystalline TiO1−xNx phase as the shell and an amorphous TiO2 phase as the core. Hereafter, we denote it as TiO2@TiO1−xNx. The TiO2@TiO1−xNx material exhibited excellent photocatalytic activities under visible-light, including the degradation of methylene blue and the production of photocurrent. The nitrogen doping in TiO1−xNx is considered to be responsible for absorbing visible light.
Experimental section
Chemicals
All chemicals were purchased from commercial suppliers and were used without further purification unless otherwise stated. Ethanol, dodecyl amine (98%, DDA), methylene blue (MB), terephthalic acid (98%), sulfuric acid (>95%), and Nafion were purchased from Sigma-Aldrich (St Louis, USA). Titanium tetraisopropoxide (TTIP) were obtained from Tokyo Chemical Industry (Tokyo, Japan).Synthesis of monodisperse TiO2 microspheres
Monodisperse TiO2 microspheres were prepared using TTIP as the titanium source. In brief, 3 mL of DDA, 4 mL of deionized water, and 300 mL of ethanol were added to a round-bottom flask, and the solution was cooled to −20 °C and stirred for 1 h. Using a syringe, a solution containing 6 mL of TTIP and 8 mL of ethanol was injected into the cold solution. After vigorous stirring for 20 h at −20 °C, white precipitates that formed gradually were separated from the solution by centrifugation. The precipitates were sintered in air at 500 °C for 6 h, yielding 1.44 g of TiO2 powders (89.2% yield based on TTIP). The powders are composed of evenly dispersed spherical microspheres and their average diameter is approximately 560 nm.Synthesis of TiO2@TiO1−xNx microspheres
The TiO2 microspheres were annealed in NH3 at 700 °C to yield the desired TiO2@TiO1−xNx microspheres. The flow of ammonia gas was controlled using a mass flow controller. The flow rate is fixed at 300 standard cubic centimeters per minute. Six different samples were prepared by controlling the annealing time. The annealing was performed at 700 °C for 1, 4, 7, 10, 24, and 48 h. The white microspheres gradually became blue when the sample was annealed under the ammonia atmosphere.Characterization
Powder X-ray diffraction (XRD) data were collected using a Rigaku DMAX 2500 diffractometer (Cu Kα; Rigaku, Japan) operating at 40 kV and 150 mA. High resolution transmission electron microscopy (TEM) was performed using a JEOL JEM-2100F microscope (JEOL, Japan). Specimens for the TEM examinations were prepared by dispersing finely ground powders of the samples in anhydrous ethanol and then allowing a drop of the suspension to evaporate on a 400 mesh carbon-coated grid. High resolution scanning electron microscopy (SEM) analyses were carried out using a Hitachi S-5500 microscope (Hitachi, Tokyo, Japan). Samples for the SEM analyses were prepared by dropping diluted samples in anhydrous ethanol on a lacey support grid. The samples were also subjected to chemical microanalyses using an Oxford Instruments INCA TEM 300 system (Oxford Instruments, Abingdon, UK) for energy dispersive X-ray (EDX) analysis. UV-visible absorption spectra of methylene blue (MB) solutions were recorded using a Perkin Elmer Lambda 950 spectrometer. The UV-visible absorption spectra of the powders were measured using an integrating sphere accessory by the diffuse reflectance method. Raman spectra were obtained at 25 °C using a LabRam HR Raman spectrometer (Horiba Jobin-Yvon) equipped with a liquid-nitrogen-cooled CCD multichannel detector. A 514 nm Ar-ion laser was used as the excitation source. Photoluminescence spectra were measured on a Hitachi F-7000 fluorescence spectrophotometer. Thermal gravimetric analysis was carried out using a TGA 2050 instrument (TA Instruments). The sample was placed on a platinum pan for each run. The data were collected in air from 25 °C to 700 °C at a rate of 5 °C min−1. Adsorption and desorption measurements were carried out at 77 K using an ASAP 2420 instrument (Micromeritics, USA) with nitrogen as the adsorptive gas. The Brunauer–Emmett–Teller (BET) surface areas were calculated using P/P0 = 0.05–0.3 from the adsorption curve using the BET equation. The pore-size distributions were obtained from the desorption curve using the density functional theory method. Prior to each sorption measurement, the sample was out-gassed at 300 °C for 24 h in vacuo to completely remove the impurities. To investigate the elemental compositions, X-ray photoelectron spectroscopy (XPS; Theta probe AR-XPS System, Thermo Fisher Scientific, UK) analysis using a mono-chromated Al Kα X-ray source (hν = 1486.6 eV) was performed at the Korea Basic Science Institute (KBSI) in Busan. The nitrogen contents of the TiO2@TiO1−xNx samples were analyzed using inductively coupled plasma atomic emission spectroscopy (ICP-AES, JY Ultima2C) at KBSI in Seoul.Results and discussion
Monodisperse titanium oxynitride (TiO2@TiO1−xNx) microspheres were prepared via two main pathways: (1) sol–gel synthesis of TiO2 microspheres and (2) nitridation of the TiO2 microspheres in NH3. Uniform TiO2 microspheres were prepared with solutions of TTIP, DDA, deionized water, and ethanol. The solution temperature was maintained at −20 °C during the sol–gel polymerization process to make TiO2 microspheres with virtually identical diameters as well as to prevent agglomeration. After separating white precipitates from the solution, white precipitates were then heated at 500 °C for 6 h, yielding white crystalline TiO2 microspheres with an average diameter of 560 nm. The TiO2 microspheres were then nitrided at 700 °C in an NH3 atmosphere for different periods of time. The white TiO2 microspheres turned blue to deep blue, depending on the annealing time. This color change implies the formation of the rock-salt TiN1−xOx phase in the TiO2 microsphere.As illustrated in Fig. 1, the TEM and SEM images of representative TiO2@TiO1−xNx microspheres clearly reveal that the size of parent TiO2 microspheres has noticeably shrunk but the spherical shape appears to be remained. The results implicate that the annealing is effective mostly on the surface of the TiO2 microspheres at 700 °C. The average diameter of TiO2@TiO1−xNx microspheres nitrided for 24 h was reduced to about 420 nm, which was estimated from the TEM images of TiO2@TiO1−xNx in Fig. 1. However, the spherical shape has almost collapsed when the sample was nitrided for 24 h at 900 °C (Fig. S1†), indicating that annealing at high temperatures completely converts the oxide phase into the nitride phase.
 | ||
| Fig. 1 TEM images of (a) parent TiO2 and (b) nitrided TiO2@TiO1−xNx microspheres where nitridation was done at 700 °C for 24 h. Corresponding SEM images of (d) TiO2 and (e) TiO2@TiO1−xNx microspheres. Photos comparing (c) white TiO2 powders and (f) dark blue TiO2@TiO1−xNx samples are given in the right panel. | ||
We first investigated the structures of the TiO2@TiO1−xNx microspheres with powder X-ray diffraction (XRD). The XRD data, given in Fig. 2, show that the anatase TiO2 phase is gradually transformed into TiN1−xOx with a cubic rock-salt structure as the nitridation time increases. The sample nitrided at 700 °C for 4 h shows that two new XRD peaks at about 37.2° and 43.2°, which can be assigned to (111) and (200) reflections in TiO1−xNx, appear along with the TiO2 peaks.20,21 This indicates that the anatase TiO2 phase coexisted with the cubic TiO1−xNx phase for a short period of time. When the nitridation was prolonged to 10 h, the two peaks corresponding to cubic TiO1−xNx became more distinctive in the XRD data while other peaks from the anatase TiO2 phase almost completely disappeared. This suggests that the cubic TiO1−xNx phase is predominantly formed on the surface of the TiO2 microspheres nitrided for longer than 10 h. The average crystallite size of TiO1−xNx synthesized by nitriding the TiO2 sample for 24 h, determined from half-peak widths of the XRD peaks by applying the Scherrer equation, is 17.1 nm. Lattice parameters were calculated using the (111) and (200) reflections in the XRD data. Table S1† lists the calculated lattice parameters for TiO1−xNx, along with the TiN and TiO standard data from the JCPDS file. Lattice constants of TiO1−xNx nitrided for 24 h are approximately in the middle of those for TiN and TiO, indicating that both O and N atoms are randomly disordered in the anion sites of the rock salt structure.
 | ||
| Fig. 2 Powder XRD patterns of TiO2@TiO1−xNx microspheres with different nitridation times. Two peaks were indexed on the basis of the JCPDS data. Peaks marked with an asterisk correspond to the TiO2 peaks. | ||
It is worth mentioning that the metallic TiN phase can be obtained only when the nitridation temperature was higher than 900 °C. This suggests that the nitridation at 700 °C results in a partial replacement of oxygen in TiO2 with nitrogen rather than complete substitution. In the course of nitridation, TiO2 is the only oxygen source. It is thus conceivable that anion exchange between nitrogen and oxygen occurs initially at the surface of TiO2. Hence, the TiO2 microspheres annealed at 700 °C are composed of partially nitrided TiO1−xNx and unreacted TiO2 phases. Presumably the nitrided phase starts forming on the outer surface of a TiO2 microsphere and gradually penetrates into the inner core via the oxygen exchange. As a result, the outer shell is predominantly composed of TiO1−xNx whereas the inner core mainly consists of TiO2, yielding the core/shell type microspheres. The XRD data suggest that the nitridation time and temperature strongly influence the formation of TiO1−xNx at the surface and the shell thickness. It is interesting to note that any XRD peaks corresponding to the TiO2 phase were not displayed in the samples nitrided for 24 and 48 h although the unreacted TiO2 phase remains in the nitrided samples. The absence of the XRD peaks suggests that TiO2 in the core part might exist as an amorphous state. Nitridation appears to induce the disordered state via the exchange between oxygen and nitrogen in the boundary, which results in the transformation of the crystalline TiO2 phase into an amorphous TiO2 state.6,22 Another plausible cause is that X-ray might not penetrate enough into the inner core of the TiO2 microsphere. The diffracted peaks are thus ascribed mainly to the TiO1−xNx phase on the surface.
Raman spectroscopy was employed to confirm the structural changes by the nitridation time (Fig. 3). The anatase phase of TiO2 has six Raman-active vibrational modes including one A1g mode at 527 cm−1, two B1g modes at 382 and 504 cm−1, and three Eg modes at 141, 154, and 654 cm−1.23 The samples nitrided at 700 °C for 7 h showed virtually identical Raman patterns owing to the anatase TiO2 phase, suggesting that the conversion of TiO2 into TiO1−xNx is not complete at this stage. However, the Eg mode at 154 cm−1 in the spectrum of the sample nitrided for 10 h was significantly blue-shifted and noticeably broadened relative to that in the TiO2 spectrum, indicating that particle sizes were changed and defects were produced by nitridation. The Raman spectrum of the TiO2 microspheres nitrided for 24 h did not show any peaks corresponding to the anatase TiO2 phase and exhibited very broad peaks at ∼210, ∼290, and ∼570 cm−1 due to the cubic TiO1−xNx phase. The first-order Raman scattering is forbidden for the crystal with a cubic rock salt structure (space group: Fm3m) because every atom is located at a site of inversion symmetry.24 Since TiO1−xNx contains a wide range of vacancies and defects, however, the forbidden rule can be relaxed and Raman peaks can thus be observed.25,26 The very broad peaks are probably associated with acoustic and optical modes of TiO1−xNx. It is worth mentioning that the unreacted TiO2 phase was not detected in the 24 h sample by Raman analysis. This indicates that structural changes occur after nitridation, resulting in oxygen disorders primarily in the TiO2 phase in the core. The Raman data obtained from the nitrided samples are consistent with the XRD results.
 | ||
| Fig. 3 Raman spectra of TiO2@TiO1−xNx microspheres with different nitridation times. For comparison, the Raman spectrum of TiO2 microspheres is displayed at the bottom. | ||
The XRD and Raman results discussed above show the formation of the cubic TiO1−xNx phase but do not provide any direct evidence on the presence of the unreacted TiO2 phase. XPS data of the nitrided samples prepared by annealing at different dwell times were examined to assess the chemical environments and oxidation states of Ti and N. In particular, two important regions including Ti 2p and N 1s were carefully investigated for each sample. Fig. 4 shows the XPS spectra in the Ti 2p region (452–462 eV) of the TiO2@TiO1−xNx samples. The Ti 2p XPS spectra of the TiO2 and nitrided samples exhibit drastic differences. The XPS data show that the binding energy was shifted from higher to lower values with increased annealing time. The sharp peak at 458.3 eV is typical for the Ti 2p3/2 in anatase TiO2, namely the Ti4+ species.27,28 A notable feature is that the peak at 458.3 eV is observed regardless of the annealing time, suggesting that the unreacted TiO2 phase is present even if the sample was nitrided for over 24 h. The broader peaks in the range of 455 to 458 eV can be attributed to Ti2+, Ti3+, and Ti4+ species.29,30
 | ||
| Fig. 4 Ti 2p XPS spectra of the TiO2@TiO1−xNx samples with different nitridation times. | ||
For the sample nitrided for 24 h, the spectral curve was fitted with five peaks centered at 455.6, 457.2, 458.3, 461.4, and 464.0 eV, which is given in Fig. 5. The Ti 2p3/2 XPS peaks are typically observed in the range of 455 to 460 eV. The lowest peak at 455.6 eV is ascribed to the Ti2+ species that could be associated with TiO. A peak centered at 457.2 eV can be assigned to the Ti3+ of TiN or N–Ti–O bonding in TiO1−xNx.31 The peak at 458.3 eV correlates with the Ti4+ in TiO2. Two peaks at 461.4 and 464.0 eV are due to the Ti 2p1/2 spectra. One peak at 464.0 eV is ascribed to the Ti4+ of TiO2 while the other at 461.4 eV is associated with the N–Ti–O bonding in the oxynitride phase such as TiO1−xNx. The Ti 2p XPS spectra suggest that the nitrided sample contains the multiple oxidation states of Ti and might be composed of TiO, TiO1−xNx, and TiO2.
 | ||
| Fig. 5 Ti 2p XPS spectra of TiO2@TiO1−xNx which was prepared by nitriding for 24 h. The black line is an experimental XPS curve. The colored curves are the fittings of experimental XPS data, which can be decomposed into a superposition of five peaks shown as colored curves. | ||
The presence of N in the nitrided sample was clearly evidenced by the N 1s XPS spectra of TiO2@TiO1−xNx as shown in Fig. 6. The N 1s peaks are more distinctive with increasing nitridation time. For the sample nitrided for 7 h, a nearly single peak is observed. However, the samples with a longer nitridation time showed a broad peak. The inset in Fig. 6 is the N 1s XPS spectrum of the 24 h sample. The red curve is the fitting data of experimental XPS data, which can be resolved into two peaks shown as blue curves. The first peak at 396.5 eV is characteristic of the N dopant which corresponds to the Ti–N bonding and the second one at 398.7 eV can be attributed to the N–Ti–O species.32
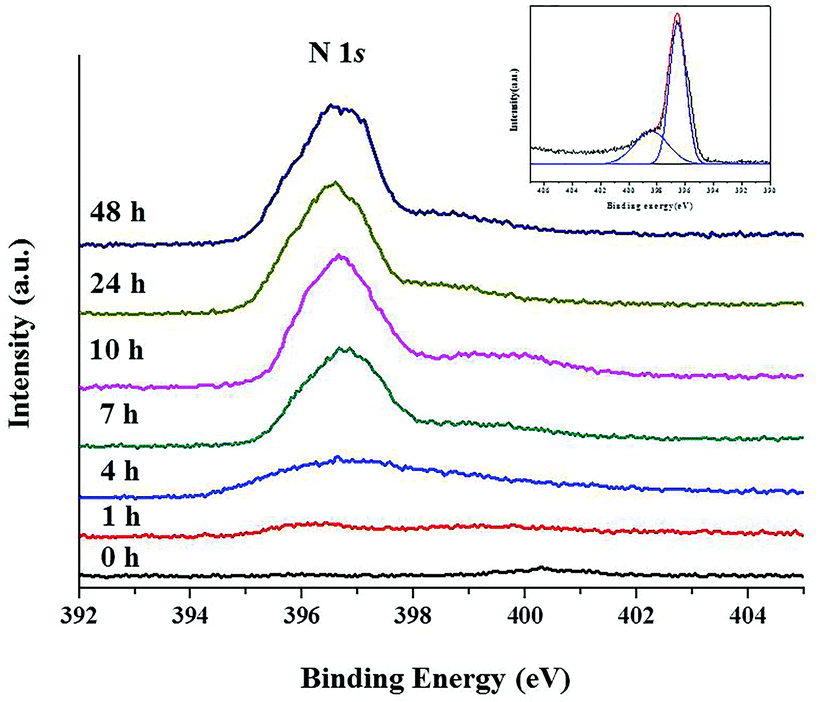 | ||
| Fig. 6 N 1s XPS spectra of the TiO2@TiO1−xNx samples with different nitridation times. The inset is the fitting of experimental XPS data for the 24 h nitridation sample, which can be decomposed into a superposition of two peaks shown as blue curves. The black curve represents XPS data. | ||
From the XRD, Raman, and XPS data, it is conceivable that the nitrided samples are composed of TiO1−xNx and TiO2. However, it is very difficult to determine how much TiO2 phase in the parent TiO2 microsphere remains intact during the nitridation procedure. Thus, TGA was employed to determine the nitrogen content, which is useful to estimate the approximate ratio of TiO1−xNx to TiO2 in TiO2@TiO1−xNx. The TGA data of the nitrided samples were collected in air as a function of temperature. For comparative purposes, commercial TiN powder was also measured under the same conditions. All the samples were transformed into the rutile TiO2 phase after measurements, which were confirmed by XRD.
As illustrated in Fig. 7, the weight percentage of TiN increases to about 129%, which is close to the theoretical value for the conversion of TiN into TiO2. In contrast, the weight gain of the sample nitrided at 700 °C for 24 h is about 115%, which is much lower than that of TiN. This clearly suggests that the unreacted TiO2 phase remains in the nitrided samples. A notable feature is that the weight gains of the samples annealed for 24 and 48 h are not significantly different, supporting that the temperature annealed at 700 °C is not sufficient to completely convert TiO2 into TiN.
 | ||
| Fig. 7 Thermogravimetric analysis (TGA) data for the samples nitrided at 700 °C for 10 and 24 h. Data were collected in air at a heating rate of 10 °C per minute. For comparison, TGA data for TiN were included. The inset is a simplified drawing of a TiO2@TiO1−xNx microsphere with a shell thickness of about 56 nm and core diameter of about 208 nm. The relative ratio of TiO1−xNx to TiO2 was determined on the basis of the TGA and EDX data for the sample nitrided at 700 °C for 24 h. | ||
Since the ratio of TiO1−xNx to TiO2 in the nitrided sample cannot be determined by the TGA data alone, the EDX elemental analysis was employed to help estimating the relative compositions in TiO2@TiO1−xNx. Although the EDX data are not completely reliable due to several factors such as surface, elements, and the specimen features, the technique provides reasonably reliable elemental compositions particularly for insoluble inorganic materials like TiO2@TiO1−xNx. Weight percentages of Ti and N of the samples nitrided for 10, 24, and 48 h were obtained directly from the EDX data and oxygen contents were calculated assuming that the samples are solely composed of Ti, N, and O. Table S2† gives weight percentages and atomic ratios for the three samples. As anticipated, the EDX data show that the nitrogen content increases as the annealing time increases.
Based on TGA and EDX data, the ratio of TiO1−xNx to TiO2 for the sample nitrided at 700 °C for 24 h is estimated to be 0.18. For this calculation, we assumed that TiO2@TiO1−xNx has a core–shell type structure because the nitridation occurs initially on the surface and gradually penetrates into the core. EDX data were taken to determine relative amounts of N and O. As illustrated in the inset of Fig. 7, the shell thickness and the core diameter of the TiO2@TiO1−xNx sample are 56 nm and 308 nm, respectively.
Fig. 8 shows elemental mapping images and EDX line scan elemental profiles of the sample nitrided at 700 °C for 24 h, clearly confirming the presence of N, O, and Ti. The Ti intensity is strong and the intensity profile along the line is nearly dome-shaped. On the other hand, the intensities of N and O are relatively weak and did not noticeably change along the line due to their low scattering power. The elemental mapping images indicate that N, O, and Ti are evenly dispersed on the spherical surface and also imply that TiO1−xNx is mostly located on the outer portion of the shell.
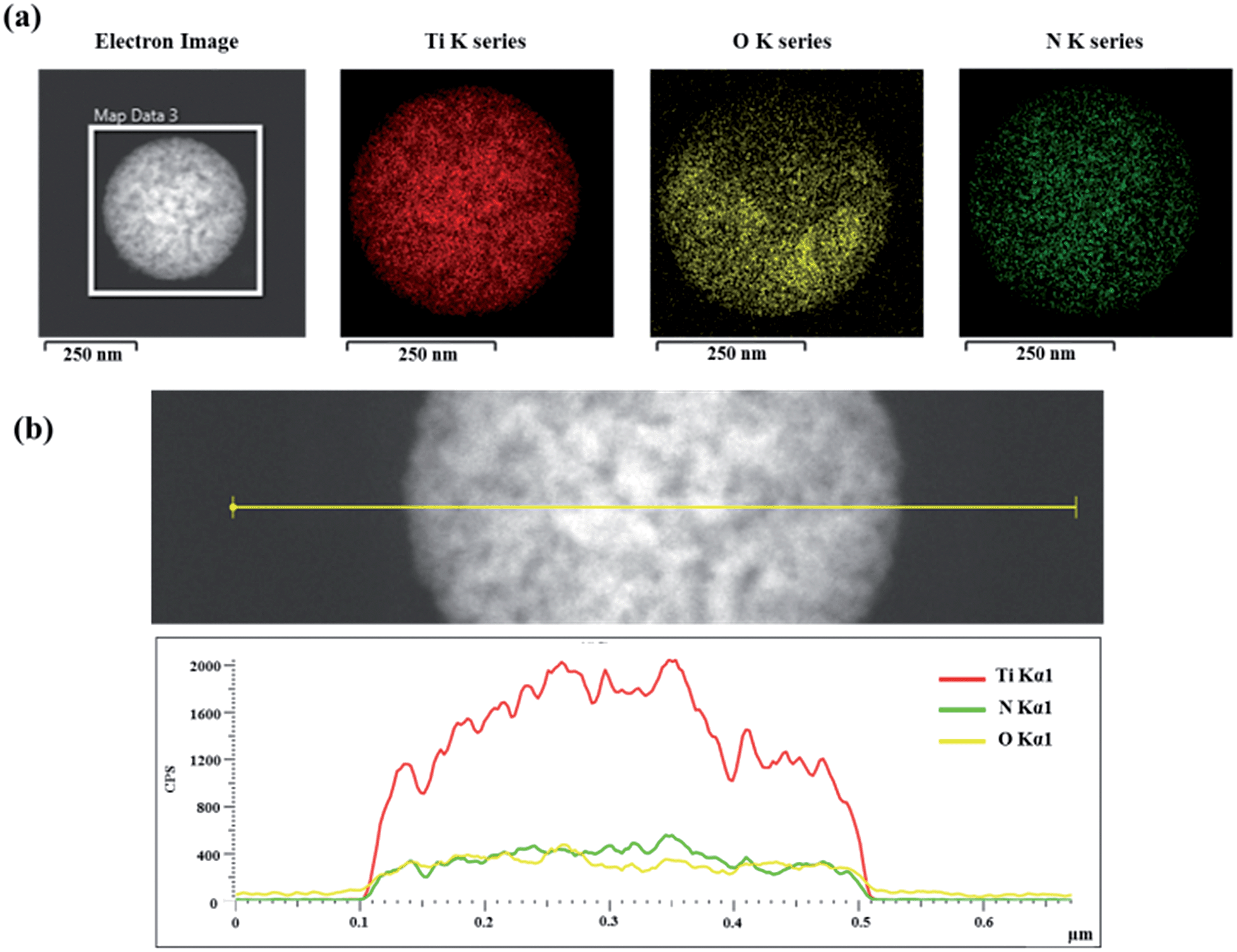 | ||
| Fig. 8 (a) Elemental mapping images of TiO2@TiO1−xNx nitrided at 700 °C for 24 h by scanning TEM. (b) EDX line profiles of N, O, and Ti across the TiO2@TiO1−xNx interface. | ||
As illustrated in Fig. 8, conventional TEM is very difficult to differentiate between core and shell components in TiO2@TiO1−xNx mainly because the L-shell peak of Ti (0.452 keV) and the K-shell peak of N (0.392 keV) were too close to distinguish their elemental intensities. We have thus investigated the structure and chemical composition of the bisected TiO2@TiO1−xNx microsphere with scanning transmission electron microscopy (STEM), electron energy loss spectroscopy (EELS), and energy-dispersive X-ray spectroscopy (EDS). A TiO2@TiO1−xNx microsphere nitrided at 700 °C for 10 h was used and approximately bisected by a focused ion beam (FIB). Fig. 9 presents the bisected STEM image and the corresponding EDS elemental mapping images of Ti, O, and N. The Ti and O signals are evenly dispersed in the core and shell parts of the bisected sphere whereas the N signals are located exclusively on the shell of the sphere, indicating that the TiO2@TiO1−xNx microsphere has the core/shell structure. To further convince the compositions of the core and shell parts, we have investigated the STEM-EDS analysis of the bisected sample. As given in Fig. S2,† the Ti, O, and N signals were observed in the shell part but only Ti and O signals were detected in the core. This clearly demonstrates that the TiO1−xNx phase is formed on the shell by the nitridation but the core TiO2 remains intact.
 | ||
| Fig. 9 (a) STEM images of a bisected TiO2@TiO1−xNx microsphere. The corresponding EELS mapping images of N ((b), green), Ti ((c), yellow), and O ((d), red). | ||
Diffuse reflectance UV-visible spectroscopy (DRUVS) was employed to determine whether the nitrided sample absorbs light effectively in the visible light region. The DRUVS spectrum of TiO2@TiO1−xNx nitrided at 700 °C for 24 h clearly shows that the sample absorbs much more light in the visible regions in comparison with that of TiO2 (Fig. S3†). Thus the photocatalytic activity of TiO2@TiO1−xNx nitrided at 700 °C for 24 h was evaluated by measuring the degradation rate of methylene blue (MB) under visible light as a function of time. The absorbance of MB at 664 nm, which is the strongest peak of MB, was used to determine the concentration change of the MB solution.33
As shown in Fig. 10(a), the strong absorption peak is continuously weakened with increasing irradiation time. This clearly indicates that the TiO2@TiO1−xNx catalyst efficiently decomposes MB under visible light. Similar efficient activities were also observed in all the samples nitrided at 700 °C. Among them, the sample nitrided for 24 h has the highest performance on the MB decomposition, which is given in Fig. 10(b). Due to the large band gap energy (Eg = 3.2 eV) of TiO2, the core TiO2 is inactive in decomposition of MB under visible light. Therefore, it is evident that the TiO1−xNx phase in the shell is the only active component in the visible range of irradiation and can be considered as a visible-light photocatalyst. This result is consistent with that of the DRUVS data.
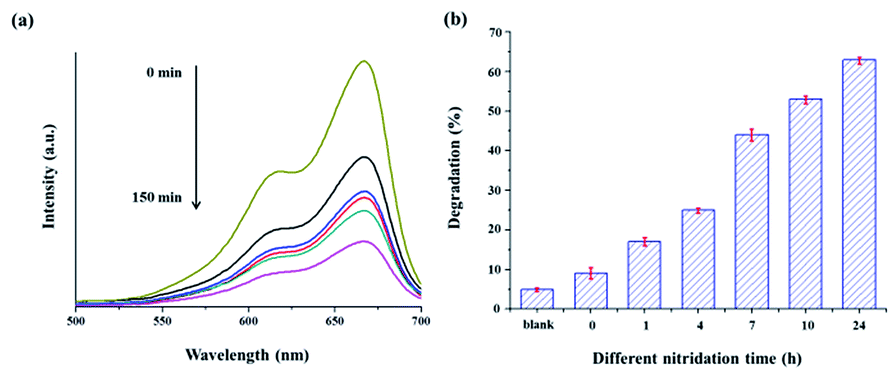 | ||
| Fig. 10 (a) UV-visible spectra of the MB solution in the presence of TiO2@TiO1−xNx nitrided at 700 °C for 24 h under visible-light irradiation (>420 nm) as a function of time of exposure to light. (b) Percentage of photocatalytic degradation of MB over the TiO2@TiO1−xNx samples with different nitridation times. | ||
To elucidate whether hydroxyl radicals (˙OH) produced from the illumination of TiO2@TiO1−xNx lead to the degradation of MB, we have employed the fluorescence technique that is usually used to estimate the formation of hydroxyl radicals on the surface of the photocatalyst.34–36Fig. 11 shows the fluorescence spectral change of the terephthalic acid in the presence of TiO2@TiO1−xNx nitrided at 700 °C for 24 h with increasing irradiation time. A new peak at about 425 nm, due to the hydroxylation product (2-hydroxyterephthalic acid), is distinctively displayed in the illuminated spectrum. The fluorescence intensity is gradually increased with increasing irradiation time. Based on these results, it is evident that the hydroxyl radicals generated from the TiO2@TiO1−xNx catalyst plays a significant role in the decomposition of MB.
 | ||
| Fig. 11 Time-dependent fluorescence spectra obtained from the illumination of a NaOH solution of terephthalic acid in the presence of TiO2@TiO1−xNx nitrided at 700 °C for 24 h. Each spectrum was recorded every 30 min of illumination. | ||
In addition to the chemical degradation of MB by the hydroxyl radicals, we also evaluated the capability of photocurrent generation of the nitrided samples under visible light. The photocurrent density was clearly observed, which was very reproducible in the repeated on/off cycles of excitation.
As illustrated in Fig. 11, the photocurrent of the nitrided samples is enhanced when the nitridation time increased. The sample nitrided for 24 h has the largest value of photocurrent density (∼170 μA cm−2), which is comparable with that of carbon nitride (C3N4).37,38 This clearly demonstrates that the TiO2@TiO1−xNx catalyst is able to generate photocurrents effectively under visible light while TiO2 does not produce photocurrents under the same conditions (Fig. 12).
 | ||
| Fig. 12 Photocurrent response of the TiO2@TiO1−xNx samples with different nitridation times under an applied potential of 0.8 V (vs. Ag/AgCl) and a periodic irradiation of visible light. | ||
Conclusions
The present study demonstrated the successful synthesis of new composite photocatalysts (TiO2@TiO1−xNx) with enhanced visible light activity, which was accomplished by a controlled nitridation of TiO2 microspheres. The nitrided products maintain a spherical shape and have a unique core/shell structure. The core and shell parts are mainly composed of amorphous TiO2 and crystalline TiO1−xNx phases, respectively. They exhibited high photocatalytic and photoelectrochemical activities under visible light irradiation. Our structural and spectroscopic results confirm that the origin of their activity is due primarily to the TiO1−xNx phase. Specifically, the nitrogen dopant in TiO1−xNx appears to be responsible for photocatalytic activity under visible-light similar to the well-documented N-doped TiO2 phase (TiO2−xNx). Our studies on the TiO1−xNx phase having a simple rock-salt structure promise a wide range of visible-light applications in both photocatalytic and photoelectrochemical systems.Acknowledgements
We express gratitude to the editorial staff at Sogang University for the careful English revision of this manuscript.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- J. Huo, Y. Hu, H. Jiang and C. Li, Nanoscale, 2014, 6, 9078–9084 RSC.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- M. Zhou and J. Yu, J. Hazard. Mater., 2008, 152, 1229–1236 CrossRef CAS PubMed.
- A. Naldoni, F. Fabbri, M. Altomare, M. Marelli, R. Psaro, E. Selli, G. Salviati and V. D. Santo, Phys. Chem. Chem. Phys., 2015, 17, 4864–4869 RSC.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. D. Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 CrossRef CAS PubMed.
- J. Graciani, S. Hamad and J. F. Sanz, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 184112/1–184112/10 CrossRef CAS.
- S. G. Seo, C. Park, H. Kim, W. H. Nam, M. Jeong, Y. Choi, Y. S. Lim, W. Seo, S. Kim, J. Y. Lee and Y. S. Cho, J. Mater. Chem. A, 2013, 1, 3639–3644 CAS.
- P. Simon, B. Pignon, B. Miao, S. C. Leconte, Y. Leconte, S. Marguet, P. Jegou, B. B-Fabre, C. Reynaud and N. H-Boime, Chem. Mater., 2010, 22, 3704–3711 CrossRef CAS.
- V. Ern and A. C. Switendick, Phys. Rev., 1965, 137, A1927–A1936 CrossRef.
- P. Zheng, J. Zhao, J. Zheng, G. Ma and Z. Zhu, J. Mater. Chem., 2012, 22, 12116–12120 RSC.
- C. Gong, C. Yan, J. Zhang, X. Cheng, H. Pan, C. Zhang, L. Yu and Z. Zhang, J. Mater. Chem., 2011, 21, 15273–15278 RSC.
- E. P. Quijorna, V. T. Costa, F. A. Rueda, P. H. Fernández, A. Climent, F. Rossi and M. M. Silván, J. Phys. D: Appl. Phys., 2011, 44, 235501/1–235501/8 CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- A. T-Zajac, M. Radecka, K. Zakrzewska, A. Brudnik, E. Kusior, S. Bourgeois, M. C. Lucas and L. Imhoff, J. Power Sources, 2009, 194, 93–103 CrossRef.
- W. Spengler, R. Kaiser and H. Bilz, Solid State Commun., 1975, 17, 19–22 CrossRef CAS.
- W. Spengler, R. Kaiser, A. N. Christensen and G. M. Vogt, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 17, 1095–1101 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–642 CrossRef CAS PubMed.
- F. Peng, L. Cai, L. Huang, H. Yu and H. Wang, J. Phys. Chem. Solids, 2008, 69, 1657–1664 CrossRef CAS.
- W. Göpel, G. Rocker and R. Feierabend, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 3427–3438 CrossRef.
- X. Song, D. Gopireddy and C. G. Takoudis, Thin Solid Films, 2008, 516, 6330–6335 CrossRef CAS.
- M. Wolff, J. W. Schultze and H. H. Strehblow, Surf. Interface Anal., 1991, 17, 726–736 CrossRef CAS.
- I. Takahashi, D. J. Payne, R. G. Palgrave and R. G. Egdell, Chem. Phys. Lett., 2008, 454, 314–317 CrossRef CAS.
- W. Cai, G. Duan and Y. Li, Hierarchical Micro/Nanostructured Materials: Fabrication, Properties and Applications, CRC Press, Taylor and Francis Group, 2014 Search PubMed.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2013, 117, 1383–1391 CAS.
- L. Sun, Y. Qin, Q. Cao, B. Hu, Z. Huang, L. Ye and X. Tang, Chem. Commun., 2011, 47, 12628–12630 RSC.
- T. P. Gujar, C. Anand, V. R. Shinde, J. Ye, K. Ariga and A. Vinu, J. Nanosci. Nanotechnol., 2010, 10, 8124–8129 CrossRef CAS PubMed.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 CAS.
- J. Liu, H. Wang, Z. P. Chen, H. Moehwald, S. Fiechter, R. Krol, L. Wen, L. Jiang and M. Antonietti, Adv. Mater., 2015, 27, 712–718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06758h |
| This journal is © The Royal Society of Chemistry 2016 |