
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structural characterisation and proton conduction of two new hydrated phases of barium ferrite BaFeO2.5−x(OH)2x†
Patrick L.
Knöchel
a,
Philip J.
Keenan
b,
Christoph
Loho
ac,
Christian
Reitz
c,
Ralf
Witte
c,
Kevin S.
Knight
d,
Adrian J.
Wright
b,
Horst
Hahn
ac,
Peter R.
Slater
b and
Oliver
Clemens
*ac
aTechnical University of Darmstadt, Joint Research Laboratory Nanomaterials, Jovanka-Bontschits-Straße 2, 64287 Darmstadt, Germany. E-mail: oliver.clemens@nano.tu-darmstadt.de; Fax: +49 6151 16 6335
bUniversity of Birmingham, School of Chemistry, Edgbaston, Birmingham B15 2TT, UK
cKarlsruhe Institute of Technology, Institute of Nanotechnology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dISIS Facility, Rutherford Appleton Laboratory, Harwell, Didcot, Oxford, OX11 0QX, UK
First published on 25th January 2016
Abstract
Materials exhibiting mixed electronic and proton conductivity are of great interest for applications ranging from electrodes for proton conducting ceramic fuel cells to hydrogen separation membranes. In this work, we report a detailed investigation of the effect of water incorporation in BaFeO2.5 on the structure and conductivity. BaFeO2.5 is shown to be topochemically transformed to two different hydrated modifications, low-water (LW-) and high-water (HW-) BaFeO2.5. A combined analysis of neutron and X-ray diffraction data was used to determine the crystal structure of LW-BaFeO2.5 (BaFeO2.33(OH)0.33), which shows a unique ordering pattern of anion vacancies for perovskite type compounds, with structural relaxations around vacancies being similar to the chemically similar compound BaFeO2.33F0.33. Approximate proton positions were determined using the bond valence method. Conductivity studies of hydrated and pure BaFeO2.5 (with additional comparison to oxidized BaFeO2.5) show a significant enhancement of the conductivity on water incorporation, which can be attributed to proton conductivity. This is the first report of significant grain proton conduction (∼10−6 to 10−7 S cm−1) in an iron based perovskite. Water uptake is further shown to be completely reversible, with reformation of BaFeO2.5 when heating the compound to temperatures above ∼450 K under Ar.
1 Introduction
Perovskite type compounds with composition AFeO2.5+y (A = Sr, Ba; 0 ≤ y ≤ 0.5) are interesting materials regarding their potential application as electrode materials for solid oxide fuel cells as well as for their magnetic properties (e.g.ref. 1 and 2). Among them, barium ferrite BaFeO2.5 has been extensively studied regarding its crystal structure, and shown to display a complex scheme of vacancy order.3–5 This vacancy order is only partly related to the well-known order found for brownmillerite type compounds ABX2.5. Recently, it was found that this material can be fluorinated to form a compound BaFeO2.33F0.33, and this prior work highlighted the vacancy relaxation mechanisms which seem to be favorable in barium ferrite type compounds.However, the vacancy chemistry of perovskite materials is not only interesting from a structure-chemical point of view. The flexibility of the anion sublattice gives rise to various properties, i.e. oxide ion conductivity as well as mobility of electrons due to the presence of a mixed valent oxidation state of the iron cation for y > 0. Apart from oxidation and fluorination reactions, anion vacancies of a perovskite matrix can be filled via the incorporation of water molecules, leading to the formation of hydroxide groups within the perovskite framework. This was then found to give rise to proton conductivity e.g. for BaZr1−xYxO3−x/2−z/2(OH)z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 or BaInO2(OH).7 The introduction of proton conductivity can then give rise to further applications, e.g. the use as proton conducting membranes for the use in Solid Oxide Electrolysis Cells (SOECs), where the use of proton conductors is of special interest due to the fact that they facilitate the formation of water-free hydrogen.8 In this respect, it is always of interest to find new materials with reasonable proton conductivity.9 In particular, one of the current limitations with regard to solid oxide fuel cells/electrolysers utilizing a proton conducting ceramic is the lack of a suitable electrode material for the air/oxygen electrode. Currently researchers are principally employing electrode materials that have been developed for use with oxide ion conducting electrolytes. While some, such as Ba0.5Sr0.5Co0.8Fe0.2O3−y, Pr2NiO4, have shown respectable performance, the performance still significantly lags behind the performance of such electrodes with oxide ion conducting electrolytes.10,11 From a consideration of the two types of fuel cell, the poor performance of traditional mixed oxide ion/electronic conducting electrodes is not that surprising, since there are significant differences in the processes occurring. In the case of a fuel cell based on an oxide ion conducting electrolyte, the cathode process is
6 or BaInO2(OH).7 The introduction of proton conductivity can then give rise to further applications, e.g. the use as proton conducting membranes for the use in Solid Oxide Electrolysis Cells (SOECs), where the use of proton conductors is of special interest due to the fact that they facilitate the formation of water-free hydrogen.8 In this respect, it is always of interest to find new materials with reasonable proton conductivity.9 In particular, one of the current limitations with regard to solid oxide fuel cells/electrolysers utilizing a proton conducting ceramic is the lack of a suitable electrode material for the air/oxygen electrode. Currently researchers are principally employing electrode materials that have been developed for use with oxide ion conducting electrolytes. While some, such as Ba0.5Sr0.5Co0.8Fe0.2O3−y, Pr2NiO4, have shown respectable performance, the performance still significantly lags behind the performance of such electrodes with oxide ion conducting electrolytes.10,11 From a consideration of the two types of fuel cell, the poor performance of traditional mixed oxide ion/electronic conducting electrodes is not that surprising, since there are significant differences in the processes occurring. In the case of a fuel cell based on an oxide ion conducting electrolyte, the cathode process is
| O2 + 4e− → 2O2− |
However for a proton conducting electrolyte system, the cathode process has additional complexity. In this case the electrode process is:
| 4H+ + 4e− + O2 → 2H2O |
Thus protons need to be transported from the electrolyte to the electrode, and the water produced needs to be removed from the electrode. Thus the exploitation of cathodes developed for oxide ion conducting electrolytes (and hence showing mainly electronic and oxide ion conduction) seems a perverse option. Rather there would appear to be a need to develop a material capable of good electronic and proton conductivity (the ability to accommodate water). Currently, apart from research into Ti/Zr based electrolyte systems,12 there is, however, a lack of studies into the ability of perovskite materials containing transition metals to accommodate water and exhibit proton conduction.
In this article, we report on the hydration of BaFeO2.5 under wet streams of argon gas, and we show that the material can be converted from a poor electron conductor into a reasonable proton conductor. The water uptake is accompanied by a change in crystal structure, which has been characterized for two different hydrated modifications of BaFeO2.5, low-water BaFeO2.5 (prepared at 423 K, LW-BaFeO2.5, ∼BaFeO2.33(OH)0.33) and high-water BaFeO2.5 (prepared at 393 K, HW-BaFeO2.5, ∼BaFeO2.25(OH)0.5).
2 Experimental
2.1 Synthesis of compounds
BaFeO2.5 was prepared from BaCO3 and Fe2O3via solid state reaction as described in a previous article.5For the preparation of the hydrated phases of BaFeO2.5 (called low- (LW-BaFeO2.5) and high-water (HW-BaFeO2.5) phases in the following), a setup as described in ref. 13 was used. The nebulizer was filled with deionized water and a very wet gas stream was produced by applying an argon flow (99.996% purity; to rule out oxidation of the powders) of 1 standard liter per minute (SLM) and fed over heated powder of BaFeO2.5. Different hydration temperatures of 120 °C, 150 °C, and 180 °C were examined, where the lower temperature led to the formation of HW-BaFeO2.5 and the two higher temperatures led to the formation of LW-BaFeO2.5.
Pellets of BaFeO2.5 were made by sintering the isostatically pressed sample (600 kN, 1 min) at 1373 K for 12 h under flowing Ar (purity 99.996%). To test the influence of oxidation, one such pellet was also heated under flowing oxygen at 773 K for 15 h. Attempts to hydrate the as-synthesized pellets did not prove to be successful and resulted in cracking (i.e. the pellets were very brittle) due to the strong volume increase upon hydration (see Section 3.3). Therefore, a pellet of LW-BaFeO2.5 was prepared by isostatically pressing an uniaxially prepressed pellet of the powder at 600 kN for 1 min (for better comparison a non-sintered pellet of non-hydrated BaFeO2.5 was also prepared using the same method). The as-prepared pellets were carefully polished and gold was sputtered on both sides to ensure good contacting for the subsequent impedance analysis. Whereas the sintered pellets of BaFeO2.5 showed high densities of ∼91% of the crystallographic density, non-sintered pellets of BaFeO2.5 as well as its hydrated compounds showed lower densities in the range of 68–83%.
2.2 Diffraction experiments
X-ray powder diffraction (XRD) patterns of the oxide and hydrated compounds were recorded on a Bruker D8 diffractometer with Bragg-Brentano geometry and a fine focus X-ray tube with Cu anode. No primary beam monochromator was attached. A VANTEC detector and a fixed divergence slit (0.1°) were used. The total scan time was 10 hours for the angular range between 5 and 130° 2θ.High-temperature X-ray powder diffraction (XRD) patterns of the hydrated compounds were recorded on the same setup (except for using a 0.3° fixed divergence slit) using an Anton Paar HTK 1200N High-Temperature Oven-Chamber in the temperature range between 50 and 550 °C.
Time of flight powder neutron diffraction (NPD) data were recorded on the HRPD high resolution diffractometer at the ISIS pulsed spallation source (Rutherford Appleton Laboratory, UK). 4 g of powdered samples of LW- and HW-BaFeO2.5 were loaded into 8 mm diameter thin-walled, cylindrical vanadium sample cans and data collected at ambient temperature (RT) and 4 K for 320 μA h proton beam current to the ISIS target (corresponding to ∼10 hours of beam time). HW-BaFeO2.5 was additionally heated at temperatures of 393, 423, 573, 773, and 1173 K to observe structure transformations of the compound, in these cases with reduced measurement times of 120 μA h proton beam current (∼3 hours of beam time). For HW-BaFeO2.5, it was observed that for the measurements recorded at 393 and 423 K (under vacuum) the compound had already transformed into LW-BaFeO2.5, whereas at all other higher temperatures as well as on the final measurement at RT, the compound had released all of the water to form monoclinic BaFeO2.5.
Refinement of the structures of the hydrated BaFeO2.5 compounds was performed with the program TOPAS Academic 5 (ref. 14 and 15) using the NPD data collected from all of the HRPD detector banks 1–3 as well as the XRD data. Details concerning the structural analysis will be extensively discussed in Section 3.2.
2.3 Thermogravimetric measurements
Thermogravimetric (TG) measurements were recorded for LW- and HW-BaFeO2.5 on a Netzsch STA 429 thermal analyser in the temperature range between 300 and 673 K using a heating rate of 5 K min−1 under a constant flow of argon gas, with the sample placed in Al2O3 crucibles. The data curves were corrected with the data curve recorded for the empty crucible.2.4 IR spectroscopy
Fourier transform infrared (IR) spectroscopy data were recorded using a Bruker Vertex 70 spectrometer (Bruker, USA). The compound was mixed with KBr in an approximate ratio of 1:20, ground in a mortar and pressed into a pellet of 15 mm diameter and 0.5 mm thickness. The spectra were recorded in a range between 400 and 4000 cm−1 with a step size of 4 cm−1.
2.5 Impedance measurements
AC impedance spectroscopy was used to determine the conductivity of the different sintered and non-sintered pellets (see Section 3.3). Pellets were sputtered with a thin layer of gold on both sides to allow for proper electrical contact. The samples were placed in a JANIS STVP-200-XG cryostat, which can be operated under a static helium atmosphere of 1 bar pressure and were investigated in variable temperature ranges between 200 and 500 K. Impedance measurements were performed using a Solartron 1260 frequency response analyzer applying an AC signal with 100 mV amplitude over a frequency range from 1 MHz to 100 mHz. The impedance data were fitted using the ZView program.163 Results and discussion
3.1 Hydration behavior of BaFeO2.5 and analysis of composition of its hydrated phases LW- and HW-BaFeO2.5
The existence of hydrated phases of BaFeO2.5 was first discovered in attempts to synthesize nano-crystalline BaFeO2.5 using a nebulized spray pyrolysis method as described in ref. 13. In this method, precursor salts are dissolved in water, nebulized using an ultrasonicator and passed through a reaction zone at temperatures usually around 1073–1273 K using argon as a carrier gas. The formed product particles are then collected on a filter heated at about 393 K. However, using this method very wet gas streams are obtained, and it was observed that although a product with a structure related to the cubic perovskite structure could be obtained, the diffraction pattern of BaFeO2.5 (ref. 5) was never observed. In further attempts, micro-crystalline powder of BaFeO2.5 prepared by classic solid state routes was then heated using the same wet flow of argon gas created by an ultrasonicator at temperatures of 393 K and 423 K. Depending on the temperature, the formation of different hydrated forms of BaFeO2.5 was observed, which will be called high water (HW) and low water (LW) BaFeO2.5 through the subsequent sections. On a further note, it was found that the formation and stability of these phases is very sensitive to the exact conditions of hydration and storage, e.g. when using a wet stream of argon made by leading argon gas through a bubbler, formation of HW-BaFeO2.5 could not be observed. Therefore, it is very important to give rather exact details on the performed experiments, since the compounds BaFeO2.5, LW- and HW-BaFeO2.5 will be shown to be transmutable into each other depending on the detailed reaction parameters.XRD patterns comparing the non-hydrated form to the different forms of BaFeO2.5 hydrated at different temperatures (423 K and 393 K) are shown in Fig. 1. From the angular shift of the center of gravity of the main group of reflections around 31° 2θ, it can be seen that hydration leads to a strong increase of cell volume. Due to the preparation under protective atmosphere, oxidation of the compounds can be effectively discounted, and indeed such oxidation was observed to take place preferably at higher temperatures (starting around 673–873 K) with longer reaction times at 1 bar O2 partial pressure.17 Furthermore, oxidation is known to be accompanied by a decrease in cell volume (rather than an increase), which can be explained by the smaller size of the Fe4+ cation compared to Fe3+.17–19 An increase of cell volume on hydration is also consistent with the situation observed for similar perovskite compounds which can incorporate large amounts of water, e.g. for the hydration of BaInO2.5 to BaInO2(OH).20
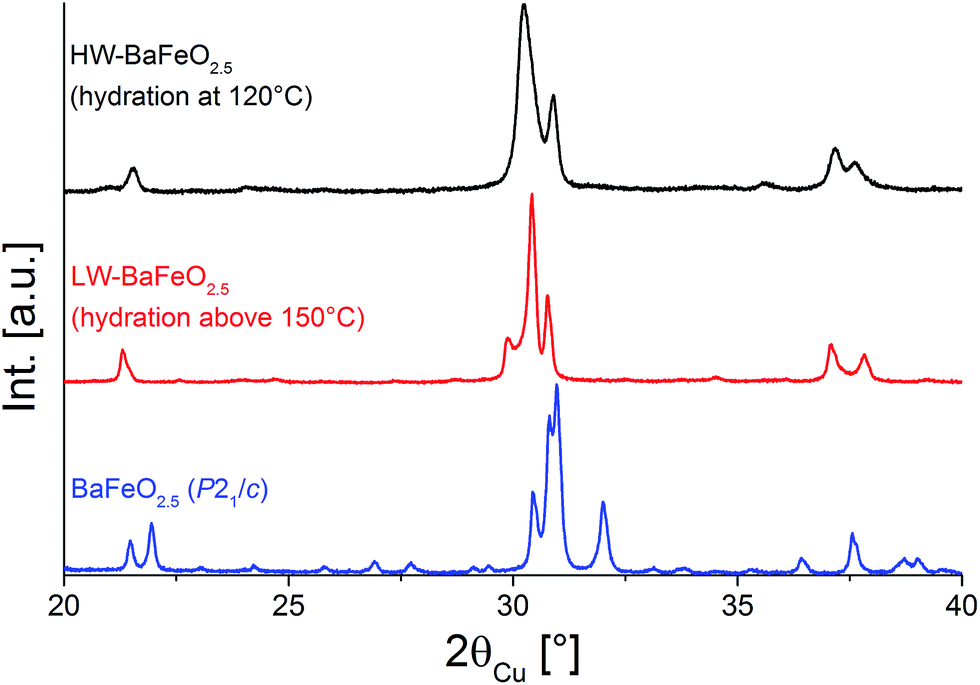 | ||
| Fig. 1 XRD patterns of BaFeO2.5 and of its two hydrated modifications LW-BaFeO2.5 and HW-BaFeO2.5. | ||
It is straightforward to conclude that the hydration reaction of BaFeO2.5 is topotactic in nature, with maintenance of the main perovskite type framework of the Ba2+ and Fe3+ cations, where only the anions show a stronger degree of structural reorganization. This topotactic nature is evident from diffraction data, where the main reflection groups are retained on dehydration of BaFeO2.5 (with changes of the splitting pattern and amount/position of superstructure reflections), and also from the fact that monoclinic BaFeO2.5 is reformed on heating the hydrated phases to elevated temperatures in a variable-temperature XRD-chamber under constant flow of dry argon gas (see Fig. 2). Additionally, the hydrated phases are not reformed on cooling due to the fact that the high-temperature chamber is constantly flushed with argon gas, which removes the water from the heating chamber, and instead pure monoclinic BaFeO2.5 is obtained after cooling down to ambient temperature. In summary, these high temperature diffraction studies provide compelling evidence that influences of redox-active reactions (e.g. uptake and release of O2) can be discounted and that the oxidation state of Fe3+ must be maintained when the compound is treated under flowing argon gas.
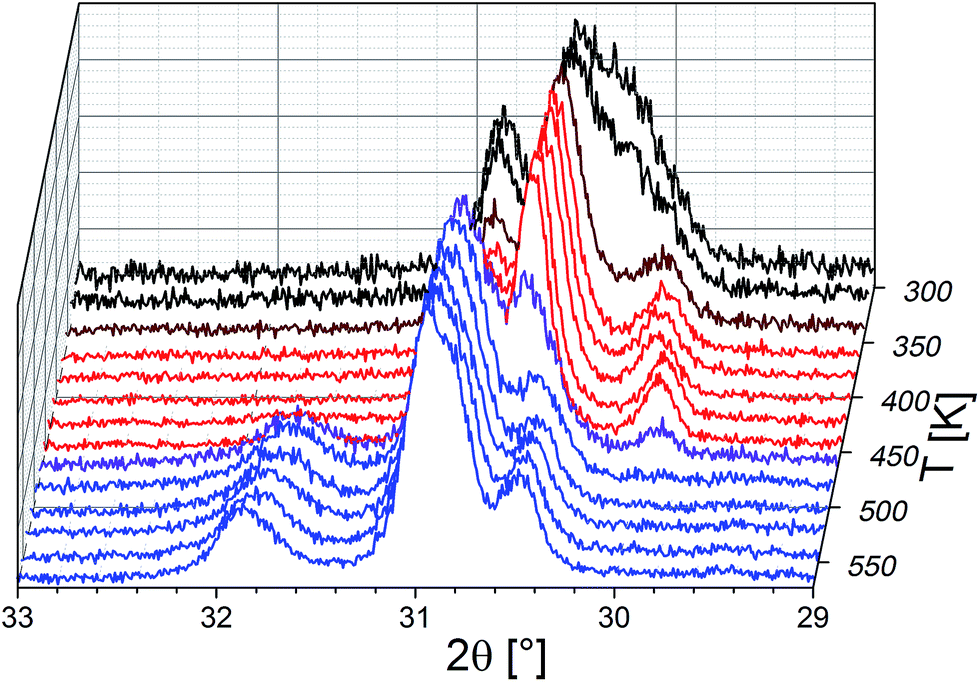 | ||
| Fig. 2 Variable temperature XRD patterns recorded between 313 and 573 K under dry flowing argon atmospheres. Diffraction patterns recorded at different temperatures with mainly HW-BaFeO2.5 (black), LW-BaFeO2.5 (red), and BaFeO2.5 (blue) are shown. Brown and purple curve indicate phase mixtures of the respective phases. The 2θ-axis was inverted to provide a better depiction of the changes. | ||
Thermogravimetric analysis was performed on both samples LW- and HW-BaFeO2.5 to determine their approximate water content (see Fig. 3). In agreement with the high temperature XRD data, LW-BaFeO2.5 was found to lose its crystal water starting around 450–500 K (the temperature difference between TG and high temperature XRD is reasonably well explained by the fact that samples were kept for 20 min at the respective measurement temperature for the XRD experiments, whereas a heating rate of 5 °C min−1 was applied for the TG measurements, i.e. the latter is a more dynamic, non-equilibrium process). Although the weight loss is quite steep, the lattice water seems not to be eliminated completely until a temperature of ∼550 K. Such a finding is in agreement with e.g. proton conductivity studies reported for other perovskite compounds (e.g. Y-doped BaZrO3 (ref. 6)) and in principal agreement with impedance studies reported in Section 3.3. In agreement with the high temperature XRD studies, HW-BaFeO2.5 starts losing its lattice water (in good agreement with the change of the diffraction pattern, see Fig. 2) already at temperatures below its hydration temperature of 393 K, which can be explained by the fact that the DTA/TG analysis was performed under a flow of dry argon atmosphere. The weight losses obtained after heating to 673 K indicate overall compositions of ∼BaFeO2.333(OH)0.333 for LW-BaFeO2.5 and ∼BaFeO2.24(OH)0.53 for HW-BaFeO2.5 respectively. Remarkably the composition BaFeX2.667 of LW-BaFeO2.5 is relatively close to what was found for the chemically similar (vacancy ordered) compound BaFeO2.333F0.333,21 and it will be shown later in the ESI† that structural similarity is indeed found between this phase and BaFeO2.333F0.333. The composition of HW-BaFeO2.5 is furthermore close to a composition of BaFeX2.75, with similar compositions also found for other anion deficient perovskite type phases (e.g. SrFeO2.75 (ref. 22)).
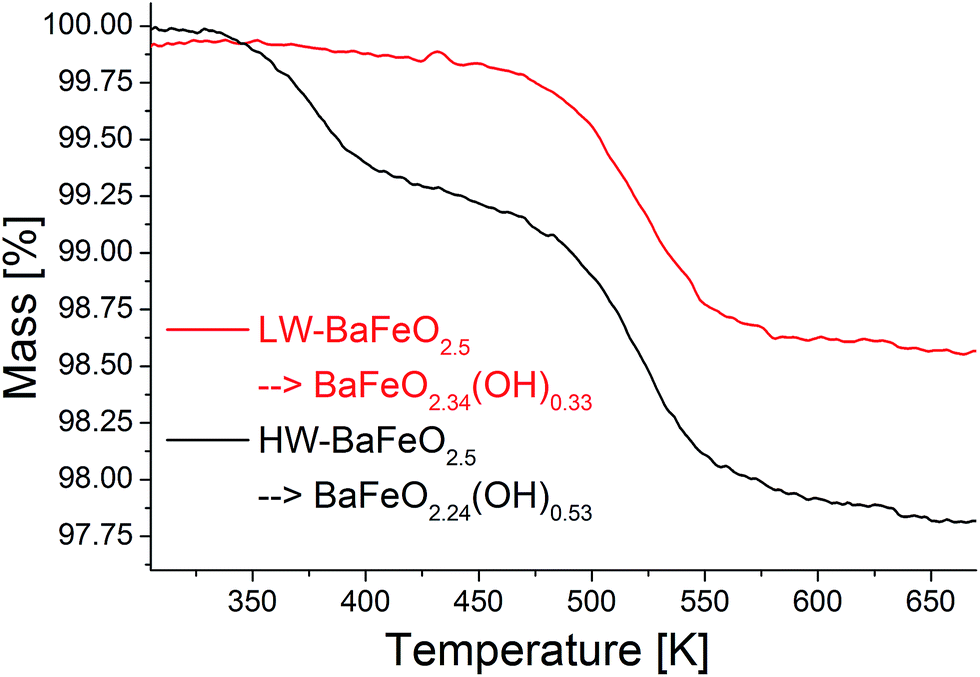 | ||
| Fig. 3 TG experiments performed on the hydrated modifications of BaFeO2.5 under flowing argon. | ||
The incorporation of protons into the structure is confirmed by FTIR measurements, which show a broad band around 3300–3400 cm−1, consistent with the OH stretching mode23 and in agreement with what has been found for other iron hydroxides (e.g.ref. 24). Apart from this, the presence of protons is also indicated by an increase of the background for the neutron diffraction experiments due to incoherent scattering from H and the consequent requirement for longer measurement times to obtain reasonable signal to noise ratios. Additionally, the loss of water molecules was independently confirmed by coupling the TG experiment to mass spectroscopy, and the temperature range is typical for the release of water within perovskite systems.
On a further note, a colour change compared to BaFeO2.5 was observed on hydration of the compounds. On hydration the colour changed from dark brown (for BaFeO2.5) to a brighter brownish tone for the hydrated modifications. This change in colour is most expressed when comparing the colour of nano-HW-BaFeO2.5 obtained by spray pyrolysis (which shows a bright yellow colour) to BaFeO2.5 obtained from solid state reaction (which shows a dark brown hue). It is also worth pointing out that on leaving LW- and HW-BaFeO2.5 standing in air, a further colour change to a very bright orange occurs. XRD analysis of this orange powder showed the presence of BaCO3, without the appearance of any further crystalline phases. This CO2 sensitivity is also known for further Ba-containing perovskite type compounds, such as e.g. BaInO2.5.9,25,26 Additionally, it gives a further support for the uptake of water, since the high basicity of the resulting BaFeO2.5−x(OH)2x powders reasonably explains the reactivity with an acidic gas such as CO2. In contrast, non-hydrated BaFeO2.5 was shown to be essentially insensitive towards CO2 uptake at ambient temperature.27,28
3.2 Structural analysis of LW- and HW-BaFeO2.5
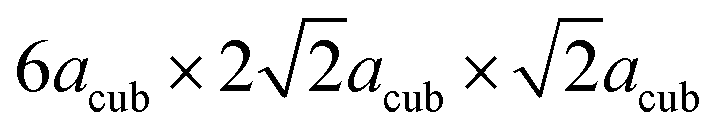 are needed to obtain an acceptable fit of the diffraction patterns (i.e. containing 24 formula units of BaFeO2.5(H2O)x per unit cell). However, both diffraction patterns of LW- as well as of HW-BaFeO2.5 can be indexed using this supercell. In particular, the large unit cell volumes are necessary to properly fit the high d-spacing superstructure reflections found at low angles in the XRD pattern (below 10° 2θ, see Rietveld analyses shown later in this section); additionally, C-centering was also indicated from the systematic absences of reflections. From our previous diffraction studies of BaFeX3−d (X = O, F),5,21 we note that barium ferrites seem to maintain their tendency to form complicated superstructures of the cubic perovskite prototype: for example BaFeO2.5 (ref. 5) crystallizes in a 28-fold vacancy-ordered superstructure, while BaFeO2.33F0.33 shows a 6-fold superstructure of the cubic perovskite structure.
are needed to obtain an acceptable fit of the diffraction patterns (i.e. containing 24 formula units of BaFeO2.5(H2O)x per unit cell). However, both diffraction patterns of LW- as well as of HW-BaFeO2.5 can be indexed using this supercell. In particular, the large unit cell volumes are necessary to properly fit the high d-spacing superstructure reflections found at low angles in the XRD pattern (below 10° 2θ, see Rietveld analyses shown later in this section); additionally, C-centering was also indicated from the systematic absences of reflections. From our previous diffraction studies of BaFeX3−d (X = O, F),5,21 we note that barium ferrites seem to maintain their tendency to form complicated superstructures of the cubic perovskite prototype: for example BaFeO2.5 (ref. 5) crystallizes in a 28-fold vacancy-ordered superstructure, while BaFeO2.33F0.33 shows a 6-fold superstructure of the cubic perovskite structure.
At this point the reader should be made aware that solving crystal structures from powder XRD data is not trivial for barium ferrites with complex structures, as was pointed out in our previous works:5,21 for BaFeO2.5, refining positional parameters (in total 7 Ba, 7 Fe, and 17 O ions on general sites x, y, z) of the prior structural model (determined on a combined analysis of neutron and X-ray diffraction data) using only X-ray diffraction data leads to a chemically implausible change of the structure due to strong correlation of parameters. Indeed, no sensible structural models of LW and HW-BaFeO2.5 could be obtained on the refinement of powder XRD data only, which is related to the fact that the substructure of the anions (and protons) is likely to be the predominant cause for the structural distortion. Apart from the large number of independent structural parameters for such large C-centered unit cells, the complexity of the structural analysis of LW- and HW-BaFeO2.5 is further exacerbated due to the fact that the compounds show strong anisotropic strain broadening (which is accounted for by using Stephens' model29 for the analysis of the XRD data and spherical harmonics for analysis of the NPD data, with the latter being reported later in this section). Therefore, we did not succeed in obtaining structurally sensible models of LW- and HW-BaFeO2.5 using powder XRD data only. However, it is worth pointing out that structural fits, using e.g. a Cmmm type superstructure with the cell parameters determined from indexing indicated that the splitting and intensity pattern of the main reflections can be well described.
To obtain a deeper structural understanding of the hydration reaction and to be able to approximately locate the additional oxygen ions and protons, neutron diffraction data have been recorded using the high resolution powder diffractometer HRPD at various temperatures between 4 and 423 K. To narrow down the number of potential space groups which must be considered for the analysis of the diffraction data, a coupled analysis of the XRD and neutron diffraction data from all the detector banks was attempted using the Pawley method. This analysis is however complicated and impeded by the fact that perovskite compounds containing only Fe3+ show magnetic ordering usually below temperatures of 600–700 K. For an accurate analysis of the correct space group from the extinction of reflections (i.e. meaningful indexing), it is therefore necessary to identify the reflections/group of reflections with strong influence from magnetic scattering. To make the subsequent discussion easier to follow, the reader must therefore be provided with some background information on magnetic ordering of related Fe3+ containing compounds: BaFeO2.5 and SrFeO2.5 both crystallize in structures derived from the cubic perovskite structure (in the case of SrFeO2.5 this is the well-known brownmillerite type structure). In both compounds, Fe coordination polyhedra are only connected by corners to neighbouring Fe polyhedra, with bond angles close enough to 180° to facilitate strong antiferromagnetic superexchange interactions between such neighbouring sites,30 resulting in G-type antiferromagnetic structures. In addition for Fe3+ containing perovskite compounds with partly hexagonal stacking of the AX3 layers (e.g. 6H- and 15R-BaFe(O/F)3−d), antiferromagnetic ordering is found between neighbouring sites connected by corner-sharing of their polyhedra,31–34i.e. between c-type stacked AX3 layers. Indeed, antiferromagnetic ordering (with potential presence of very small canting of the magnetic moments) of the compounds is in excellent agreement with very low magnetic moments per formula unit found in magnetic measurements, which are exemplarily shown for LW-BaFeO2.5 (see Fig. 4). The remaining small ferromagnetic component is below 0.002 μB per formula unit and could be explained by a small magnetic moment resulting from magnetic canting and/or the presence of a very small amount of a further impurity phase.
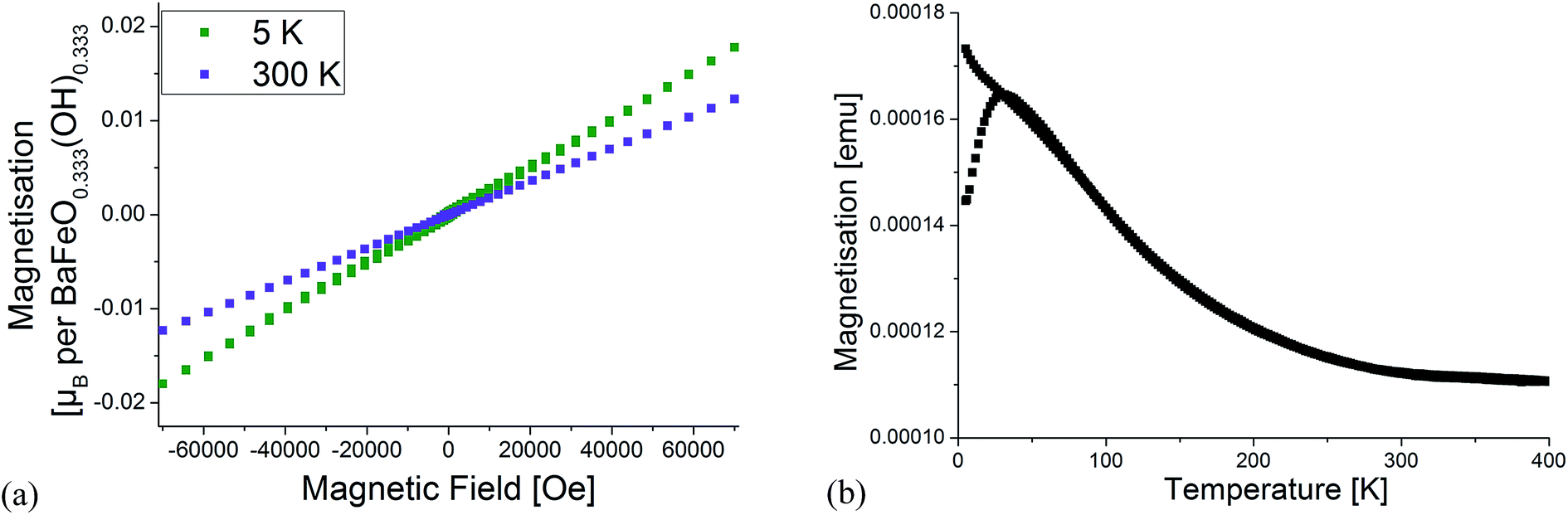 | ||
| Fig. 4 Magnetic characterisation of LW-BaFeO2.5. Field dependent measurements at 5 K and 300 K (a) as well as temperature dependence of magnetisation (b). | ||
Fig. 5 and 6 give an overview of the neutron diffraction data recorded in different detector banks of the HRPD diffractometer. A strong contribution from magnetic scattering was observed for all the measurements recorded for HW-BaFeO2.5, whereas the contribution of magnetic scattering for LW-BaFeO2.5 was only high for the measurement taken at 4 K. At 393 and 423 K, the contribution of magnetic scattering is lowered to a minimum contribution for LW-BaFeO2.5. In addition it was found that the magnetic reflection groups predominately appear (also in the 90° and backscattering detector banks 2 and 1) at the same position as the magnetic reflections observed for G-type antiferromagnetically ordered BaFeO2.5. At a later stage of our analysis, it was found that the intensity pattern of the magnetic reflections is indeed accurately described by such G-type antiferromagnetic arrangement of magnetic moments (∼ 4 μB at 4 K), agreeing with what was found for other Fe3+ containing compounds5,31,32,35–37 and what would be expected for cations with d5 high spin configuration in coordination polyhedra with corner sharing only. In order to facilitate the first structural analyses, neutron diffraction data of LW-BaFeO2.5 recorded at RT were used to obtain an approximate solution of the structure of LW-BaFeO2.5 due to the fact that most of the magnetic scattering had nearly disappeared for this compound. We would also like to make the reader aware that additional reflections can be found in the patterns recorded at 4 K (Fig. 6), which are present for both LW- and HW-BaFeO2.5 at exactly the same positions in the high resolution back-scattering bank 1, but not in bank 2. Due to the additional fact that they appear at exactly the same position for both compounds (which strongly differ in cell parameters, see later in this article), they can be ruled out as belonging to the perovskite phase, and instead are attributed to the cryostat.
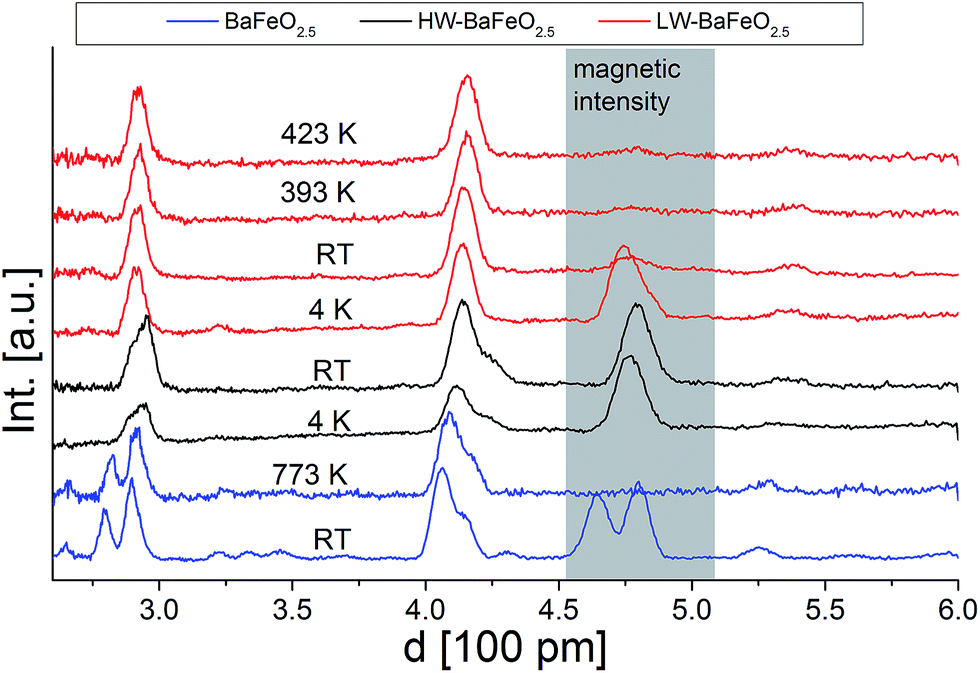 | ||
| Fig. 5 Neutron diffraction data (normalized to the most intense reflection) recorded in the low angle detector bank 3 of the HRPD diffractometer (ISIS, UK). The d-range with a high amount of magnetic scattering is highlighted. | ||
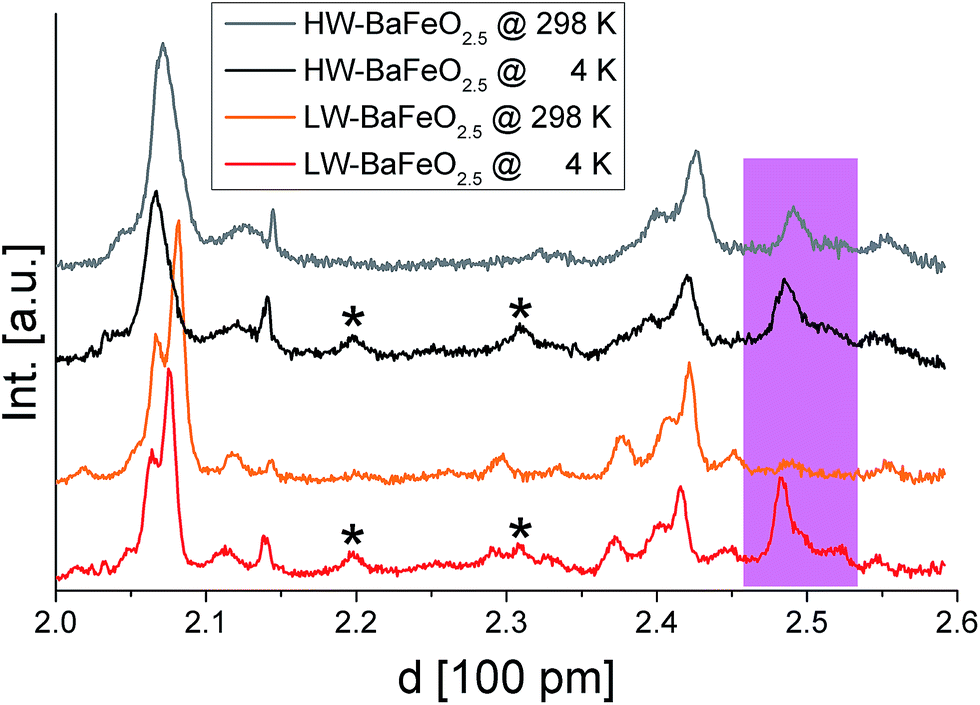 | ||
| Fig. 6 Neutron diffraction data recorded for HW- and LW-BaFeO2.5 at ambient temperature and 4 K in the high resolution detector bank 1 of the HRPD diffractometer. Reflections from the instrumental setup or potential small amounts of impurity phases are marked by an asterisk, reflections resulting from G-type antiferromagnetic ordering are coloured pink. | ||
In order to determine possible space group symmetries for these systems, a coupled Pawley-analysis was performed on LW-BaFeO2.5 using the ambient temperature XRD as well as the neutron data recorded at RT of all the detector banks. For this analysis, positions with a strong influence of magnetic scattering on cooling to lower temperatures have been neglected. For such Pawley fits with large unit cell, it is important to optically investigate small but significant peak dependent differences in qualities of the fit by the naked eye instead of relying solely on residual values.38 From this analysis, C-centering of the unit cell with dimensions of 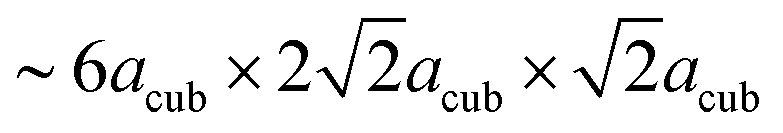 22 could be confirmed (however, magnetic intensities appear at positions where nuclear reflections prohibited by C-centering would occur). Such a large unit cell size has also been reported for a series of perovskite type compounds La1−xSrxMnO3−(0.5+x)/2 (0.67 ≤ x ≤ 1; primitive space group 57).39 The large unit cell size exacerbates the analysis of the Pawley fits further, and special care needs to be taken on the relatively high d-spacings of detector banks 1 and 2 of the neutron diffraction data. Further reduction of space group symmetry using primitive orthorhombic unit cells does not give a significant additional improvement of the fit. It is worth adding that refining the patterns using the structural model of La1−xSrxMnO3−(0.5+x)/2 (ref. 39) was not successful, and such primitive orthorhombic cells were therefore discarded for all analyses.
22 could be confirmed (however, magnetic intensities appear at positions where nuclear reflections prohibited by C-centering would occur). Such a large unit cell size has also been reported for a series of perovskite type compounds La1−xSrxMnO3−(0.5+x)/2 (0.67 ≤ x ≤ 1; primitive space group 57).39 The large unit cell size exacerbates the analysis of the Pawley fits further, and special care needs to be taken on the relatively high d-spacings of detector banks 1 and 2 of the neutron diffraction data. Further reduction of space group symmetry using primitive orthorhombic unit cells does not give a significant additional improvement of the fit. It is worth adding that refining the patterns using the structural model of La1−xSrxMnO3−(0.5+x)/2 (ref. 39) was not successful, and such primitive orthorhombic cells were therefore discarded for all analyses.
Among the centrosymmetric orthorhombic space groups with C-centering, Cmmm does not cause further systematic absences of reflections due to the presence of glide planes or screw axes. An overview of the different C-centered orthorhombic space groups with different extinction rules and the results of the different Pawley fits of the patterns is given in Table S1 of the ESI.† This analysis showed that the space group symmetries Ccca, Ccmm, Cccm, Cmce, and Cmma can be effectively ruled out following a systematic investigation of the performed Pawley fits. Instead it was strongly indicated that attempts to solve the structure should focus on the centrosymmetric space groups Cmcm and Cmmm (see Fig. S1 of the ESI† for a Pawley fit of the room temperature data using the Cmcm model). The space groups Ama2 and Cmc21 as well as Amm2, Cmm2, and C222 are translationengleiche non-centrosymmetric subgroups of Cmcm and Cmmm, and due to the problems regarding the structural analysis reported throughout this article, such subgroups were not considered for potential further improvement of the as-determined model.
Using the tools provided on the Bilbao Crystallographic Server,40–42 structural models of the compounds were built for both space groups (see Fig. S2a of the ESI†). For both space groups, two different structures within the same group can be obtained (see Fig. S2b of the ESI†), depending on the choice of shift of origin on lowering of the symmetry (next to obviously structurally equivalent descriptions with respect to the additional generators of the orthorhombic space groups43). Those different structures are distinguished by the interchange of the Ba and Fe sites within the space groups, resulting in two different splitting schemes for both types of atoms. Those four structural models were analyzed and the model which resulted in the best fit is reported in this article.
Structural analysis of LW-BaFeO2.5 was first performed by a combined analysis of the room temperature XRD and neutron diffraction data (using the data from all three detector banks) to avoid correlations resulting from interference with magnetic scattering. At this temperature, it is unlikely that protons can be located, but it will be shown that the analysis will result in a reasonable principal description of the anion vacancy pattern. Use of the structural models within the space groups Cmcm resulted in significantly better fitting of the patterns compared to the Cmmm structural models according to Hamilton's significance test44 (the number of structural parameters are almost the same for both structural models). Comparing the two structural models with symmetry of Cmcm, a significantly better fit was obtained when using structural model 1 (see discussion provided in the ESI†) in comparison to structural model 2 (with a decrease of the Rwp from ∼9.6% to 9.0%). Furthermore, model 1 provided more reliable bond distances, with structural relaxations around vacancies which are also found for other barium ferrite systems (see discussion provided in the ESI†).
Since the structural model determined at RT resulted in a reasonably good description of the patterns, attempts were then made to couple the ambient temperature XRD data to the neutron data recorded at 4 K (including an additional G-type antiferromagnetic model to describe the magnetic ordering as discussed at the beginning of this section) to obtain a deeper understanding of the structure and to determine potential ordering of the protons. Attempts to limit the number of refined parameters as much as possible were first made by using a constraint in such a way that an overall isotropic thermal displacement parameter was refined for each type of atom. This strategy proved to be very useful for stabilizing the analysis of the structure of BaFeO2.5,5 which was shown to be unstable otherwise due to the high number of structural parameters (7 Ba, 7 Fe and 17 O crystallographically independent sites on general positions x, y and z) with an excellent fit to the observed pattern. However, in the case of the structure of LW-BaFeO2.5 significant intensity misfits were still obtained when using such a model, indicating the need to include further structural parameters. Attempts to locate the protons using the Fourier difference method were not, however, successful. Rather it was found that employing independent anisotropic thermal displacement parameters proved successful for obtaining a good final fit of the pattern. It should be noted, however, that the very high values of those parameters are not physically possible with regards to describing thermal motions of the atoms, but rather indicate a further degree of structural disorder for LW-BaFeO2.5, which might also result from a distribution of locations of the protons around those anions. The fit to the diffraction patterns at 4 K is shown in Fig. 7. The refined structural data are given in Table 1, bond distances are listed in Table 2 and a depiction of the crystal structure is shown in Fig. 8.
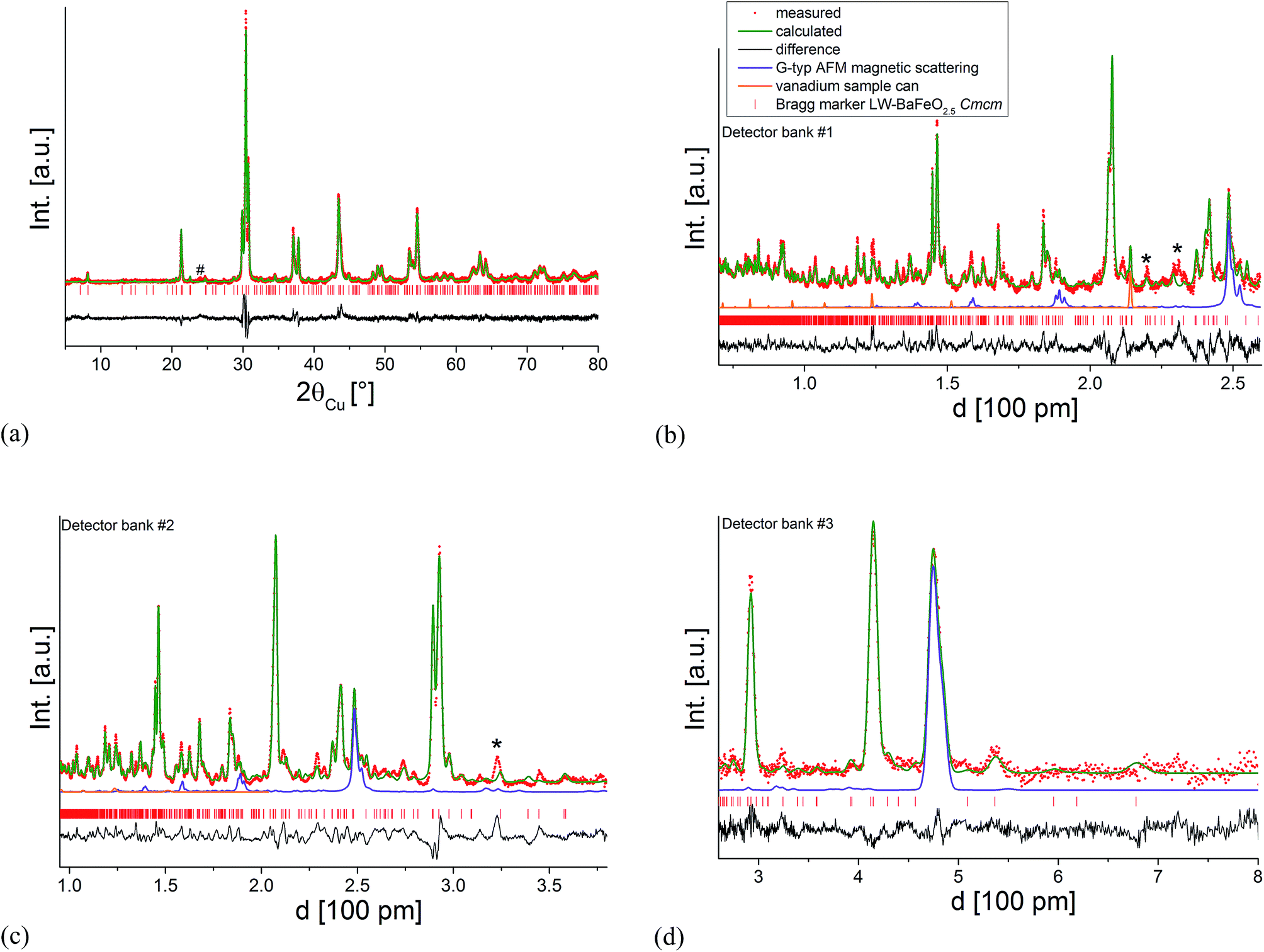 | ||
| Fig. 7 Combined Rietveld analysis of LW-BaFeO2.5 using the XRD data recorded at ambient temperature (a) neutron diffraction data recorded at 4 K (b–d). Reflections from the instrumental setup (see Fig. 6) are marked by an asterisk (*), a small impurity of BaCO3 resulting from partial decomposition due to the long measurement time of the compound in air is marked by a hash (#). | ||
| Site label | Atom type | Site | x | y | z | Occ. | BVS | CN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ba1 | Ba2+ | 4c | 0 | 0.6119(6) | 1/4 | 1 | 1.94 | 12 | ||||||||
| Ba2 | Ba2+ | 4c | 0 | 0.1620(5) | 1/4 | 1 | 2.55 | 12 | ||||||||
| Ba3 | Ba2+ | 8g | 0.8224(2) | 0.1228(5) | 1/4 | 1 | 1.53 | 9 + 2* | ||||||||
| Ba4 | Ba2+ | 8g | 0.3369(2) | 0.1199(5) | 1/4 | 1 | 2.34 | 10 | ||||||||
| Fe1 | Fe3+ | 8g | 0.7445(3) | 0.3527(5) | 1/4 | 1 | 2.65 | 4 | ||||||||
| Fe2 | Fe3+ | 8g | 0.5915(4) | 0.3521(9) | 1/4 | 1 | 2.23 | 6 | ||||||||
| Fe3 | Fe3+ | 8g | 0.0779(3) | 0.4032(4) | 1/4 | 1 | 2.67 | 5 + 1* | ||||||||
| O1 | O2−/OH− | 4c | 0 | 0.8858(2) | 1/4 | 1 | 0.99 | |||||||||
| O2 | O2− | 4c | 0 | 0.3780(9) | 1/4 | 1 | 2.15 | |||||||||
| O3 | O2− | 8g | 0.805(3) | 0.444(2) | 1/4 | 1 | 1.91 | |||||||||
| O4 | O2− | 8g | 0.3318(6) | 0.3668(1) | 1/4 | 1 | 1.94 | |||||||||
| O5 | O2− | 8d | 1/4 | 1/4 | 0 | 1 | 2.08 | |||||||||
| O6 | O2− | 8e | 0.4136(4) | 0 | 0 | 1 | 2.26 | |||||||||
| O7 | O2−/OH− | 8e | 0.9190(5) | 0 | 0 | 1 | 0.93 | |||||||||
| O8 | O2− | 16h | 0.0833(7) | 0.2537(11) | 0.032(5) | 1 | 1.48 | |||||||||
| a [Å] | 24.742(2) | b [Å] | 11.909(1) | c [Å] | 5.7833(3) | |||||||||||
| GOFoverall | 8.1 | R wp,overall [%] | 7.6 | R Bragg (NPDbank1) [%] | 3.1 | |||||||||||
| Cation | Distances to anions [Å] | Cation | Distances to anions [Å] | Cation | Distances to anions [Å] |
|---|---|---|---|---|---|
| Fe1 | 1.85(6) (1 × O3) | Ba1 | 2.79(1) (1 × O2) | Ba3 | 2.755(4) (2 × O5) |
| 1.90(2) (1 × O4) | 2.894(1) (2 × O2) | 2.904(1) (2 × O4) | |||
| 1.899(4) (2 × O5) | 2.905(8) (4 × O6) | 3.06(1) (1 × O4) | |||
| 3.08(2) (4 × O8) | 3.08(2) (2 × O8) | ||||
| 3.26(2) (1 × O1) | 3.15(1) (2 × O7) | ||||
| 3.80(6) (1 × O3) | |||||
| 3.85(3) (1 × O3) | |||||
| Fe2 | 1.91(2) (1 × O4) | Ba2 | 2.57(1) (1 × O2) | Ba4 | 2.24(3) (1 × O3) |
| 2.07(3) (2 × O8) | 2.65(2) (4 × O8) | 2.780(8) (2 × O6) | |||
| 2.294(8) (2 × O7) | 2.947(4) (2 × O1) | 2.94(1) (1 × O4) | |||
| 2.30(1) (1 × O1) | 3.135(9) (4 × O7) | 2.97(2) (2 × O8) | |||
| 3.29(2) (1 × O1) | 3.019(5) (2 × O5) | ||||
| 3.09(2) (2 × O3) | |||||
| Fe3 | 1.861(3) (2 × O6) | ||||
| 1.951(8) (1 × O2) | |||||
| 2.19(2) (2 × O8) | |||||
| 2.94(7) (1 × O3) |
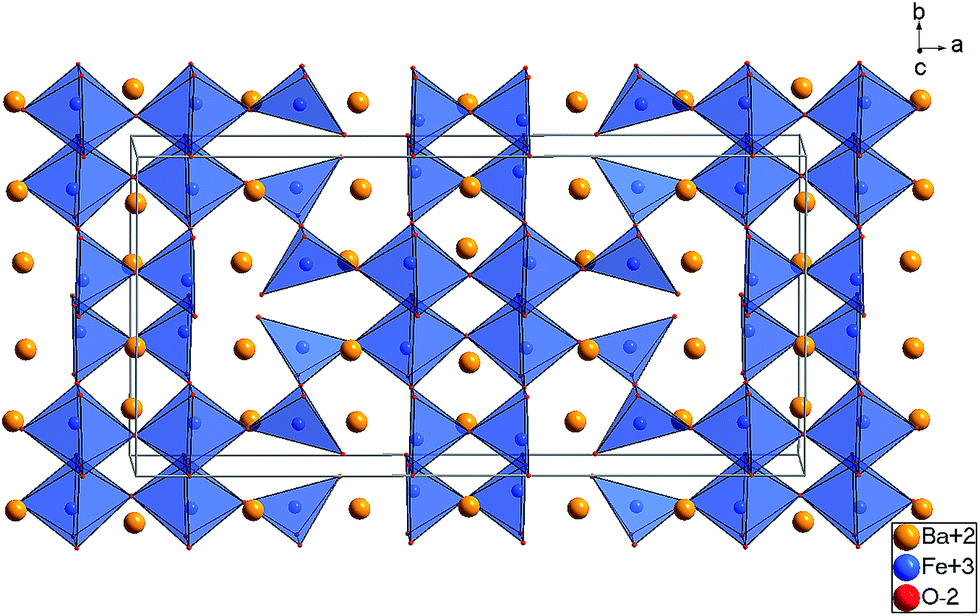 | ||
| Fig. 8 Substructure of the Ba, Fe, and O ions found for LW-BaFeO2.5 at 4 K (Cmcm, Structure 1). | ||
The refined structure of the Ba, Fe, and O ions of LW-BaFeO2.5 is in excellent agreement with a composition BaFeO2.33(OH)0.33 determined by TG experiments (see Section 3.1) and is closely related to two similar Fe3+ containing compounds. It is worth noting that the vacancy pattern is similar to what is found for La1/3Sr2/3FeO8/3 with every third layer showing tetrahedral coordination of the Fe3+ ions.45 For La1/3Sr2/3FeO8/3, the structure relaxes around the vacancies via tilting of the octahedra within the remaining two layers and tilting of the tetrahedra in directions perpendicular to the direction along which octahedral and tetrahedral layers stack. However, in a previous article, we highlighted that this type of tilting is not favourable for barium ferrites, since it results in a lowering of the coordination number of the A-site cation, which must be considered to be unfavourable for the large Ba2+ ions. Therefore, the local structural relaxation around the tetrahedral cations results in shift of one of the corners shared with an octahedron towards the tetrahedron, resulting in a change of the coordination from octahedral to square pyramidal.
| Fe[4]O4/2 + Fe[6]O6/2 → Fe[4]O3/2O1/1 + Fe[5]O5/2 |
This structural relaxation mechanism in the present study is therefore effectively identical to what is found mainly in BaFeO2.5 (ref. 5) as well as exclusively in BaFeO2.33F0.33.21
The structures determined at ambient temperature and 4 K are closely related in terms of the vacancy pattern, polyhedra tilting, as well as the principal structural relaxations. As described above, identification of the location of the protons at 4 K did not prove successful. However, further analysis of the as determined Ba, Fe and O ionic substructure can give information about which oxide ions are most likely to be protonated. In this respect, bond valence sums (BVS)46,47 are powerful tools whenever combinations of anions such as O2−, F− and OH−, which are indistinguishable by means of diffraction methods, are co-located in a crystal structure (see e.g.ref. 31, 33, 34 and 48). Regarding the BVS values given in Table 1, it is found that O1 and O7 show values relatively close to −1, which might indicate that protons are mainly located close to those anions around 4 K. Those ions together form one face of the octahedron surrounding the Fe2 ion. This coordination situation is therefore relatively similar to what is found in the hexagonal type perovskites 6H- and 15R–BaFeO2F,31,32 where Fe ions are found to be located in a FeF3[FACE1]O3[FACE2] type coordination. For those compounds, the Fe3+ ions shift towards the O2−. Such shifts of the Fe2 ion away from the O1 and O7 ions is also indicated for LW-BaFeO2.5 (see Fig. 9, indicated by green arrows). In contrast to the F− ion, the hydroxide groups carry a positive dipolar moment. Therefore, cations will be repelled from this dipolar moment. Pointing of the protons towards the Fe3+ cations within the octahedron is implausible since this would imply very short Fe–H distances. In contrast, Ba2+ is a soft cation and more tolerant towards distorting its coordination sphere. Indeed, such strong repulsion and shift is indicated for the Ba2 cations (see Fig. 9, indicated by orange arrows). This results in a relatively large rectangular cavity along the c-direction (indicated by an orange cloud in Fig. 9), which is most likely to contain the protons. Whereas for BaFeO2.33F0.33, the lower charged fluoride ions were indicated to be located at one edge of the tetrahedron,21 for LW-BaFeO2.5, the location of the hydroxide group at one corner of the tetrahedrally coordinated iron ions is not suggested from the BVS values. This might be explained by the fact that the additional (weak) bonds formed by this group (see Fig. 9, indicated by green and red lines) saturate the coordination environment around this ion and therefore seem to prohibit bonding to a further proton.
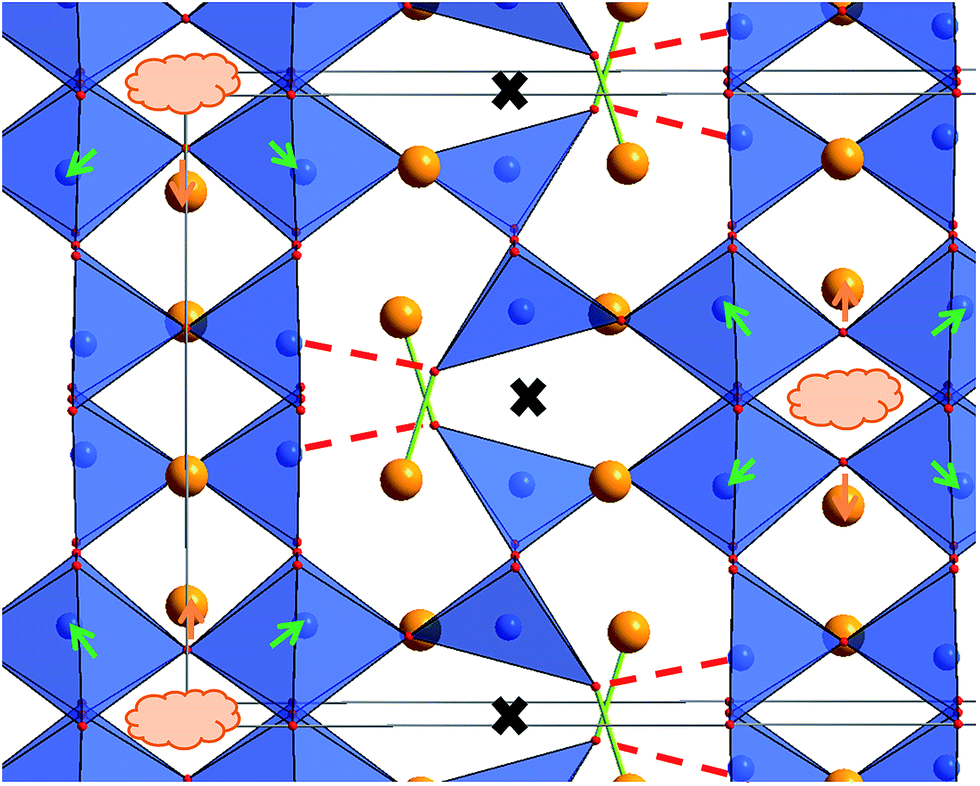 | ||
| Fig. 9 Structure excerpt of LW-BaFeO2.5 at 4 K. The weak bond between the strongly shifted O3 ion to the Fe ion is shown as a dashed red line, the shortened bond to a Ba ion is shown as a green line. The position of the vacancies along the cubic [1 1 0]cub direction is indicated by black crosses. Strong shifts of Fe and Ba ions are shown as green and orange arrows. | ||
It should be noted that the obtained structural model still shows some imperfections regarding the obtained bond distances and bond valence sums (also regarding the global instability index) of the Fe cations, and in terms that the obtained fit of the diffraction patterns still contains obvious intensity misfits. This can be attributed to problems described within this section (such as strong strain broadening and low symmetry resulting in strong reflection overlap, in addition to the difficulty in locating the protons, and a relatively poor signal to noise ratio due to strong incoherent scattering of the protons) that result in a lowering of the number of independent observables which can be relied on for the structural analysis. Nevertheless at this stage, we believe that the structural model provided represents the best current solution to what is a clearly complex system (the neutron diffraction data are provided in the ESI†). In this respect, it should be also pointed out that the exact determination of symmetry for even the well-known orthorhombic brownmillerite type compounds is also known to be problematic e.g. even for SrFeO2.5 a range of structure descriptions with different space groups (Icmm, Ibm2, Pcmn) are found in literature,49–51 with all structures being similar and only distinguishing in the detailed type of polyhedra tilting. Additionally, the vacancy pattern found for LW-BaFeO2.5 is consistent with structural relaxation mechanisms found for other barium ferrite perovskite type compounds, and a more detailed discussion of vacancy order in barium ferrite compounds is being presented in the ESI† as well as in ref. 21.
| Compound | a [Å] | b [Å] | c [Å] | a/6 [Å] | b/80.5 [Å] | c/20.5 [Å] | V [Å3] | (Vf.u.)1/3 [Å] |
|---|---|---|---|---|---|---|---|---|
| HW-BaFeO2.5 | 25.321(4) | 11.769(2) | 5.787(1) | 4.220 | 4.161 | 4.092 | 1724.5 | 4.157 |
| LW-BaFeO2.5 | 24.742(2) | 11.909(1) | 5.7833(3) | 4.124 | 4.211 | 4.089 | 1704.0 | 4.141 |
| BaFeO2.5* | — | — | — | — | — | — | — | 4.075 |
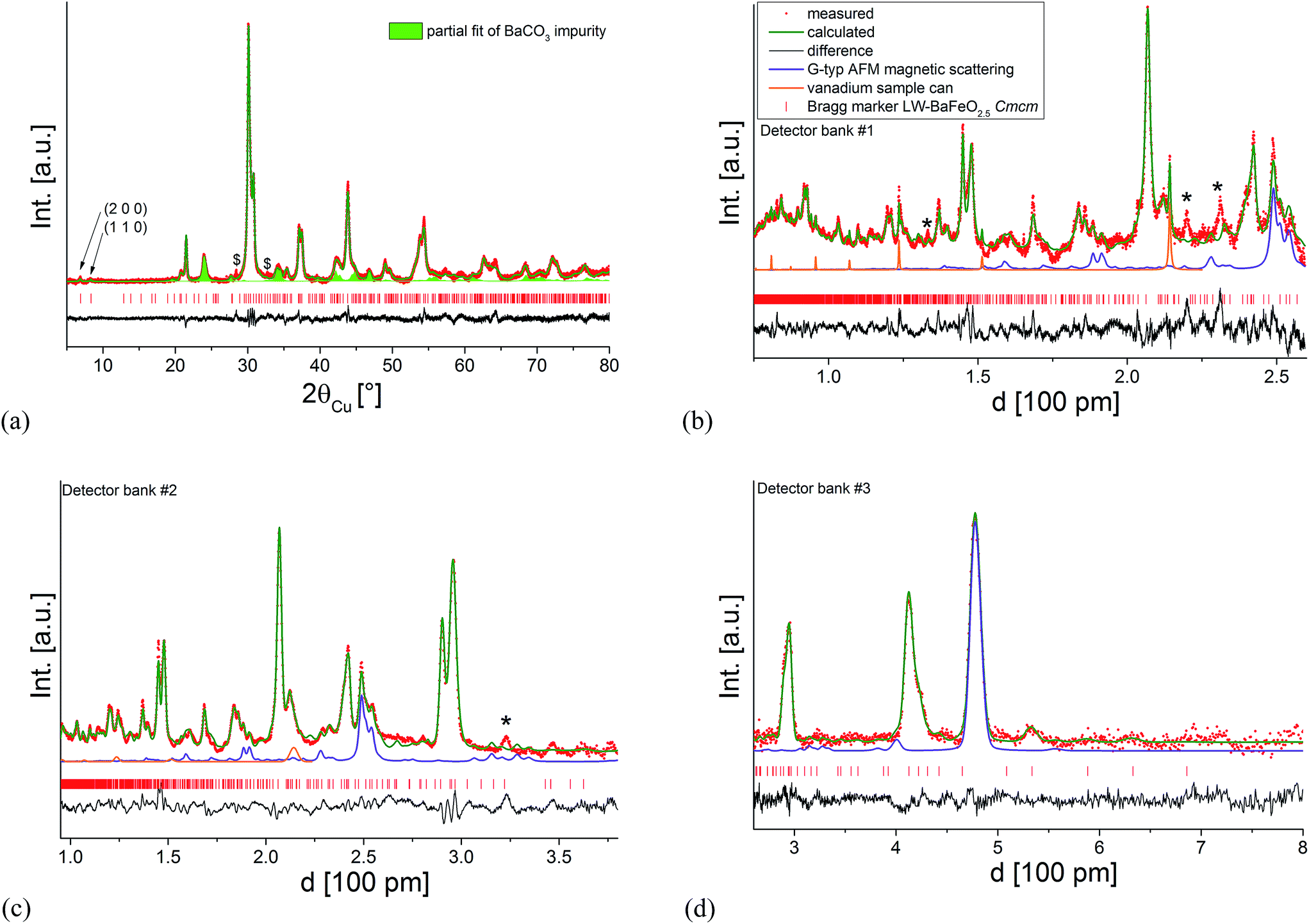 | ||
| Fig. 10 Combined Rietveld analysis of HW-BaFeO2.5 using the XRD data recorded at ambient temperature (a) neutron diffraction data recorded at 4 K (b–d). Reflections from the instrumental setup or potential small amounts of impurity phases are marked by an asterisk (*), the partial fit curve BaCO3 resulting from partial decomposition due to the long measurement time of the compound in air is shown as a green filled curve, a further unknown impurity with significantly sharper reflections is mark with a dollar sign ($). | ||
It was found that the diffraction data can be reasonably described (see Fig. 10) by refining the structural model of LW-BaFeO2.5 (refining the occupancy as well as positional parameters of the anion site which is vacant in LW-BaFeO2.5) together with a G-type antiferromagnetic structure. This resulted in an increase in the occupancy of the formerly vacant O9 site (8e) to 48%, which would be in close agreement to the composition determined by thermal analysis. Additionally, strong shifts were also observed for the Ba and Fe cations as well as the O3 ion closer towards the position they would adopt in a pseudocubic structural setting. The latter is explained by the observation that superstructure reflections are far less pronounced for the XRD as well as for the neutron diffraction data. However, although the pattern can be refined using such a structural model together with anisotropic displacement parameters (again mainly describing a further overall disorder of the ionic structure), an overall unique reliable structural model could not be determined. In the case of HW-BaFeO2.5, similar qualities of fit can be obtained by allowing vacancies also for some of the other anion sites, and this would be in principal agreement with the finding of increased disorder in this compound.
Acknowledging the fact that determining an appropriate structural model of HW-BaFeO2.5 was not successful, structural data of this compound will not be given here. The lattice parameters determined for LW- and HW-BaFeO2.5 are given and compared in Table 3. Remarkably, a change between the relative axes' lengths is found for HW-BaFeO2.5 with the a-axis being the longest axis with respect to a pseudocubic length. Additionally, a significant increase in cell volume is found for HW-BaFeO2.5, in good agreement with what would be expected for further uptake of water molecules.
3.3 Conductivity studies of BaFeO2.5, its hydrated phases LW- and HW-BaFeO2.5, and comparison to oxidized phases BaFeO2.5+δ
In contrast to perovskite-type compounds such as BaInO2.5 and BaZr1−xYxO3−x/2, containing B-site cations which cannot easily be oxidized/reduced, it is not trivial to study the (proton) conductivity of ferrites by means of impedance spectroscopy since the electronic contribution to the overall electrical conductivity needs to be ruled out first. Partial oxidation of the compound is known to cause the formation of BaFeO2.5+y (i.e. a mixed valent Fe3+/Fe4+ compound, with y in the order of ≫0.1 (ref. 52)), for which significant electronic conductivity must be expected. Therefore, to draw proper conclusions about the proton conductivity in the hydrated modifications of BaFeO2.5, the pure compounds BaFeO2.5 and BaFeO2.5+y need to be characterized first in the temperature range under concern (which to the best of our knowledge has not been reported in literature so far, although a recent study on BaFeO2.5 in the temperature range between 873 and 1173 K exists53). Therefore, two types of pellets have been investigated: a pellet of BaFeO2.5 sintered at 1373 K under flowing nitrogen to rule out strong oxidation of the compound as well as an as-prepared pellet which has been treated under flowing oxygen atmosphere at 773 K in a subsequent step.Furthermore, conductivity studies on the hydrated samples are exacerbated due to the strong volume increase of the compound on hydration. This results in the fact that a sintered pellet of BaFeO2.5 could not be hydrated without breaking the pellet. Therefore, pellets of preformed LW-BaFeO2.5 and HW-BaFeO2.5 had to be prepared which were then compacted by means of isostatic pressing without further sintering. Obviously, the total conductivity of such porous pellets will drop in comparison to sintered pellets due to a strong influence of the grain boundaries. In order to allow for an easier comparison to non-hydrated BaFeO2.5, a porous pellet of BaFeO2.5 was also similarly prepared: before pressing, the powder of BaFeO2.5 was additionally heated at 423 K under dry flowing argon gas to rule out a possible influence of the oxidation of the compound by small traces of oxygen (i.e. the same conditions under which LW-BaFeO2.5 was made except for the use of a wet argon stream).
Fig. 11 shows the temperature dependence of the total conductivity of pure (sintered and non-sintered pellet) as well as oxidized BaFeO2.5+y, Fig. 12 exemplarily shows the NYQUIST plots for the three compounds recorded at 400 K, and an overview of the determined total conductivity data is given in Table 4.
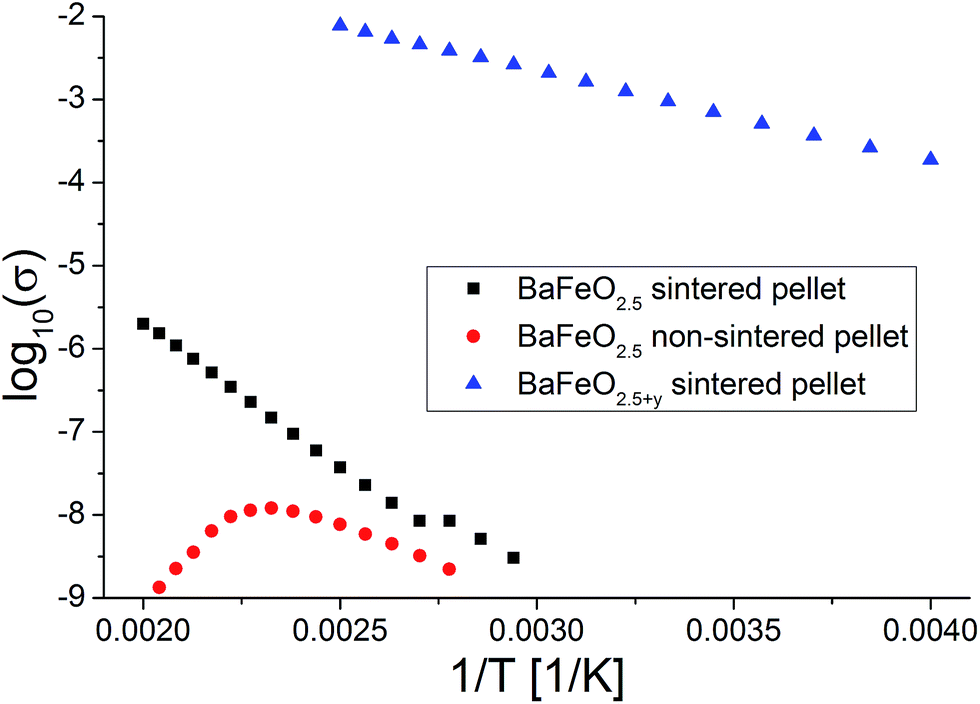 | ||
| Fig. 11 Temperature dependence of conductivity for sintered and non-sintered pellets of BaFeO2.5 as well as of a sintered pellet of BaFeO2.5+y. | ||
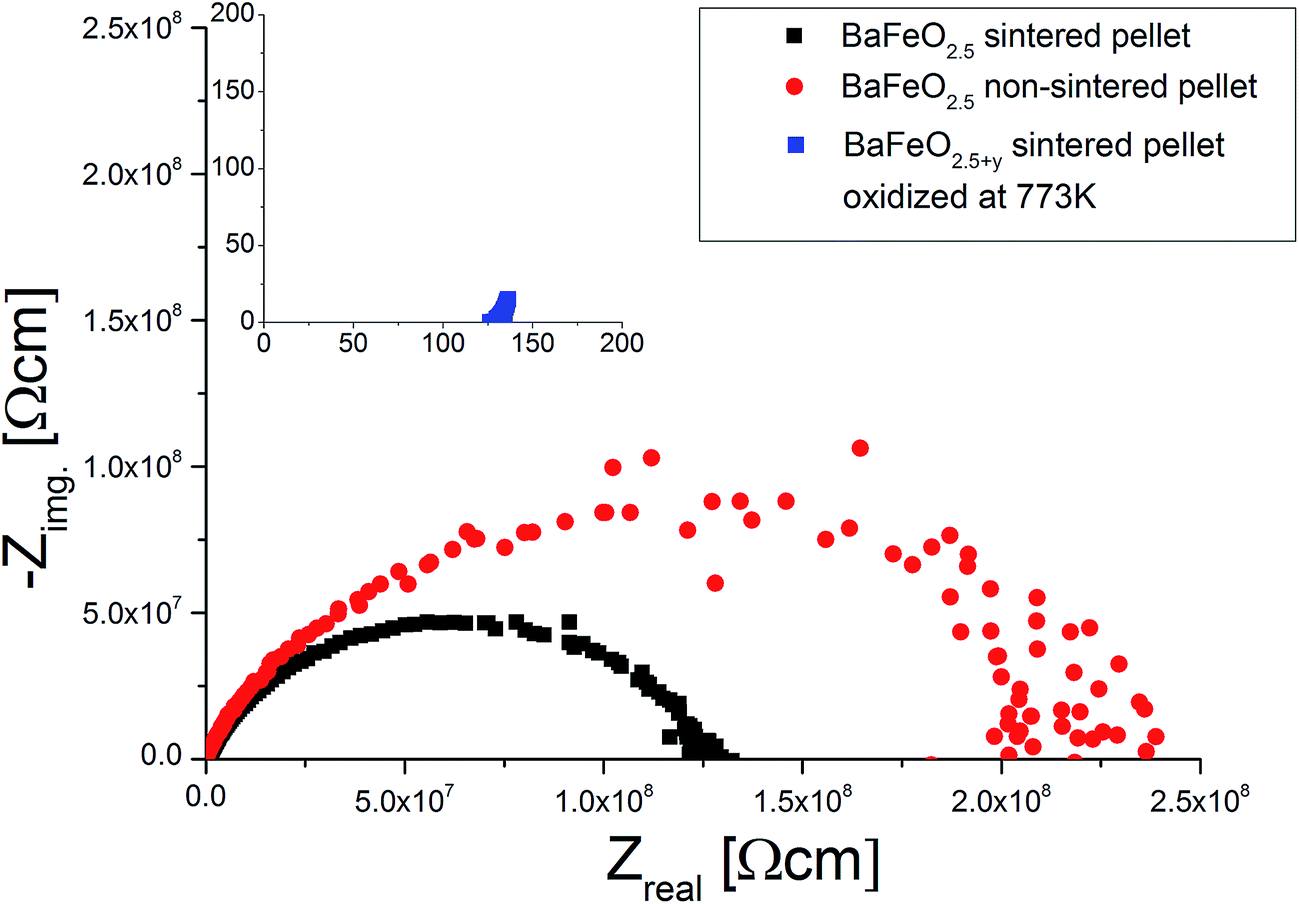 | ||
| Fig. 12 Comparison of NQUIST plots at 400 K (frequency range 1 MHz to 1 Hz) of a sintered and non-sintered pellet of BaFeO2.5 to a sintered pellet of BaFeO2.5 which has been oxidized under a flow of dry O2. | ||
| Material | E A,total [eV] | σ 0,total [S cm−1] | ε r | σ RT,tot [S cm−1] |
|---|---|---|---|---|
| BaFeO2.5, sintered pellet, heated at 1373 K | 0.70(1) | 2.2 × 101 | 20–30 (g) 160 (gb) | 10−11 |
| BaFeO2.5, non-sintered pellet, made of powder treated at 423 K under dry flowing argon | 0.37(1) | 3.5 × 10−3 | 20–30 | 10−11 |
| BaFeO2.5+y, sintered pellet, oxidized at 773 K under flowing O2 | 0.20(1) | 2.2 × 10 | — | 10−3 |
The conductivity of oxidized BaFeO2.5+y is found to be nearly independent of frequency (see inlay of Fig. 12), which can be explained by the material being a reasonably good p-type electron conductor. The conductivity of this compound is of the order of 10−3 S cm−1 at ambient temperatures, with the impedance being dominated by the real part. This behavior is well explained by the presence of mobile electrons, resulting from a mixed valency of Fe3+/Fe4+ and conduction via a hopping mechanism between neighbouring Fe3+/Fe4+ sites.
For the sintered and non-sintered pellet of BaFeO2.5, the NYQUIST plot shows one large highly depressed semicircle, which was found to be best described by a series of two RQ units. Compared to partially oxidized BaFeO2.5+y, the conductivity of pure BaFeO2.5 is very low and of the order of ∼10−11 S cm−1 at temperatures close to ambient temperature. The assumption of a poor electron conductor is also indicated by the fact that the low frequency part of the semicircle basically intersects with the Zreal axis in the NYQUIST plot, and there is no electrode response typical for an ionic conductor.
The total conductivity of BaFeO2.5 further decreases when comparing the non-sintered pellet to the pellet sintered at 1373 K, which is reasonably explained by the lower density and an increased amount of less conducting grain boundaries for this compound. In contrast to the sintered pellet of BaFeO2.5, it was found that the conductivity strongly decreases on heating above temperatures of 420 K. Comparing to what was found for the hydrated phases (see subsequent paragraphs), one can assume that this might be explained by the easy uptake of very small traces of water during the preparation of the pellet at the grain boundaries (which is done in air), such that the as-prepared pellet contains a small amount of water, which is lost on heating. This assumption would be in agreement with findings of Zou et al.,4 who observed that the grain boundary of BaFeO2.5 behaves different than the bulk compound. On cooling, the conductivity is not recovered and remains extremely low (orders of magnitude below 10−10 S cm−1 at 350 K, which is the limit of detection for the impedance spectrometer used). In contrast, the sintered pellet (which shows a relatively high density of ∼90% of the crystallographic density), can be considered to be less prone to such an uptake of water due to a higher density of >90%, and the heating as well as cooling curve are basically identical for sintered pellets of BaFeO2.5.
For LW-BaFeO2.5, the impedance spectra consist of two relatively well separated semicircles. The first semicircle corresponds to a relative dielectric permittivity of ∼90–100 (see Table 5). Therefore, this semicircle can be most likely assigned to grain (bulk) conductivity with maybe a contribution from the grain boundaries. The second semicircle does not approach the Zreal-axis, even for the smallest frequencies measured (0.1 Hz), and shows a very high relative dielectric permittivity of ∼106 (again see Table 5). Therefore, this unfinished semicircle most likely corresponds to (respectively contains) an electrode response, however, final assignment still remains unclear, and it may include a contribution from the grain boundaries. The difficulty in the conclusive assignment of the data lies in the fact that the pellets could not be sintered. The second semicircle (although it cannot be recorded fully) was included in the calculation of the total conductivity (which we calculated from the sum of the resistances of the first and the second semicircle). Therefore, we would like to point out that the total conductivity could be even higher, i.e. of an order of magnitude as the conductivity which is indicated by the high frequency semicircle (which we assigned to the grain/grain boundary semicircle). An increase of the total conductivity by ∼3 orders of magnitudes compared to the sintered pellet and by ∼4 orders magnitude was found compared to the non-sintered pellet of BaFeO2.5 (see Fig. 14) at room temperature. From the comparison to the non-sintered pellet of BaFeO2.5 (again, for which the powder of BaFeO2.5 was pretreated under the same conditions used for the hydration to form LW-BaFeO2.5, except for the use of a flow of dry argon gas), as well as to the sintered pellet of BaFeO2.5, which both show a significantly reduced total conductivity, we conclude that the total conductivity for LW-BaFeO2.5 can be considered to be dominated by the proton contribution, and that electronic contribution can be neglected to contribute significantly to the total conductivity. Additionally, it was found that the conductivity of LW-BaFeO2.5 decreases when the sample is heated above ∼400 K, which can be explained by a partial release of the crystalline water or changes at the grain boundary, which influence the grain as well as the grain boundary contribution to the total impedance. Since the measurements were performed under a static helium atmosphere of 1 bar pressure, the released water cannot be easily taken up on cooling again due to kinetic effects. In contrast, when heated to significantly lower temperatures of ∼350 K to avoid water release, the difference in conductivity between heating and cooling was found to be marginal, in agreement with the TG data as well as variable temperature diffraction studies reported in Section 3.1. It is also interesting to note that the resistance of the second semicircle increases when heating the compound to elevated temperatures (see Fig. 13). This might therefore be explained by either a change in the grain boundary behaviour or by decreased contact to the sputtered gold electrode due to the partial release of water.
| Material | E A,g [eV] | σ 0,g [S cm−1] | ε r,g | E A,gb [eV] | σ 0,gb [S cm−1] | ε r,gb | σ RT,tot [S cm−1] |
|---|---|---|---|---|---|---|---|
| LW-BaFeO2.5 (non-sintered pellet, dry He atmosphere) | 0.44(1) | 3.5 × 10 | 70–130 | 0.27(1) | 5 × 10−4 | 0.5 to 4 × 106 | 10−8 |
| BaZrO3 (Y-doped, sintered pellet, p(H2O) = 0.031 atm)6 | 0.46–0.47 | 2.2 to 3.1 × 104 | 99–150 | 0.66–0.68 | 0.4 to 1.9 × 106 | 0.97 to 2.2 × 104 | 10−6 |
| BaCeO3 (Gd-doped, sintered pellet, water saturated argon)54 | 0.56 | 7.4 × 104 | 37 | 0.67 | 3.0 × 106 | 1.6 × 104 | 10−5 |
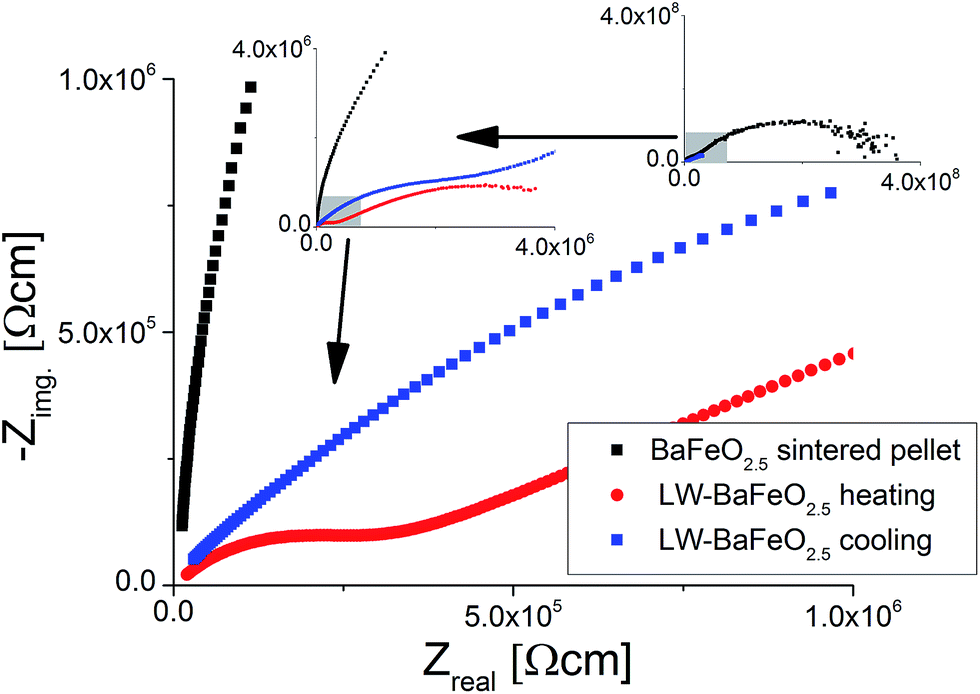 | ||
| Fig. 13 NYQUIST plots of impedance data recorded for BaFeO2.5 and LW-BaFeO2.5 at a temperature of 350 K. For LW-BaFeO2.5 a decrease of conductivity is found on cooling the pellet after heating to 400 K. | ||
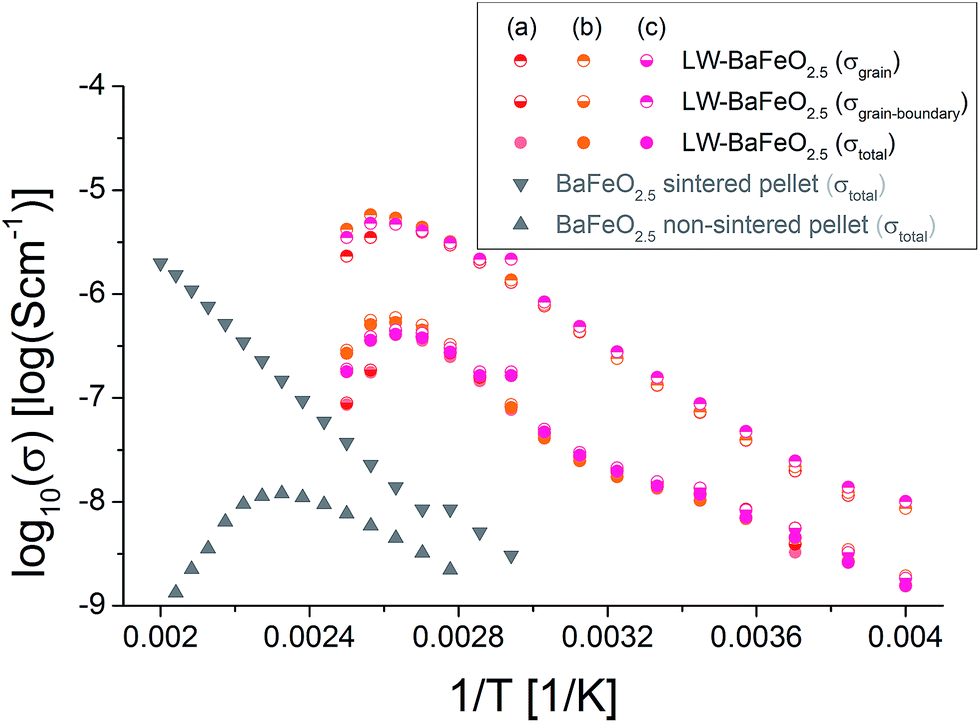 | ||
| Fig. 14 Arrhenius plot of conductivity data recorded for heating of LW-BaFeO2.5 in comparison to different samples of BaFeO2.5. As described in the text, the reader should be aware that the grain contribution could be alternatively assigned to a grain/grain-boundary contribution and that the grain-boundary contribution most likely includes an electrode response. | ||
Table 5 summarizes the conductivity data of LW-BaFeO2.5 determined in the temperature range between 260 and 330 K. If one compares the values to the grain and grain boundary contributions as reported for BaZr1−xYxO3−x/2 (ref. 6) and BaCe1−xGdxO3−x/2 (ref. 54) (which have however been obtained by measurement in wet argon atmospheres), similar activation energies are found for the grain contribution, in agreement with the assumption of mobile protons in LW-BaFeO2.5. Overall the room temperature grain conductivity (10−6 to 10−7 S cm−1) is, however, about 2–4 orders of magnitude lower compared to BaZr1−xYxO3−x/2 (ref. 6) and BaCe1−xGdxO3−x/2,54 but the materials show a significantly increased conductivity of a few orders of magnitudes in comparison to other proton conductors, such as BaInO2.5 (ref. 55) or La(Nb,Ta)O4 or LaBaGaO4.9 In addition, the use of non-sintered pellets must be assumed to result in a significant lowering of conductivity by at least one order of magnitude. Therefore, this instability of a sintered pellet towards hydration establishes a limit of the study presented here which cannot be easily overcome. In future work, thin film technologies will be explored to investigate if determination of more precise values for the bulk conductivity will be possible on such samples.
An explanation why LW-BaFeO2.5 shows a reduced conductivity compared to the similar perovskite type compounds Y-doped BaZrO3 and Gd-doped BaCeO3 could lie in the fact that vacancy order is likely to show a negative impact on the conductivity. This is explained by such lowering of symmetry causing a nonequivalence of the oxide ions (e.g. some of them are shared between two octahedra, some of them between an octahedron and a tetrahedron), and such nonequivalence would have to result in different values of acidity/basicity of the ion (also expressed in terms of the bond valence sums, see Table 1). This then results in the fact that oxide ions with a BVS closer to −1 can be considered as an energetic minimum, rendering transfer of the protons to oxide ions with a BVS closer to −2 less likely. In contrast, a higher symmetry can establish energetically equivalent protonation of oxygen ions, enabling more facile proton transport through the structure.
It is worth noting that attempts were also made to measure the conductivity of HW-BaFeO2.5. Proton conductivity could also be deduced for this compound (see Fig. 15). The total conductivity is similar compared to LW-BaFeO2.5, however it was found that the contribution of the first semicircle to the overall impedance spectrum is lower compared to the LW compound. The fact that a slightly reduced conductivity compared to LW-BaFeO2.5 is found (instead of increased conductivity which could be assumed for a compound with higher disorder) might be explained by the observation that HW-BaFeO2.5 shows an increased sensitivity towards CO2 uptake and decomposition under formation of BaCO3, which is likely to be of crucial importance for the investigation of this compound, or a change of the electrode behavior. Furthermore, HW-BaFeO2.5 did not show a strong decrease of conductivity around 400 K, which might be explained by a partial release of water already taking place at temperatures above 300 K, transforming the material into LW-BaFeO2.5 and adding water to the helium atmosphere in the cryostat (see thermogravimetric measurements reported in Section 3.1).
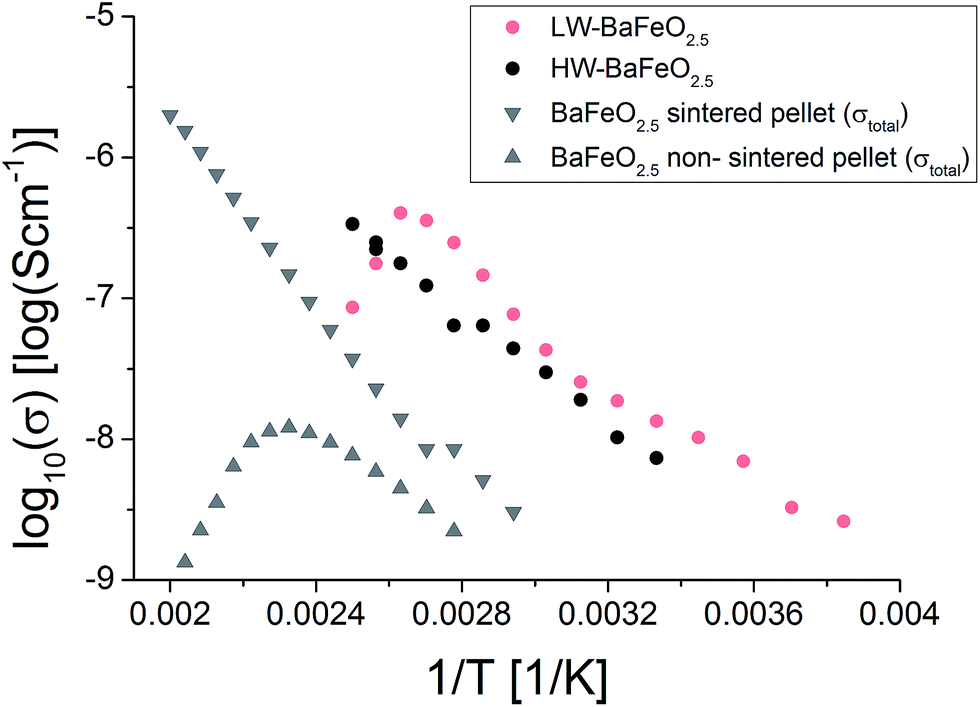 | ||
| Fig. 15 Total conductivities of BaFeO2.5 and its hydrated modifications. | ||
4 Conclusions and outlook
We have shown that BaFeO2.5 can be reversibly transformed into two different hydrated modifications, LW-BaFeO2.5 and HW-BaFeO2.5, depending on the detailed hydration temperature used, transforming the compound from a poor electron conductor into a proton conductor with a grain proton conductivity in the range of ∼10−6 to 10−7 S cm−1 in dry atmospheres around ambient temperatures. This represents the first report of proton conductivity in a Fe based perovskite, and raises potential applications in solid oxide fuel cells and/or electrolysers and H2 separation membranes.In order to take this work further, thin film studies are required to eliminate the grain boundary contribution. Also, the conductivity is intended to be studied under variable oxidizing and non-oxidizing, wet and dry, atmospheres to obtain a deeper understanding of the conditions where proton conductivity can be regarded to be superior compared to the electronic contribution. Furthermore, chemical substitution of the A- and B-site cations might be expected to allow for material optimization with regards to the magnitude of the proton and electronic contribution to the conductivity, as well as the stability.
Acknowledgements
Neutron diffraction beam time at ISIS was provided by the Science and Technology Facilities Council (STFC).References
- M. Kuhn, S. Hashimoto, K. Sato, K. Yashiro and J. Mizusaki, Solid State Ionics, 2011, 195, 7–15 CrossRef CAS.
- N. Hayashi, T. Yamamoto, H. Kageyama, M. Nishi, Y. Watanabe, T. Kawakami, Y. Matsushita, A. Fujimori and M. Takano, Angew. Chem., Int. Ed., 2011, 50, 12547–12550 CrossRef CAS PubMed.
- M. Parras, L. Fournes, J. C. Grenier, M. Pouchard, M. Vallet, J. M. Calbet and P. Hagenmuller, J. Solid State Chem., 1990, 88, 261–268 CrossRef CAS.
- X. D. Zou, S. Hovmoller, M. Parras, J. M. Gonzalez-Calbet, M. Vallet-Regi and J. C. Grenier, Acta Crystallogr., Sect. A: Found. Crystallogr., 1993, 49, 27–35 Search PubMed.
- O. Clemens, M. Groeting, R. Witte, J. Manuel Perez-Mato, C. Loho, F. J. Berry, R. Kruk, K. S. Knight, A. J. Wright, H. Hahn and P. R. Slater, Inorg. Chem., 2014, 53, 5911–5921 CrossRef CAS PubMed.
- Y. Yamazaki, R. Hernandez-Sanchez and S. M. Haile, Chem. Mater., 2009, 21, 2755–2762 CrossRef CAS.
- G. B. Zhang and D. M. Smyth, Solid State Ionics, 1995, 82, 153–160 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- A. Orera and P. R. Slater, Chem. Mater., 2010, 22, 675–690 CrossRef CAS.
- L. Bi, E. Fabbri and E. Traversa, Solid State Ionics, 2012, 214, 1–5 CrossRef CAS.
- J. Dailly, S. Fourcade, A. Largeteau, F. Mauvy, J. C. Grenier and M. Marrony, Electrochim. Acta, 2010, 55, 5847–5853 CrossRef CAS.
- K. D. Kreuer, S. Adams, W. Münch, A. Fuchs, U. Klock and J. Maier, Solid State Ionics, 2001, 145, 295–306 CrossRef CAS.
- R. Djenadic, M. Botros, C. Benel, O. Clemens, S. Indris, A. Chodhary, T. Bergfeldt and H. Hahn, Solid State Ionics, 2014, 263, 49–56 CrossRef CAS.
- A. A. Coelho, TOPAS-Academic, http://www.topas-academic.net, (accessed 20th of October 2014) Search PubMed.
- Topas V4.2, General profile and structure analysis software for powder diffraction data, User's Manual, Bruker AXS, Karlsruhe, Germany, 2008 Search PubMed.
- D. Johnson, ZView: a Software Program for IES Analysis, 2.8; Scribner Associates, Inc.: Southern Pines, NC, 2008.
- O. Clemens, C. Rongeat, M. A. Reddy, A. Giehr, M. Fichtner and H. Hahn, Dalton Trans., 2014, 43, 15771–15778 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Mori, J. Am. Ceram. Soc., 1966, 49, 600–605 CrossRef CAS.
- W. Fischer, G. Reck and T. Schober, Solid State Ionics, 1999, 116, 211–215 CrossRef.
- O. Clemens, J. Solid State Chem., 2015, 225, 261–270 CrossRef CAS.
- J. P. Hodges, S. Short, J. D. Jorgensen, X. Xiong, B. Dabrowski, S. M. Mini and C. W. Kimball, J. Solid State Chem., 2000, 151, 190–209 CrossRef CAS.
- L. Truong, M. Howard, O. Clemens, K. S. Knight, P. R. Slater and V. Thangadurai, J. Mater. Chem. A, 2013, 1, 13469–13475 CAS.
- M. Gotić and S. Musić, J. Mol. Struct., 2007, 834–836, 445–453 CrossRef.
- J. F. Shin, A. Orera, D. C. Apperley and P. R. Slater, J. Mater. Chem., 2011, 21, 874–879 RSC.
- J. F. Shin and P. R. Slater, J. Power Sources, 2011, 196, 8539–8543 CrossRef CAS.
- F. Fujishiro, Y. Kojima and T. Hashimoto, J. Am. Ceram. Soc., 2012, 95, 3634–3637 CrossRef CAS.
- F. Fujishiro, K. Fukasawa and T. Hashimoto, J. Am. Ceram. Soc., 2011, 94, 3675–3678 CrossRef CAS.
- P. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- J. B. Goodenough, Magnetism and the Chemical Bond, Interscience Publisher, New York, 1963 Search PubMed.
- O. Clemens, F. J. Berry, J. Bauer, A. J. Wright, K. S. Knight and P. R. Slater, J. Solid State Chem., 2013, 203, 218–226 CrossRef CAS.
- O. Clemens, A. J. Wright, F. J. Berry, R. I. Smith and P. R. Slater, J. Solid State Chem., 2013, 198, 262–269 CrossRef CAS.
- M. Sturza, S. Daviero-Minaud, H. Kabbour, O. Gardoll and O. Mentré, Chem. Mater., 2010, 22, 6726–6735 CrossRef CAS.
- M. Sturza, H. Kabbour, S. Daviero-Minaud, D. Filimonov, K. Pokholok, N. Tiercelin, F. Porcher, L. Aldon and O. Mentré, J. Am. Chem. Soc., 2011, 133, 10901–10909 CrossRef CAS PubMed.
- O. Clemens, F. J. Berry, A. J. Wright, K. S. Knight, J. M. Perez-Mato, J. M. Igartua and P. R. Slater, J. Solid State Chem., 2015, 226, 326–331 CrossRef CAS.
- O. Clemens, R. Kruk, E. A. Patterson, C. Loho, C. Reitz, A. J. Wright, K. S. Knight, H. Hahn and P. R. Slater, Inorg. Chem., 2014, 53, 12572–12583 CrossRef CAS PubMed.
- O. Clemens, F. J. Berry, A. J. Wright, K. S. Knight, J. M. Perez-Mato, J. M. Igartua and P. R. Slater, J. Solid State Chem., 2013, 206, 158–169 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louer and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- E. Dixon, J. Hadermann and M. A. Hayward, Chem. Mater., 2012, 24, 1486–1495 CrossRef CAS.
- M. I. Aroyo, A. Kirov, C. Capillas, J. M. Perez-Mato and H. Wondratschek, Acta Crystallogr., Sect. A: Found. Crystallogr., 2006, 62, 115–128 CrossRef PubMed.
- M. I. Aroyo, J. M. Perez-Mato, C. Capillas, E. Kroumova, S. Ivantchev, G. Madariaga, A. Kirov and H. Wondratschek, Z. Kristallogr., 2006, 221, 15–27 CAS.
- M. I. Aroyo, J. M. Perez-Mato, D. Orobengoa, E. Tasci, G. de la Flor and A. Kirov, Bulg. Chem. Commun., 2011, 43, 183–197 CAS.
- U. Müller, Symmetry Relationships between Crystal Structures: Applications of Crystallographic Group Theory in Crystal Chemistry, Oxford University Press, Oxford, 2013 Search PubMed.
- W. Hamilton, Acta Crystallogr., 1965, 18, 502–510 CrossRef CAS.
- P. D. Battle, T. C. Gibb and P. Lightfoot, J. Solid State Chem., 1990, 84, 237–244 CrossRef CAS.
- I. D. Brown, The chemical bond in inorganic chemistry: the bond valence model, Oxford University Press Inc., New York, 2002 Search PubMed.
- I. D. Brown and K. K. Wu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1957–1959 CrossRef.
- O. Clemens and P. R. Slater, Rev. Inorg. Chem., 2013, 33, 105–117 CrossRef CAS.
- M. Harder and H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 1980, 464, 169–175 CrossRef CAS.
- C. Greaves, A. J. Jacobson, B. C. Tofield and B. E. F. Fender, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 641–646 CrossRef.
- M. Schmidt and S. J. Campbell, J. Solid State Chem., 2001, 156, 292–304 CrossRef CAS.
- S. Mori, J. Am. Ceram. Soc., 1965, 48, 165 CrossRef CAS.
- F. Fujishiro, Mater. Chem. Phys., 2015, 153, 5–8 CrossRef CAS.
- S. M. Haile, D. L. West and J. Campbell, J. Mater. Res., 1998, 13, 1576–1595 CrossRef CAS.
- J. F. Shin, D. C. Apperley and P. R. Slater, Chem. Mater., 2010, 22, 5945–5948 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Neutron powder diffraction data of LW- and HW-BaFeO2.5 recorded at different temperatures, explanations regarding the indexing procedure and symmetry considerations as well as regarding the structural relaxation mechanisms found in barium ferrite type compounds. See DOI: 10.1039/c5ta06383c |
| This journal is © The Royal Society of Chemistry 2016 |