
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics of liquid–liquid phase separation in protein solutions exhibiting LCST phase behavior studied by time-resolved USAXS and VSANS†
Stefano
Da Vela
 a,
Michal K.
Braun
a,
Andreas
Dörr
a,
Alessandro
Greco
a,
Johannes
Möller
b,
Zhendong
Fu
c,
Fajun
Zhang
*a and
Frank
Schreiber
a
a,
Michal K.
Braun
a,
Andreas
Dörr
a,
Alessandro
Greco
a,
Johannes
Möller
b,
Zhendong
Fu
c,
Fajun
Zhang
*a and
Frank
Schreiber
a
aInstitut für Angewandte Physik, Universität Tübingen, Auf der Morgenstelle 10, 72076 Tübingen, Germany. E-mail: fajun.zhang@uni-tuebingen.de; Fax: +49 7071 29 5110; Tel: +49 7071 29 78670
bEuropean Synchrotron Radiation Facility, 71 avenue des Martyrs, 38043 Grenoble Cedex 9, France
cForschungszentrum Jülich GmbH, JCNS@MLZ, Lichtenbergstrasse 1, 85747, Garching, Germany
First published on 3rd November 2016
Abstract
We study the kinetics of the liquid–liquid phase separation (LLPS) and its arrest in protein solutions exhibiting a lower critical solution temperature (LCST) phase behavior using the combination of ultra-small angle X-ray scattering (USAXS) and very-small angle neutron scattering (VSANS). We employ a previously established model system consisting of bovine serum albumin (BSA) solutions with YCl3. We follow the phase transition from sub-second to 104 s upon an off-critical temperature jump. After a temperature jump, the USAXS profiles exhibit a peak that grows in intensity and shifts to lower q values with time. Below 45 °C, the characteristic length scale (ξ) obtained from this scattering peak increases with time with a power of about 1/3 for different sample compositions. This is in good agreement with the theoretical prediction for the intermediate stage of spinodal decomposition where the growth is driven by interface tension. Above 45 °C, ξ follows initially the 1/3 power law growth, then undergoes a significant slowdown, and an arrested state is reached below the denaturation temperature of the protein. This growth kinetics may indicate that the final composition of the protein-rich phase is located close to the high density branch of the LLPS binodal when a kinetically arrested state is reached.
1 Introduction
Understanding liquid–liquid phase separation (LLPS) in protein solutions allows to gain insight on phase separation mechanisms in biological systems.1,2 This process is of relevance, for instance, to protein crystallization or protein condensation related diseases, where subtle changes of the protein structure alter the effective interactions and lead to a phase transition.3–5 LLPS is also of technological relevance for the formulation of biopharmaceuticals.6In colloidal and protein systems, it has been established that a short-ranged attraction leads to a metastable LLPS into a liquid dense and a dilute phase. In this case, the gelation line often cuts the phase boundary near the critical point, leading to an arrested phase transition. Such arrested LLPS has been reported in many colloid and protein systems.7–19 Studies on lysozyme solutions indicate that systems undergoing an arrested spinodal decomposition have a bicontinuous structure with a protein-poor fluid interpenetrating a dense glassy protein network.9,10,12,20,21 The LLPS in these systems is normally enthalpy-driven, i.e. it features an upper critical solution temperature (UCST) phase behavior. A glass line can be predicted by using mode coupling theory. However, the exact location of the glass line is still debated.7–9,22–27 Cardinaux et al.9 experimentally determined the glass line in a lysozyme solution in the presence of NaCl, featuring UCST phase behavior. It was shown that the glass line enters the two-phase region. Thus, for deep quenches, the phase transition arrests at the early stage of spinodal decomposition.9,10,12 The arrest at the early stage implies that only density fluctuations occur. Once the density of the protein-rich phase reaches the one of the glassy state, the system becomes arrested. The characteristic length thus stays constant in time before coarsening starts. A similar phase behavior with the fluid–fluid coexistence intersected by an arrest line has been reported recently for γb-crystallin.28 However, other experimental, theoretical and simulation work on colloid–polymer systems with short-range attraction indicates that the glass line may also follow the binodal for equilibrium liquid–gas phase separation. In this case the arrest occurs during the coarsening process, i.e. the coarsening process of spinodal decomposition generates large clusters that span the system and arrest dynamically.8,29 In particular, simulation30 and experimental31 work using “patchy” systems has shown evidence for the latter scenario. Studies on the effects of hydrodynamic interactions on the glass formation in colloidal systems also suggested that the hydrodynamic interactions during the coarsening process enhance the formation of open structures which favor percolation and dynamic arrest.32,33 Moreover, the relation between the glass line and the phase boundaries, as well as their impact on the formation of the arrested state may be sensitive to the features of the attractive potential between the particles.25,34
One possible way to distinguish these two different routes to the arrested state is to follow the growth kinetics in both the initial and the coarsening stage. The kinetics of the phase transition is best characterized by following the large-scale structural features of the dense network as a function of time. The recent developments of ultra-small angle X-ray scattering (USAXS) and very small angle neutron scattering (VSANS) beamlines allow to cover a minimum q of 9 × 10−4 nm−1 and 4.3 × 10−4 nm−1 respectively, i.e. maximum length scales up to 7.0 μm for USAXS and 14.6 μm for VSANS. This opens new opportunities for studying the kinetics of this phase transition and its arrest. VSANS and USAXS have the advantages of high transmission and of reduced multiple scattering effects which are often the limiting factors when employing ultra-small angle light scattering (USALS) and optical microscopy, especially for concentrated or turbid samples.10,35 The combination of USAXS and VSANS can be used to study the structural evolution in a large range of length scales during the phase transition. The knowledge gained from the time-resolved structural characterization of the spinodal decomposition has the potential to provide new insights into related processes, such as protein crystallization, protein condensation, formation of arrested states and possibly even the formation of biological photonic structures.36 Technologically, a deeper understanding of these phenomena impacts the production of novel food gels and glasses.12
In this work, we aim to follow the kinetics of LLPS and distinguish the exact route leading to the arrested state. We have studied the phase behavior of bovine and human serum albumin (BSA and HSA) in the presence of trivalent salt as a function of salt concentration and temperature.37,38 BSA (HSA is closely related) is a globular protein, roughly heart-shaped, with a radius of gyration of 29.8 Å.39 Protein–protein interactions in the presence of trivalent salts have been characterized previously for these systems.40,41 A reentrant condensation phase behavior has been established with a LLPS occurring within the condensed regime in a closed area. The phase behavior was rationalized in terms of an ion-activated patchy interaction.40,42 We have recently extended the phase diagram along the temperature axis. The LLPS binodal has been determined for BSA with YCl3, which leads to a lower critical solution temperature (LCST) phase behavior.43
Here we use USAXS and VSANS to study the kinetics of LCST–LLPS in the dense phases of the BSA–YCl3 system. First we aim to establish a new method for studying the kinetics of the phase transition based on the advantages of USAXS and VSANS. Second, we aim to explore how an arrested state can be reached in our system by a temperature jump to higher temperatures. The limiting condition for this system is that protein thermal denaturation (around 60 °C) sets the upper limit of the temperature window. Furthermore, from the high quality growth kinetics data obtained from USAXS, we aim to distinguish the exact route to the dynamically arrested state in our system.
2 Experimental
2.1 Materials and sample preparation
Bovine serum albumin (BSA, A7906) and yttrium chloride (YCl3, 451363) were purchased from Sigma-Aldrich and used as received. Salt and protein stock solutions were prepared by dissolving the desired amount of protein and salt in degassed Milli-Q water (Merck Millipore 18.2 MΩ cm). The stock solutions were used to prepare protein samples in the two-phase region of the phase diagram at room temperature (22 °C). To obtain these samples, concentrations were adjusted by dilution with pure Milli-Q water. No buffer was used to avoid the effect of other co-ions. In the following, the concentrations of salt and protein required to obtain the phase separated samples are referred to as “parent solutions”, having a fixed protein concentration of 175 mg mL−1 and varying salt concentrations in the range of 36–46 mM. After preparation, the two phases were separated by centrifugation. The dense phases were kept cold before USAXS and VSANS measurements. For 175 mg mL−1 parent solutions in a comparable salt concentration range (35 to 42 mM), the BSA concentration of the dense phases after equilibration at room temperature shows little change, varying between 270 ± 6 mg mL−1 and 289 ± 31 mg mL−1. The preferential partitioning of YCl3 was not determined in this work.2.2 Determination of the binodal of LLPS
To determine the binodal of LLPS, parent solutions with constant protein and salt concentration were prepared under a series of temperatures. A water bath (Thermo Scientific) was used to control the temperature with a resolution of ±0.1 °C. The resulting two phases were separated by a brief centrifugation step (2 min at 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 470 × g) to sharpen the interface between the dense and dilute phases. The concentration of the dilute phase was determined by UV-vis absorption using E280 = 0.667 mg−1 mL cm−1. The BSA concentration in the corresponding dense phase was calculated using the lever rule. Samples prepared at the higher temperatures feature turbid and gel-like dense phases. The centrifugation step was applied to all the samples used to determine the binodal in Fig. 1. For temperatures below 40 °C, where full LLPS occurs, long time sedimentation or a brief centrifugation gave the same results. Above 40 °C, the nonequilibrium state of the system may slightly affect the determination of the binodal.
470 × g) to sharpen the interface between the dense and dilute phases. The concentration of the dilute phase was determined by UV-vis absorption using E280 = 0.667 mg−1 mL cm−1. The BSA concentration in the corresponding dense phase was calculated using the lever rule. Samples prepared at the higher temperatures feature turbid and gel-like dense phases. The centrifugation step was applied to all the samples used to determine the binodal in Fig. 1. For temperatures below 40 °C, where full LLPS occurs, long time sedimentation or a brief centrifugation gave the same results. Above 40 °C, the nonequilibrium state of the system may slightly affect the determination of the binodal.
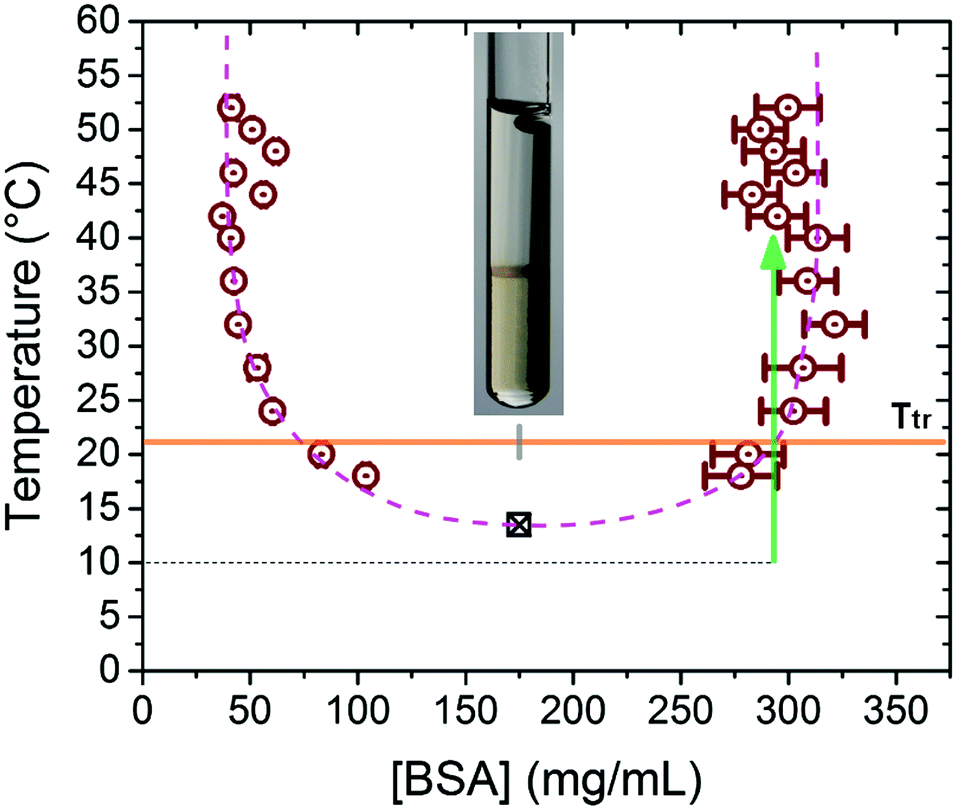 | ||
| Fig. 1 Binodal of the LLPS for a parent solution with 175 mg mL−1 BSA and 42 mM YCl3. The purple dashed line is a guide to the eye, the green arrow represents a typical temperature jump from 10 to 40 °C. The orange line marks the preparation temperature, at which the parent solutions phase separated to yield the dense phases used in scattering experiments. The square symbol is the LCST value of the parent solution, the error bar is smaller than the symbol. The inset shows a sample tube after LLPS and sedimentation at room temperature, with a yellowish dense and a transparent dilute phase. | ||
2.3 Ultra-small angle X-ray scattering (USAXS)
USAXS experiments were performed at the ESRF beamline ID0244 (Grenoble, France) at an X-ray energy of 12.46 keV, which corresponds to a wavelength of λ = 0.995 Å. The sample-to-detector distance was set to 30.7 m, covering a q range of 9 × 10−4 nm−1 to 7 × 10−2 nm−1, with q being the scattering vector defined as q = (4π/λ)·sinθ where 2θ is the scattering angle. The data were collected by a fast-readout low-noise (FReLoN) fibre-optic coupled CCD detector in a 2 × 2 binning mode. For each USAXS experiment, the sample capillary (1.0 mm in diameter) was first equilibrated at 10 °C. Then, the temperature was quickly increased to the desired temperature with a heating rate of 80 K min−1, using a Linkam heating stage. Successive exposures (5 to 50 ms depending on the acquisition rate) were collected at intervals varying between 0.3 s to 5 s in order to capture the time evolution of the scattering intensity during the temperature jumps, covering a time span of 10 minutes from the start of each jump. At the end of each experiment, the sample was cooled back to 10 °C in order to check its stability and the reversibility of the process (see Fig. S1, ESI†). Each measurement was repeated 2 or 3 times, changing the position of the capillary at the end of each USAXS run to reduce the radiation damage. The scattering profile collected before the temperature jump from the sample in the single-phase state at 10 °C was used for background correction. The first scattering profile at t = 0 was collected after a waiting time to compensate for the time needed to reach the phase transition temperature given the heating rate. The time intervals for the other profiles were deduced by difference from the time stamps of the scattering profiles or by the known acquisition rate for the high-rate exposures used to cover the early stage of the phase separation. The peak position in the background subtracted data was located using a computer script based on signal smoothing and search for the maximum in the smoothed data.
2.4 Very small angle neutron scattering (VSANS)
VSANS experiments were performed in order to follow the growth kinetics at the later stages of the phase separation. We employed the focusing mirror instrument KWS-3 operated by JCNS at the Heinz Maier-Leibnitz Zentrum (MLZ), Garching, Germany.45 The neutron wavelength was 12.8 Å with Δλ/λ = 20%, and the sample-to-detector distance was 9.5 m, corresponding to a q-range from 4.3 × 10−4 to 1.1 × 10−2 nm−1. A 6Li-scintillation detector with pixel size of 0.116 × 0.116 mm2, size 3 × 3 cm2 and a deadtime of 2.7 μs was used to record the scattered intensity, using no beamstop. Experiments were performed in a temperature-controlled sample environment for 1 mm quartz cells. The beam size at the entrance aperture was 0.7 × 0.7 mm2. Each sample was equilibrated for 20 min at 10 °C, then heated to 35 °C. The scattered intensity was collected in twelve 10 min exposures for 2 h. At the end of the collection at 35 °C, the sample was cooled back to 10 °C and the scattering intensity was collected for further 2 h to check that the scattering had returned to background level. The peak position was found on the radially integrated data by means of a local Gaussian fit using the data points in the vicinity of the maximum (justified, since these are well-developed pronounced peaks). VSANS data reduction and absolute intensity calibration was performed using the program QtiKWS provided by JCNS.463 Results and discussion
3.1 Binodal of LCST–LLPS for a given protein–salt composition
The binodal in the temperature-protein concentration plane was determined by allowing the parent solutions with constant compositions to equilibrate at different temperatures. We have determined the binodal for a system with 175 mg mL−1 BSA and 42 mM YCl3. The resulting phase boundary is shown in Fig. 1, clearly displaying the LCST phase behavior. At pH = 7.0 and at moderate ionic strengths, the denaturation temperature of BSA is above 60 °C,47–49 preventing the extension of the phase diagram to higher temperatures.We note that for temperatures higher than 45 °C, the dense phases became gel-like and the determined protein concentrations on the high density side of the binodal are smaller than those at lower temperatures. Cardinaux et al. observed a significant reduction of protein concentration for lysozyme solutions when the dense phase became gel-like, which was interpreted as the glass line entering the co-existence region.9 From Fig. 1, we cannot determine whether the dense phases at higher temperatures are arrested or not, nor whether the arrested state follows the equilibrium binodal. In the following we thus employ USAXS to follow the kinetics of phase separation to gain a better understanding of the system.
In order to study the kinetics of phase separation, samples located on the high protein concentration branch of the binodal were obtained from a first LLPS of parent solutions with 175 mg mL−1 BSA and YCl3 in the range of 36 to 46 mM. Such “dense phase” samples are all clear solutions at the preparation temperature of ∼22 °C. The preparation temperature of the samples thus corresponds to the binodal at the phase transition temperature Ttr. Increasing the temperature to a temperature above Ttr, the dense phases will undergo a new LLPS as the samples are again brought into the two-phase region above the binodal. Ttr is also used to define the quench depth for the temperature jumps with a final temperature Tjump as ΔT = Tjump − Ttr.
Upon the temperature jump, these high density samples are expected to undergo an off-critical quench in the two-phase region (see green arrow in Fig. 1). While for small quench depths the temperature jump may end up in the metastable zone between the binodal and spinodal line, for sufficiently deep quenches the phase separation mechanism is expected to be spinodal decomposition. A study on LLPS in lysozyme solutions by Shah et al.50 indicates that the spinodal and binodal lines are relatively close to each other, and only for shallow temperature quenches (ΔT below 5 °C), a pure nucleation and growth scenario can be observed. In our experiments, the quench depths are in the range ΔT = 8 to 35 °C. The samples will thus further phase separate into a new dense phase (majority phase) and a new dilute phase (minority phase). There is then the possibility that the system may produce a sample-spanning majority phase which can dynamically arrest during the phase transition.
3.2 Classical spinodal decomposition for Tjump ≤ 40 °C
 | ||
| Fig. 2 Typical time series of USAXS profiles for a sample obtained from a parent solution with 175 mg mL−1 BSA and 42 mM YCl3, after a temperature jump to Tjump = 35 °C in log (a) and linear (b) scale. The scattering profiles were obtained at an acquisition rate of 3.1 s−1. Only the curves for t < 13 s are shown. A plot including later stages of the peak evolution is shown in Fig. S2, ESI.† The peak position, qmax is inversely proportional to the characteristic length, ξ, between the structures forming in the phase separating system. | ||
Although the dense liquid phases we used for the temperature jump are located on the high volume fraction side of the binodal (Fig. 1), the spontaneous evolution (Fig. 2a) and the bi-continuous morphology observed by optical microscopy (Fig. S3, ESI†) in the early stage suggest that the overall kinetics of the phase transition follows a spinodal decomposition.
The USAXS profiles allow to characterize the time evolution of μm-size structures in the phase-separating sample. The low-q scattering peak associated to these structures evolves in time as the phase transition proceeds. It is known to feature distinct scaling behaviors depending on the starting composition of the phase separating mixture. In general, the time evolution of the scattering intensity is described as resulting from the application of a time-dependent characteristic length to a time-independent scaling function. By making use of the dynamic similarity scaling operation,51,52 we normalize the scattering curves with the maximum intensity Imax and the corresponding q value qmax. As can be seen in Fig. 3, while the scattering curves do not fully collapse onto a master curve in the first few seconds, the later data up to 400 s coincide reasonably well. In order to double-check the off-critical nature of the quench, we also tried to normalize the data using a dynamic scaling operation of the kind I·qmax3 with or without the normalization by a Porod invariant-like integral (example plots are shown in Fig. S4, ESI†). The latter scaling operation is valid for critical quenches. The data do not overlap at all as expected, implying that we are not facing a critical quench.
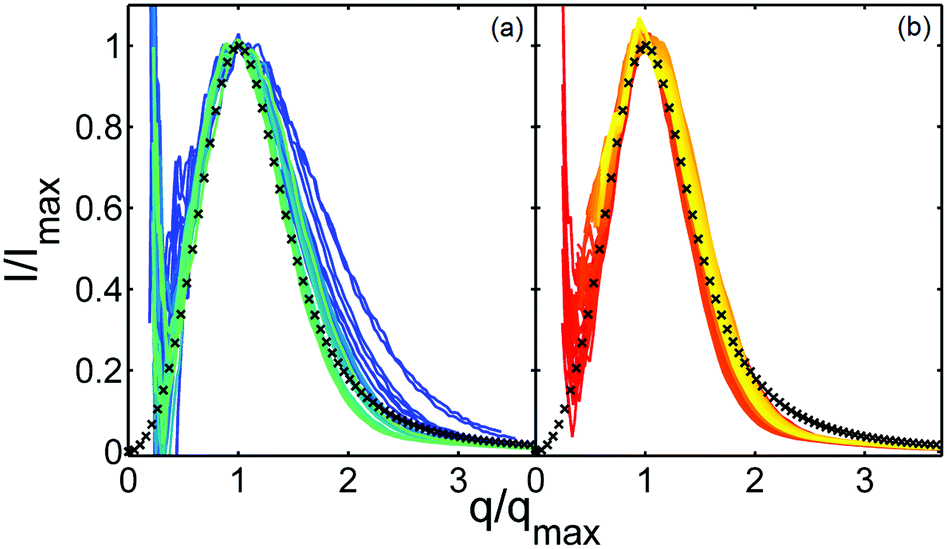 | ||
| Fig. 3 Dynamic similarity scaling of USAXS data by Imax and qmax, for the same sample and temperature jump as in Fig. 2. (a) Data from 2.3 to 10.2 s after the temperature jump. (b) Data from 10.6 to 385.5 s after the temperature jump. In both figures lighter colors refer to later times, the function depicted by black crosses is the off-critical Furukawa scaling function (eqn (1)). In agreement with theory, the scaling function describes the data reasonably well for later times. | ||
According to Furukawa,53 the off-critical scaling function for a three-dimensional system can be described as:
 | (1) |
 | ||
| Fig. 4 Plot of the characteristic length determined by USAXS as a function of time for different salt concentrations and Tjump. Dense phases from 175 mg mL−1 BSA with YCl3 (a) 44 mM; (b) 42 mM and (c) 40 mM. As the identification of a clear peak is not always possible for the earliest stage, the plots can differ in the starting time point. | ||
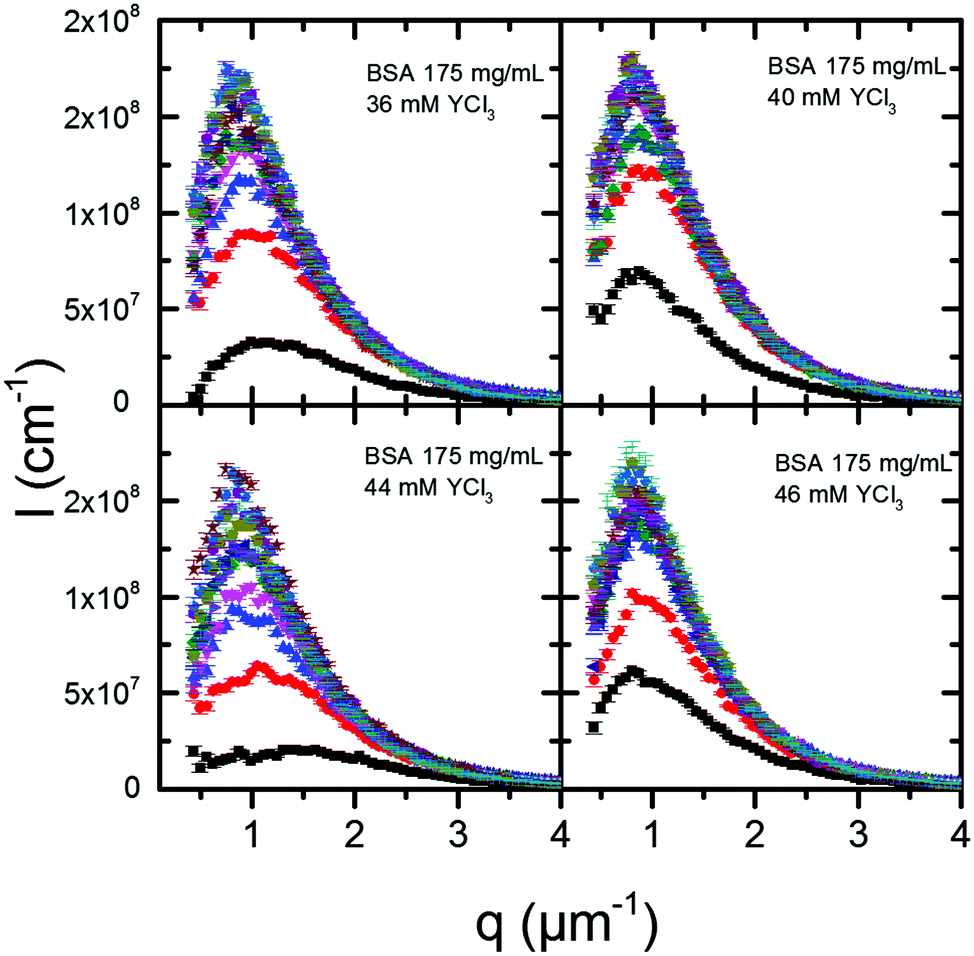 | ||
| Fig. 5 VSANS profiles of the late stage (10 min to 120 min) coarsening process of dense liquid phases after a temperature jump to Tjump = 35 °C. The caption in each picture specifies the composition of the parent solution from which the dense phases were obtained. The VSANS profiles were collected every 10 min. | ||
Combining USAXS and VSANS measurements, we can cover a larger time range for the kinetics of LLPS in our system. Fig. 6 shows the data for several samples undergoing the same temperature jump to 35 °C followed by USAXS or VSANS. The VSANS data clearly show the slowdown of the development of the characteristic length with time. Here the system shows deviation from theory,54 which would predict a power law increase of ξ ∼ t for the later stage of coarsening. The reason for the abrupt change of the kinetics is not entirely clear. Such a slowdown after the t1/3 trend is established, has been observed in molecular dynamics simulations for spinodal decomposition of glass-forming liquids.57 In those simulations, a logarithmic late stage growth of the characteristic length is found and ascribed to an intermittent coarsening mechanism. The intermittent coarsening sets in for quenches sufficiently deep as the dense phase becomes glassy, causing the dynamics to slow down and eventually resulting in an extremely slow ageing process. For the samples used in this work, however, this late time slowdown is more likely to result from other effects, such as sedimentation or confinement from the sample container, hindering the coarsening. In fact, the time evolution of analogous samples characterized by optical microscopy (Fig. S3, ESI†) shows how large droplets will eventually emerge from the phase-separating mixture after some hours. From this observation and from evidence of arrest occurring much earlier for deeper quenches (above 40 °C) presented in the next section, the significant slowdown of the coarsening in the VSANS data may be due to gravity-induced sedimentation.
 | ||
| Fig. 6 Plot of the characteristic length ξ determined by USAXS and VSANS as a function of time for different salt concentrations. All samples underwent the same temperature jump to Tjump = 35 °C. | ||
3.3 Arrested phase transition for Tjump > 40 °C
Employing the same method, time-resolved USAXS data were collected for temperature jumps to higher temperatures up to 57.5 °C. Typical USAXS profiles and selected 2D patterns are shown in Fig. S5 and S6, ESI.† In general, the structure factor peak behaves similarly as for lower temperatures in the initial stage. However the shift to lower q of the structure factor peak becomes gradually slower as the temperature is increased. For higher temperatures, after the start of the intermediate stage, the peak stops evolving altogether. After this, successive scattering profiles substantially overlap, and the system is kinetically arrested.The growth kinetics for a typical sample (dense phase of BSA 175 mg mL−1 with 44 mM YCl3) with different temperature jumps are shown in Fig. 7. Between 30 and 40 °C, the characteristic length increases with time following a power law of t1/3. At 45 °C, a significant slow-down is already visible. Above 50 °C, the characteristic length increases only in the initial stages (up to about 30 s) and then stays at a constant value indicating an arrested state. We note that below 55 °C, the samples are reversible even if the arrested state is reached, i.e. upon cooling the arrested state dissolves and the dense phase becomes clear again. Above 55 °C, the transition is not reversible, most likely due to effects related to protein denaturation in these conditions. We note that also for this irreversible condition, the structures formed in the arrested state are preserved.
 | ||
| Fig. 7 Plots of the characteristic length as a function of time for a dense liquid phase prepared from a 175 mg mL−1 BSA with 44 mM YCl3 parent solution at room temperature. The values of Tjump are shown in the figure. Above 40 °C a significant slowdown of the kinetics is observed and an arrested state is reached. | ||
Interestingly, our data show that a power law growth of the characteristic length is still present before it comes to a halt when the arrested phase transition occurs. This observation indicates that the arrest occurs during the coarsening instead that at the early stage of spinodal decomposition. This is consistent with a glass line closely following the equilibrium boundary. It can be seen in our experimentally established binodal in Fig. 1, that the high density branch tilts slightly towards the coexistence region for temperatures above 45 °C, but the deviation from the expected equilibrium boundary is small.
However, as remarked in a recent study on the coarsening kinetics of polymer blends with dynamic asymmetry,58 there could be situations in which the dense and dilute phase are out of equilibrium during coarsening, in spite of the existence of a sharp interface between the two phases. Nevertheless we emphasize that, in our system, the solutions undergo off-critical quenches, whereas in order to have so-called interface quench effects59 leading the system to metastable concentrations, a critical (or symmetrical) quench is needed. Moreover, earlier simulation work on spinodal decomposition with formation of a glassy phase,60,61 results in an arrested state with the two phases out of equilibrium precisely because the glass line was assumed to cross the binodal for the simulation. The apparent reprise of the coarsening in the arrested samples, for the longest times investigated, can be interpreted as arising from an ageing mechanism. This later stage behavior could be due to migration and coalescence of minority phase droplets through a glassy phase60 or rearrangements of the majority phase due to mechanical stress relaxation.57,62
Olais-Govea et al. have recently proposed a non-equilibrium theory of arrested spinodal decomposition,63 which reproduces the main experimental observations in colloid and protein systems.9,10,12,64 In their framework, the dynamic arrest of a solution undergoing LLPS can lead to three scenarios, depending on the quench depth. For shallow quenches (Region I) LLPS proceeds to completion, for intermediate quenches (Region II) the dynamic arrest prevents the full LLPS, and for very deep quenches (Region III) a homogeneous attractive glass is formed. The data we presented, featuring the kinetic arrested state formed during coarsening, may correspond to quenches just entering Region II trough the predicted non-sharp crossover boundary with Region I. For these quench depths then, the volume fraction at the glass line would still be close to the equilibrium binodal.
4 Conclusions
We have shown that USAXS and VSANS are adequate tools to follow the kinetics of LLPS in protein solutions. The relevant range of length and time scales can be monitored during the phase separation. Using our model system of BSA with YCl3 which exhibits an LLPS–LCST phase behavior, we have studied the kinetics of spinodal decomposition upon a temperature jump. The characteristic length as a function of time for different temperature jumps are analyzed. Below 45 °C, the USAXS profiles exhibit a scattering maximum at a finite qmax and its position shifts to lower q with time. In the early stage of phase transition (up to 300 seconds), the growth of the characteristic length ξ = 2π/qmax follows a power law ∼t1/3, which is consistent with the theoretical prediction of the coarsening process driven by the surface tension and limited by the diffusion of the protein. The growth kinetics show only a weak dependence on the starting concentration of YCl3 in the parent solutions. The post-coarsening process followed by VSANS for non-arrested samples significantly deviates from a power law growth. On this time scale (from 600 s up to 104 s), this slowdown may be due to ageing or sedimentation of the dense phase.Above 45 °C, the growth of the characteristic length in the early stage (below 30 s) still follows the t1/3 power law, but is then followed by a significant slowdown. At 50 °C and 55 °C, the characteristic length stays constant after the initial growth, indicating an arrested state. We note that below 55 °C, the samples can be cooled back to the clear, single-phase state. Above 55 °C, the transition is not reversible due to protein denaturation. The kinetics of the arrested phase transition indicate that the arrest occurs during the coarsening stage instead that at the early stage of spinodal decomposition, which suggests that the gelation line follows more closely the equilibrium boundary of the binodal.
Acknowledgements
We gratefully acknowledge financial support from DFG, the allocation of beamtimes from ESRF and Forschungszentrum Jülich/FRM2 (Munich), and the financial support provided by JCNS to perform the neutron scattering measurements at the Heinz Maier-Leibnitz Zentrum (MLZ), Garching, Germany. We would like to thank J. K. G. Dhont for the helpful discussion.References
- L.-P. Bergeron-Sandoval, N. Safaee and S. W. Michnick, Cell, 2016, 165, 1067–1079 CrossRef CAS PubMed.
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Jülicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS PubMed.
- Y. Wang, A. Lomakin, T. Hideshima, J. P. Laubach, O. Ogun, P. G. Richardson, N. C. Munshi, K. C. Anderson and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13359–13361 CrossRef CAS PubMed.
- J. Gunton, A. Shiryayev and D. Pagan, Protein Condensation: Kinetic Pathways to Crystallization and Disease, Cambridge University Press, 2007 Search PubMed.
- O. Galkin, K. Chen, R. L. Nagel, R. E. Hirsch and P. G. Vekilov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8479–8483 CrossRef CAS PubMed.
- A. S. Raut and D. S. Kalonia, Mol. Pharmaceutics, 2016, 13, 1431–1444 CrossRef CAS PubMed.
- P. N. Segrè, V. Prasad, A. B. Schofield and D. A. Weitz, Phys. Rev. Lett., 2001, 86, 6042–6045 CrossRef PubMed.
- P. J. Lu, E. Zaccarelli, F. Ciulla, A. B. Schofield, F. Sciortino and D. A. Weitz, Nature, 2008, 453, 499–503 CrossRef CAS PubMed.
- F. Cardinaux, T. Gibaud, A. Stradner and P. Schurtenberger, Phys. Rev. Lett., 2007, 99, 118301 CrossRef PubMed.
- T. Gibaud and P. Schurtenberger, J. Phys.: Condens. Matter, 2009, 21, 322201 CrossRef PubMed.
- T. Gibaud, F. Cardinaux, J. Bergenholtz, A. Stradner and P. Schurtenberger, Soft Matter, 2011, 7, 857–860 RSC.
- T. Gibaud, N. Mahmoudi, J. Oberdisse, P. Lindner, J. S. Pedersen, C. L. P. Oliveira, A. Stradner and P. Schurtenberger, Faraday Discuss., 2012, 158, 267 RSC.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- A. Zaccone, J. J. Crassous and M. Ballauff, J. Chem. Phys., 2013, 138, 104908 CrossRef PubMed.
- B. Barton, P. Graham and A. McHugh, Macromolecules, 1998, 31, 1672–1679 CrossRef CAS.
- Y. Gao, J. Kim and M. E. Helgeson, Soft Matter, 2015, 11, 6360–6370 RSC.
- M. J. Glassman and B. D. Olsen, Biomacromolecules, 2015, 16, 3762–3773 CrossRef CAS PubMed.
- P. D. Godfrin, S. D. Hudson, K. Hong, L. Porcar, P. Falus, N. J. Wagner and Y. Liu, Phys. Rev. Lett., 2015, 115, 228302 CrossRef PubMed.
- R. Vavrin, J. Kohlbrecher, A. Wilk, M. Ratajczyk, M. P. Lettinga, J. Buitenhuis and G. Meier, J. Chem. Phys., 2009, 130, 154903 CrossRef CAS PubMed.
- S. Manley, H. Wyss, K. Miyazaki, J. Conrad, V. Trappe, L. Kaufman, D. Reichman and D. Weitz, Phys. Rev. Lett., 2005, 95, 238302 CrossRef CAS PubMed.
- H. Sedgwick, K. Kroy, A. Salonen, M. Robertson, S. Egelhaaf and W. Poon, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 16, 77–80 CrossRef CAS PubMed.
- A. I. Campbell, V. J. Anderson, J. S. van Duijneveldt and P. Bartlett, Phys. Rev. Lett., 2005, 94, 208301 CrossRef PubMed.
- K. Kroy, M. E. Cates and W. C. K. Poon, Phys. Rev. Lett., 2004, 92, 148302 CrossRef CAS PubMed.
- M. Miller and D. Frenkel, Phys. Rev. Lett., 2003, 90, 135702 CrossRef PubMed.
- A. P. Eberle, N. J. Wagner and R. Castañeda-Priego, Phys. Rev. Lett., 2011, 106, 105704 CrossRef PubMed.
- N. E. Valadez-Pérez, Y. Liu, A. P. R. Eberle, N. J. Wagner and R. Castañeda-Priego, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 060302 CrossRef PubMed.
- G. Foffi, C. D. Michele, F. Sciortino and P. Tartaglia, Phys. Rev. Lett., 2005, 94, 078301 CrossRef CAS PubMed.
- S. Bucciarelli, L. Casal-Dujat, C. De Michele, F. Sciortino, J. Dhont, J. Bergenholtz, B. Farago, P. Schurtenberger and A. Stradner, J. Phys. Lett., 2015, 6, 4470–4474 CAS.
- J. Conrad, H. Wyss, V. Trappe, S. Manley, K. Miyazaki, L. Kaufman, A. Schofield, D. Reichman and D. Weitz, J. Rheol., 2010, 54, 421 CrossRef CAS.
- F. Sciortino and E. Zaccarelli, Curr. Opin. Solid State Mater. Sci., 2011, 15, 246–253 CrossRef CAS.
- B. Ruzicka, E. Zaccarelli, L. Zulian, R. Angelini, M. Sztucki, A. Moussaïd, T. Narayanan and F. Sciortino, Nat. Mater., 2011, 10, 56–60 CrossRef CAS PubMed.
- C. P. Royall, J. Eggers, A. Furukawa and H. Tanaka, Phys. Rev. Lett., 2015, 114, 258302 CrossRef PubMed.
- A. Furukawa and H. Tanaka, Phys. Rev. Lett., 2010, 104, 245702 CrossRef PubMed.
- L. J. Teece, M. A. Faers and P. Bartlett, Soft Matter, 2011, 7, 1341–1351 RSC.
- N. A. Verhaegh, J. S. van Duijneveldt, J. K. Dhont and H. N. Lekkerkerker, Phys. A, 1996, 230, 409–436 CrossRef CAS.
- E. R. Dufresne, H. Noh, V. Saranathan, S. G. J. Mochrie, H. Cao and R. O. Prum, Soft Matter, 2009, 5, 1792–1795 RSC.
- F. Zhang, M. W. A. Skoda, R. M. J. Jacobs, S. Zorn, R. A. Martin, C. M. Martin, G. F. Clark, S. Weggler, A. Hildebrandt, O. Kohlbacher and F. Schreiber, Phys. Rev. Lett., 2008, 101, 148101 CrossRef CAS PubMed.
- F. Zhang, S. Weggler, M. J. Ziller, L. Ianeselli, B. S. Heck, A. Hildebrandt, O. Kohlbacher, M. W. A. Skoda, R. M. J. Jacobs and F. Schreiber, Proteins, 2010, 78, 3450–3457 CrossRef CAS PubMed.
- J. W. Anderegg, W. Beeman, S. Shulman and P. Kaesberg, J. Am. Chem. Soc., 1955, 77, 2927–2937 CrossRef CAS.
- F. Zhang, R. Roth, M. Wolf, F. Roosen-Runge, M. W. A. Skoda, R. M. J. Jacobs, M. Sztucki and F. Schreiber, Soft Matter, 2012, 8, 1313–1316 RSC.
- M. Wolf, F. Roosen-Runge, F. Zhang, R. Roth, M. W. Skoda, R. M. Jacobs, M. Sztucki and F. Schreiber, J. Mol. Liq., 2014, 200, 20–27 CrossRef CAS.
- F. Roosen-Runge, F. Zhang, F. Schreiber and R. Roth, Sci. Rep., 2014, 4, 7016 CrossRef CAS PubMed.
- O. Matsarskaia, M. K. Braun, F. Roosen-Runge, M. Wolf, F. Zhang, R. Roth and F. Schreiber, J. Phys. Chem. B, 2016, 120, 7731–7736 CrossRef CAS PubMed.
- P. Van Vaerenbergh, J. Léonardon, M. Sztucki, P. Boesecke, J. Gorini, L. Claustre, F. Sever, J. Morse and T. Narayanan, Proceedings of the 12th International Conference on Synchrotron Radiation Instrumentation-Sri2015, 2016, p. 030034 Search PubMed.
- V. Pipich and Z. Fu, Journal of Large-Scale Research Facilities, 2015, 1, 31 CrossRef.
- V. Pipich, QtiKWS: user-friendly program for reduction, visualization, analysis and fit of SA(N)S data, http://www.qtikws.de, 2012 Search PubMed.
- G. Barone, S. Capasso, P. Del Vecchio, C. De Sena, D. Fessas, C. Giancola, G. Graziano and P. Tramonti, J. Therm. Anal., 1995, 45, 1255–1264 CrossRef CAS.
- C. Giancola, C. De Sena, D. Fessas, G. Graziano and G. Barone, Int. J. Biol. Macromol., 1997, 20, 193–204 CrossRef CAS PubMed.
- Y. Moriyama, E. Watanabe, K. Kobayashi, H. Harano, E. Inui and K. Takeda, J. Phys. Chem. B, 2008, 112, 16585–16589 CrossRef CAS PubMed.
- M. Shah, O. Galkin and P. G. Vekilov, J. Chem. Phys., 2004, 121, 7505–7512 CrossRef CAS PubMed.
- J. K. Dhont, J. Chem. Phys., 1996, 105, 5112–5125 CrossRef CAS.
- J. K. G. Dhont, An Introduction to Dynamics of Colloids, Elsevier Science B. V., 1996 Search PubMed.
- H. Furukawa, Phys. A, 1984, 123, 497–515 CrossRef.
- E. D. Siggia, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 20, 595–605 CrossRef CAS.
- H. Tanaka, J. Chem. Phys., 1997, 107, 3734–3737 CrossRef CAS.
- S. Bhat, R. Tuinier and P. Schurtenberger, J. Phys.: Condens. Matter, 2006, 18, L339 CrossRef CAS PubMed.
- V. Testard, L. Berthier and W. Kob, J. Chem. Phys., 2014, 140, 164502 CrossRef PubMed.
- W. Shi, W. Liu, J. Yang, Z. He and C. C. Han, Soft Matter, 2014, 10, 2649–2655 RSC.
- H. Tanaka, Phys. Rev. Lett., 1994, 72, 3690 CrossRef CAS PubMed.
- D. Sappelt and J. Jäckle, EPL, 1997, 37, 13 CrossRef CAS.
- D. Sappelt and J. Jäckle, Phys. A, 1997, 240, 453–479 CrossRef CAS.
- I. Zhang, C. P. Royall, M. A. Faers and P. Bartlett, Soft Matter, 2013, 9, 2076–2084 RSC.
- J. M. Olais-Govea, L. López-Flores and M. Medina-Noyola, J. Chem. Phys., 2015, 143, 174505 CrossRef PubMed.
- H. Guo, S. Ramakrishnan, J. L. Harden and R. L. Leheny, J. Chem. Phys., 2011, 135, 154903 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm01837h |
| This journal is © The Royal Society of Chemistry 2016 |