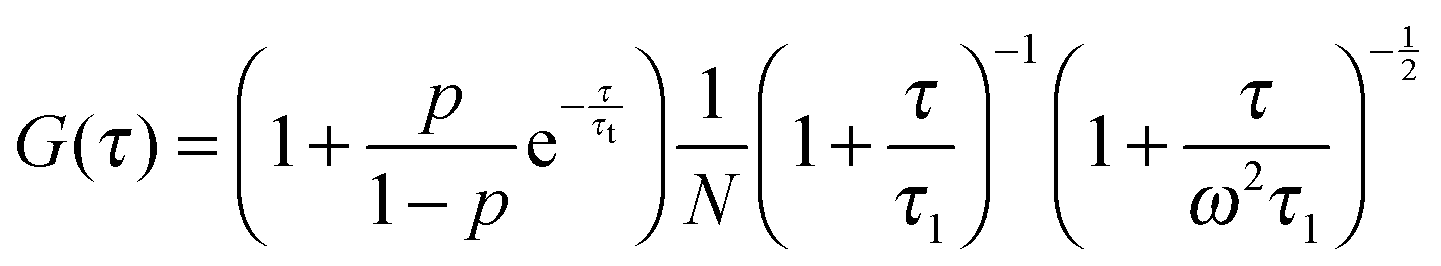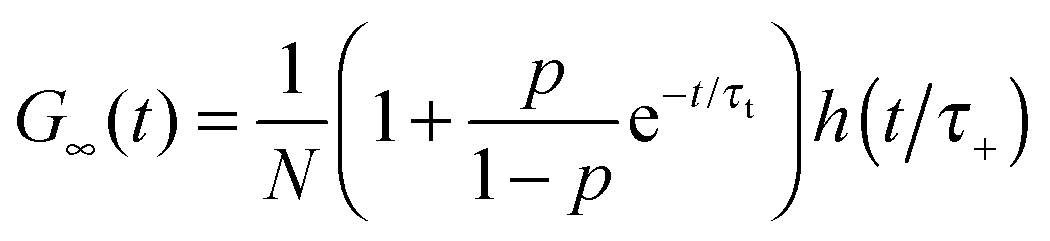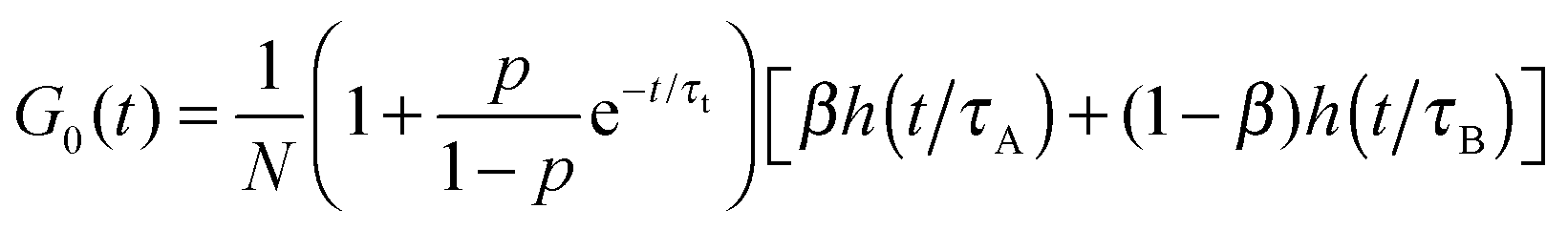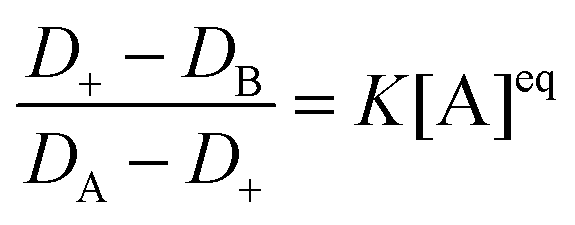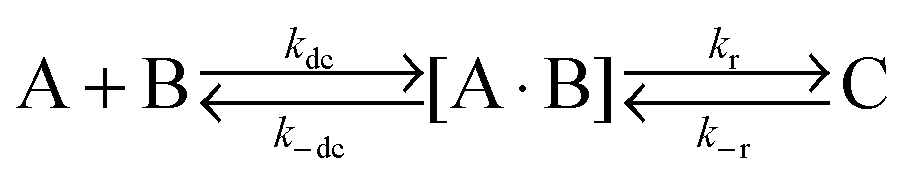DOI:
10.1039/C6SM01791F
(Paper)
Soft Matter, 2016,
12, 8186-8194
Determination of equilibrium and rate constants for complex formation by fluorescence correlation spectroscopy supplemented by dynamic light scattering and Taylor dispersion analysis†
Received
3rd August 2016
, Accepted 2nd September 2016
First published on 2nd September 2016
Abstract
The equilibrium and rate constants of molecular complex formation are of great interest both in the field of chemistry and biology. Here, we use fluorescence correlation spectroscopy (FCS), supplemented by dynamic light scattering (DLS) and Taylor dispersion analysis (TDA), to study the complex formation in model systems of dye–micelle interactions. In our case, dyes rhodamine 110 and ATTO-488 interact with three differently charged surfactant micelles: octaethylene glycol monododecyl ether C12E8 (neutral), cetyltrimethylammonium chloride CTAC (positive) and sodium dodecyl sulfate SDS (negative). To determine the rate constants for the dye–micelle complex formation we fit the experimental data obtained by FCS with a new form of the autocorrelation function, derived in the accompanying paper. Our results show that the association rate constants for the model systems are roughly two orders of magnitude smaller than those in the case of the diffusion-controlled limit. Because the complex stability is determined by the dissociation rate constant, a two-step reaction mechanism, including the diffusion-controlled and reaction-controlled rates, is used to explain the dye–micelle interaction. In the limit of fast reaction, we apply FCS to determine the equilibrium constant from the effective diffusion coefficient of the fluorescent components. Depending on the value of the equilibrium constant, we distinguish three types of interaction in the studied systems: weak, intermediate and strong. The values of the equilibrium constant obtained from the FCS and TDA experiments are very close to each other, which supports the theoretical model used to interpret the FCS data.
Introduction
Determination of the equilibrium and rate constants of reagents that form noncovalent complexes is crucial for the understanding of various biochemical processes and chemical reactions,1–3 such as drug activities in vivo,4–6 formation of supermolecular structures and their dynamics,7–9etc. However, the main methods used to study the kinetics of noncovalent interactions, e.g. spectroscopy, NMR and titration experiments, sometimes give inconsistent results.6,10,11 For instance, the association rate constants between small molecules and cyclodextrins measured by Berezovski et al. are five orders of magnitude smaller than the results of Al-Soufi's group.6,9 The binding constants of DNA–doxorubicin interactions obtained by Garcia et al. are almost two orders of magnitude higher than those measured by Giustini.12,13 Therefore, further development of advanced techniques and theories to study quantitatively the formation of molecular complexes is still needed.
Fluorescence correlation spectroscopy (FCS) was first introduced by Magde and Elson in the early 1970s to determine the chemical kinetic constants of the interaction between DNA and ethidium bromide.14–16 Since the advent of confocal microscopy illumination, FCS experienced a renaissance in the 1990s.17–19 In FCS, we monitor the fluctuations of the fluorescence signals originating from molecules diffusing through the focal volume. The autocorrelation analysis of these fluctuations provides information on the diffusion coefficients of the molecules, their concentrations and structures,20 singlet–triplet dynamics, etc. Recent advances in theoretical studies make it possible to determine the equilibrium and rate constants of noncovalent interactions by fitting the experimental autocorrelation function of FCS to a theoretical model.21 For example, McNally et al. proposed a new model of the autocorrelation function for analyzing the association and dissociation rates of DNA binding in live cells.1 Al-Soufi et al. introduced a general correlation function to investigate the supermolecular dynamics9,22 and dye–micelle exchanging dynamics.23,24 However, the values of the equilibrium and rate constants obtained from these models usually disagree with the results of other works even by a few orders of magnitude. These huge discrepancies may come from unproven assumptions in the theoretical models or too many free parameters in the fitting procedures. In general, the existing models of the autocorrelation function focus on fast reactions,9,22–25 which may be the reason that the rate constants obtained from FCS are inconsistent with those obtained from other techniques.6,8,13,26 Therefore, an analytical theory for the FCS autocorrelation function is demanded for probing the interaction dynamics of diffusants in soft matter and biology.27
In this work, we study the interactions between fluorescent dyes and surfactant micelles by FCS, in particular, the kinetics of the dye–micelle complex formation. We use three surfactants with different charges: C12E8 (neutral), SDS (negative) and CTAC (positive), which have been extensively studied previously.28–32 Their physicochemical properties regarding the micelle formation and diffusion are well-established. As fluorescent probes we choose two zwitterionic dyes: rhodamine 110 and ATTO-488 (see Fig. 1). Their diffusional properties in the three surfactant solutions of various concentrations are obtained by FCS. To determine the rate of the dye–micelle complex formation, we apply an approximate form of the autocorrelation function, which is derived in the accompanying theoretical paper33 on the basis of Magde's theory.14–16 A sketch of the deviation is also presented in the ESI†. This new formula works for both slow and fast reactions and can be useful in the studies of chemical kinetics of noncovalent reactions, which we demonstrate here. In the case of a slow reaction, equilibrium is established outside the focal volume since the characteristic time of the reaction is much longer than the typical time of diffusion through the focal volume. Therefore, we observe two diffusing species with different diffusion times: the fluorescent reactant and the complex. In contrast, the characteristic time of a fast reaction is much shorter than the diffusion time through the focal volume and only a single effective diffusion is observed. For fast reactions, our autocorrelation function reduces to the known expression for single-component effective diffusion. Since the effective diffusion coefficient is related to the equilibrium constant of the dye–micelle interaction, we can use this relation to determine the latter by FCS. To test this method, we also determine the equilibrium constant by Taylor dispersion analysis (TDA).34–36 Then, we use FCS to investigate the kinetics of complex formation, i.e., the association and dissociation rates. It is feasible if the chemical relaxation rate (definition in the Materials and methods section) is reduced below the limit of fast reaction, which can be usually achieved in the case of diluted micellar solutions because the relaxation rate depends linearly on the concentration of micelles. To determine the relaxation rate, we apply our approximate model of the autocorrelation function with a small number of fitting parameters. Therefore, to reduce this number, we first determine the equilibrium diffusion coefficient of surfactant micelles in water by an independent technique, i.e., dynamic light scattering (DLS). The diffusion coefficients of the dyes are also known from the literature. Then, the relaxation rate, together with two parameters of the triplet-state correction, are the only free parameters in the autocorrelation function to be fitted. Finally, the association and dissociation rate constants are evaluated from the dependence of the relaxation rate on the micellar concentration.
 |
| | Fig. 1 (A–E) The chemical structures of rhodamine 110, ATTO-488, C12E8, SDS and CTAC. (F) FCS setup. The light from an argon-ion laser (488 nm) passes through a water immersion microscope objective to excite the fluorescent dye in the micellar solutions. The emitted fluorescence signals are collected by the detector and further fed to a TCSPC unit. Kinetic information such as the equilibrium and rate constants for the dye–micelle systems can be derived from our approximate autocorrelation function. | |
Materials and methods
Materials
The cationic surfactant CTAC (purity: 99%) and anionic surfactant SDS (purity: 99.9%) were purchased from TCI and Roth, respectively. The non-ionic surfactant C12E8 (purity: 99%) was purchased from Fluka. Fluorescent dyes rhodamine 110 and ATTO-488 were purchased from Sigma-Aldrich and ATTO-TEC GmbH, respectively. All chemicals were used without further purification.
Formation of dye–micelle complexes
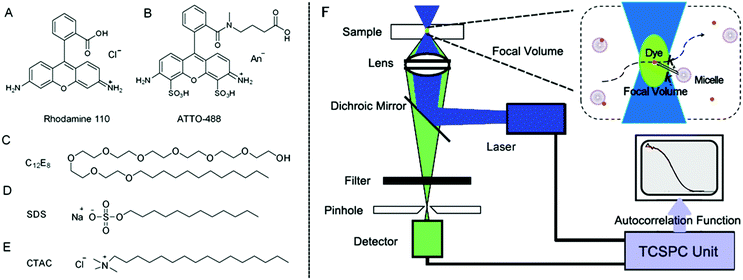
Surfactant molecules in an aqueous solution aggregate to form micelles denoted as A, when their concentration is above the critical micelle concentration (CMC). Above the CMC, the concentration of micelles [A] can be determined from the surfactant concentration [S] as follows:| | 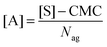 | (1) |
where Nag is the mean number of aggregated surfactant molecules. In dilute solution Nag is constant since the shape and size of micelles are fixed. The values of Nag and CMC for all surfactants used in this work are listed in Table 1.
Table 1 Diffusion coefficients, hydrodynamic radii, electric charges, critical micelle concentrations and the number of aggregated surfactant molecules of the studied samples. All values are obtained from the references or DLS measurements at 25 °C
| |
D (×10−10 m2 s−1) |
R
h (nm) |
Charge |
CMC (mM) |
N
ag
|
|
Values obtained from our DLS measurements at 25 °C.
|
| Rh110 |
(4.7 ± 0.4)37 |
(0.52 ± 0.05) |
Zwitterionic |
|
|
| ATTO-488 |
(4.0 ± 0.1)38 |
(0.62 ± 0.02) |
Zwitterionic |
|
|
| C12E8 |
(0.34 ± 0.01)a |
(7.2 ± 0.02)a |
Neutral |
0.08230 |
9529 |
| SDS |
(0.92 ± 0.02)a |
(2.7 ± 0.06)a |
Negative |
8.231 |
6031 |
| CTAC |
(0.80 ± 0.01)a |
(3.1 ± 0.04)a |
Positive |
1.132 |
8032 |
When dye molecules diffuse in a micellar solution the noncovalent bonding between a dye and micelle is treated as a chemical reaction with the equilibrium constant K:
| | 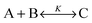 | (2) |
where B and C denote the dyes and dye–micelle complexes, respectively. The equilibrium constant is related to the association,
k+, and dissociation,
k−, rate constants and to the equilibrium concentrations of the components, [A]
eq, [B]
eq, and [C]
eq, as follows:
| | 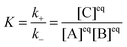 | (3) |
The relaxation rate
R of the pseudo-first-order reaction, which describe the rate of a reaction's return to equilibrium, is defined as
15| | | R = k+([A]eq + [B]eq) + k− | (4) |
Because the micelle concentration is always much higher than the dye concentration in our FCS experiments,
i.e., [A]
eq ≫ [B]
eq ≈ 10
−9 M, we use the approximation:
Thus, we can control the relaxation rate to some extent by changing the micelle concentration. Combining
eqn (4) and (5), we get
FCS: experiment and theory
The FCS setup used in our experiments was a commercial inverted NIKON EZ-C1 confocal microscope. The focal setup was additionally equipped with a PicoHarp 300 FCS setup made by PicoQuant. The experiments were conducted at 25 °C using a 488 nm argon-ion laser for illumination. A water immersion objective with a numerical aperture of 1.2 and magnification of 60 was used in FCS measurements. Before each measurement a drop of filtered, de-ionized water was used as the immersion medium between the objective and sample container (an 8-chambered coverglass, Lab-Tek®). During measurements the laser power was set at a constant level and the focal volume was at a constant distance of 10 μm from the edge of the coverglass. An avalanche photo diode was used for detection. All surfactant solutions were prepared with a probe concentration of ∼10−9 M. 200 μl of the solution was transported into the sample container and analyzed by FCS. Each measurement (duration 60 s) was repeated at least ten times and the autocorrelation function curves were further analyzed by the SymPhoTime program and Gnuplot version 4.5.
In FCS experiments, the distribution of the laser light intensity, I, in the focal volume is often approximated by a three-dimensional Gaussian: I(x, y, z) = I0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−2(x2 + y2)/L2 − 2z2/H2], where L is the transverse radius of the focal volume and H is its height. Fluctuations of the fluorescence intensity, δF(t), are analyzed by means of the autocorrelation function: G(τ) = 〈δF(t)δF(t + τ)〉/〈F(t)〉2, where δF(t) = F(t) − 〈F(t)〉 and “〈〉” denotes the average over time t. In the case of a three-dimensional isotropic single-component diffusion with the triplet-state correction, G(τ) is given by39
exp[−2(x2 + y2)/L2 − 2z2/H2], where L is the transverse radius of the focal volume and H is its height. Fluctuations of the fluorescence intensity, δF(t), are analyzed by means of the autocorrelation function: G(τ) = 〈δF(t)δF(t + τ)〉/〈F(t)〉2, where δF(t) = F(t) − 〈F(t)〉 and “〈〉” denotes the average over time t. In the case of a three-dimensional isotropic single-component diffusion with the triplet-state correction, G(τ) is given by39
| | 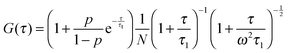 | (7) |
where
p is the fraction of dye molecules in the triplet state,
τt is the triplet lifetime,
N is the average number of molecules in the focal volume, and
ω =
H/
L. The characteristic diffusion time through the focal volume is defined as
τ1 =
L2/4
D, where
D denotes the diffusion coefficient. The transverse radius
L was obtained from the calibration measurement before each experiment. The free diffusion of rhodamine 110 (standard sample) in water was measured at 25 °C for calibration and we obtained the value
DRh110 = 4.7 × 10
−10 m
2 s
−1 for the diffusion coefficient.
37 The typical diffusion time through the focal volume,
τRh110, is around 20 μs. The calculated value of
L is around 0.2 μm and the value of
ω is about 5 in our FCS setup.
Approximate form of the autocorrelation function
Due to the reaction of dyes with micelles there are three diffusional components in the system: micelles (A), dyes (B) and dye–micelle complexes (C), whose diffusion coefficients are denoted as DA, DB and DC, respectively. To investigate the kinetics of the dye–micelle interaction, which is characterized by the equilibrium constant K and relaxation rate R, we use an approximate form of the FCS autocorrelation function, Ga(t), derived in the framework of Magde's theory.14–16 The full expression for Ga(t) is presented in the accompanying paper,33 but here we use a slightly simplified form (see formula (8)). The derivation of Ga(t) is based on the following two assumptions. (1) The diffusion coefficient of the micelle is constant and much smaller than that of the dye, hence the diffusion coefficient of the dye–micelle complex can be approximated by DC = DA ≪ DB. (2) The quantum yield of the dye fluorescence, QB, does not change after binding to the micelle, i.e., QB = QC = Q, which has been confirmed by the quantum yield measurements (ESI†). Taking into account the intrinsic triplet states of fluorescent dyes, we obtain the following approximate formula for the autocorrelation function:| | 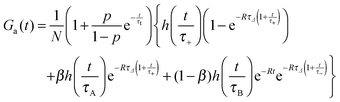 | (8) |
where τA = L2/4DA, τB = L2/4DB, τ± = L2/4D±, τΔ = L2/4|Δ|, and Δ = DA − DB < 0. The parameter β = k+[A]eq/R = K[A]eq/(1 + K[A]eq) and D+ = DAβ + DB(1 − β), D− = DA(1 − β) + DBβ are the effective diffusion coefficients. The function h(![[t with combining macron]](https://www.rsc.org/images/entities/i_char_0074_0304.gif) ) = (1 + )−1(1 + /ω2)−1/2 is the normalized autocorrelation function for the diffusion of one component and denotes the dimensionless lag time. The parameters characterizing the focal volume: H, L and ω are known from the calibration measurements of reference fluorescent dyes with known diffusion coefficient DB. DA is the diffusion coefficient of micelles determined by DLS. The equilibrium constant can be determined by an independent technique, e.g., TDA.34–36 The rest of the parameters (τA, τB, τΔ, τ± and β) whose values depend on the structure of the focal volume and the micellar concentration in formula (8) can be exactly calculated and then fixed during the fitting procedure. The target quantity R is the only free model parameter in Ga(t) to fit, besides the triplet-state parameters p and τt. In the case of highly concentrated micellar solutions, in which the dye–micelle association is extremely fast, the limit of fast reaction (R → ∞) can be applied in formula (8). Then, Ga(t) reduces to the known formula for the single-component diffusion (cf.eqn (7)):
) = (1 + )−1(1 + /ω2)−1/2 is the normalized autocorrelation function for the diffusion of one component and denotes the dimensionless lag time. The parameters characterizing the focal volume: H, L and ω are known from the calibration measurements of reference fluorescent dyes with known diffusion coefficient DB. DA is the diffusion coefficient of micelles determined by DLS. The equilibrium constant can be determined by an independent technique, e.g., TDA.34–36 The rest of the parameters (τA, τB, τΔ, τ± and β) whose values depend on the structure of the focal volume and the micellar concentration in formula (8) can be exactly calculated and then fixed during the fitting procedure. The target quantity R is the only free model parameter in Ga(t) to fit, besides the triplet-state parameters p and τt. In the case of highly concentrated micellar solutions, in which the dye–micelle association is extremely fast, the limit of fast reaction (R → ∞) can be applied in formula (8). Then, Ga(t) reduces to the known formula for the single-component diffusion (cf.eqn (7)):| | 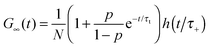 | (9) |
In the limit of fast diffusion (R = 0), formula (8) yields| | 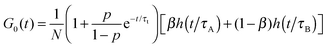 | (10) |
which is the autocorrelation function for the two-component model of diffusion.39
Results and discussion
A dye molecule in a micellar solution can be either in the free-motion state or in the bound-motion state, where the latter is due to the molecular interaction with micelles.23,24,40 In our experimental studies, we record the diffusion of rhodamine 110 and ATTO-488 in different surfactant solutions of various concentrations by FCS. In the case of rhodamine 110 in the CTAC solution, we observe a gradual shift of the autocorrelation curves towards longer diffusion times when the surfactant concentration increases (see Fig. 2). However, the viscosity of a very diluted solution should not change noticeably with the surfactant concentration. Thus, the increase in the diffusion time of rhodamine 110 implies that the free diffusion of the dye is gradually hindered due to the formation of dye–micelle complexes. The more dye–micelle complexes are formed in the solution, the longer the diffusion time observed. Such a hindered diffusion was also reported in the work of Zettl et al.,40 in which they focused on the investigation of micelle formation. They analyzed their experimental autocorrelation curves by a two-component model for FCS with the assumption that the relaxation rate of the dye–micelle reaction was much longer than the typical diffusion time through the focal volume. However, without a proper analysis of the dynamics of the reaction that occur in the system, the two-component model may give meaningless values of the diffusion time and fraction of the second component, i.e., micelles. As a result, the size of CTAC micelles probed by different tracers was not equal if the curves were fitted by the two-component model.40 The two-component model could only be applied to bound and freely moving tracers, but not for the analysis of dynamics of molecular interaction.
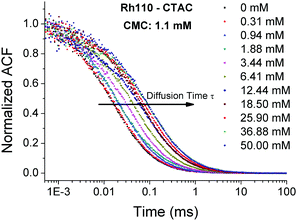 |
| | Fig. 2 Normalized experimental autocorrelation function curves of rhodamine 110 diffusing in solutions of CTAC with various surfactant concentrations. The diffusion times of rhodamine 110 shift gradually to the right as a function of the CTAC concentration. | |
Analysis of FCS data using Ga(t)
The autocorrelation function, given by formula (8), is suitable to study the kinetics of intermediate interaction (i.e., Rh110–CTAC system) whose relaxation rate is smaller than the diffusion time through the focal volume. In the Rh110–CTAC system, the size and concentration of rhodamine 110 (r ≈ 0.5 nm, at 5 nM concentration) are too small to be observed by DLS, compared to the much bigger micelles at much higher concentrations (R ≈ 3 nm, in the μM concentration range). Therefore, we only measured the collective diffusion coefficient (Dc) of the CTAC micelles in aqueous solution as a function of the micellar concentration at 25 °C. DC measured in DLS reduces to D0 (self-diffusion coefficient) when extrapolated to zero concentration. The extremely low concentration of the studied dye–micelle complexes in FCS experiments (in the nM range) can be approximately assumed to be equal to the zero concentration in DLS experiments. Hence, extrapolating the micelle concentration to zero concentration in DLS, we obtain the D0 of the CTAC micelles with a value of 0.80 × 10−10 m2 s−1 (for details see Fig. S2 in the ESI†), which is consistent with the published results.32 We used the D0 for all the micelles in formula (8), because we only observe the diffusion of individual dye–micelle complexes in FCS and D0 is concentration-independent.41 Our previous small angle neutron scattering (SANS) experiments and literature data also have showed that the size and structure of micelles do not change in the dilute region.42,43
The equilibrium constant of the Rh110–CTAC interaction has been determined by the TDA method. The principle of TDA is based on the difference between the diffusion coefficients of rhodamine 110 in water and in the CTAC micellar solution as a function of the CTAC micellar concentration. The obtained value of the equilibrium constant for the Rh110–CTAC system is 4.28 × 104 M−1 (see the ESI† for details).
Having determined the equilibrium constant K by TDA and the diffusion coefficient of CTAC micelles by DLS, we use them to calculate the values of parameters τΔ, τ± and β that appear in Ga(t) (see formula (8) and Table 2). The parameters of triplet states, p and τt, will be discussed later. Fig. 3 (upper panel) shows that the FCS autocorrelation curves for rhodamine 110 in the micellar solution of CTAC are well fitted by Ga(t). The values of R obtained from this fitting are presented in Fig. 3 (lower panel) as a function of the CTAC micellar concentration. Then, we use eqn (6) to determine the association rate k+. The K in eqn (6) is known from TDA measurements and fixed at 4.28 × 104 M−1 during the fitting process. The fitted value of k+ for the Rh110–CTAC complex formation amounts to 2.46 × 108 M−1 s−1, which is one order of magnitude more than that in the case of the DNA–EtBr interaction studied by Magde et al.15 The value of dissociation rate k− calculated according to eqn (3) amounts to 5.75 × 103 s−1.
Table 2 Calculated values of the parameters appearing in formula (8) for the CTAC–Rh110 system, based on the results obtained from DLS and TDA measurements. See the Materials and method section and ESI for more details
| Conc. (μM) |
β
|
τ
+ (μs) |
D
+ (×10−10 m2 s−1) |
τ
− (μs) |
D
− (×10−10 m2 s−1) |
τ
Δ
(μs) |
D
Δ
(×10−10 m2 s−1) |
| 1.88 |
0.07 |
17.3 |
4.3 |
70.1 |
1.1 |
19.6 |
3.8 |
| 11.6 |
0.33 |
22.4 |
3.4 |
36.4 |
2.1 |
19.6 |
3.8 |
| 25.3 |
0.52 |
28.6 |
2.6 |
27.0 |
2.8 |
19.6 |
3.8 |
| 40.9 |
0.64 |
34.4 |
2.2 |
23.2 |
3.2 |
19.6 |
3.8 |
| 60.5 |
0.72 |
40.4 |
1.9 |
21.1 |
3.6 |
19.6 |
3.8 |
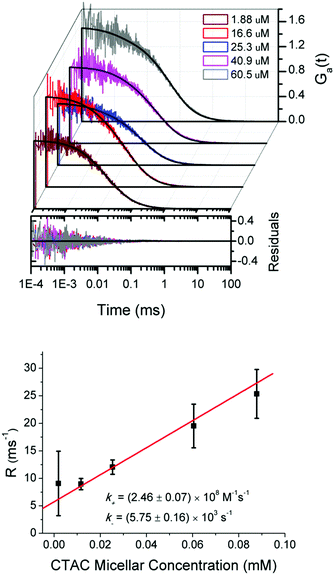 |
| | Fig. 3 Upper panel: Experimental autocorrelation curves for rhodamine 110 diffusing in diluted CTAC micellar solutions and the fitting curves of formula (8). The plotted residuals correspond to the fits of experimental data using formula (8). Lower panel: The fitted values of R plotted versus the CTAC micellar concentration according to eqn (6) with K known from TDA measurements. The association rate constant k+ is obtained from the slope of the red line, and then the dissociation rate k− is calculated according to eqn (3). | |
Determination of the equilibrium constant by FCS
Since R increases with the micelle concentration we expect that at sufficiently high concentrations the limit R → ∞ can be taken in formula (8), i.e., G∞(t) given by eqn (9) can be used to fit the FCS data. Then, we determine the equilibrium constant from the dependence of the effective diffusion coefficient D+ on the concentration of micelles according to the definition of D+ and β (in the Materials and method section):| | 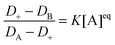 | (11) |
where D+ is calculated from the relation D+ = L2/4τ+,44 and τ+ is the effective diffusion time of dyes and dye–micelle complexes through the focal volume obtained from the fitting of experimental curves by G∞(t).
To check this possibility, we study rhodamine 110 in a series of CTAC micellar solutions at relatively high concentrations of micelles. Fig. 4 shows that the experimental curves are well fitted by G∞(t). Then, by substituting D+ obtained from FCS and the diffusion coefficient of micelles, DA, from DLS into eqn (11), we obtain the equilibrium constant K from the slope of the linear fit shown in Fig. 4. For the Rh110–CTAC system the equilibrium constant determined using eqn (9) is 4.32 × 104 M−1, which agrees with the value of 4.28 × 104 M−1 obtained from TDA measurements.
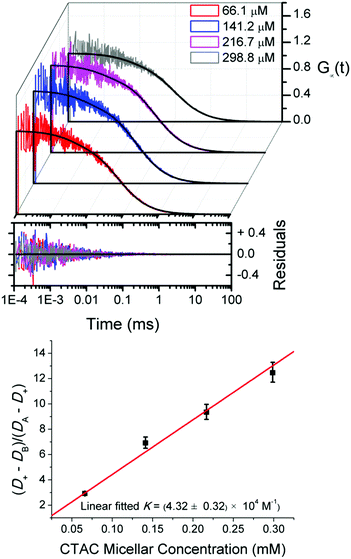 |
| | Fig. 4 Upper panel: Experimental FCS autocorrelation curves for rhodamine 110 in CTAC solutions at relatively high concentrations of micelles fitted by eqn (9). Lower panel: The equilibrium constant K of the Rh110–CTAC interaction obtained from the linear fit by eqn (11). | |
Weak and strong interactions
We have also used FCS to study the diffusion and molecular interactions of ATTO-488 in C12E8 and SDS solutions at surfactant concentrations both below and above the CMC. We have not observed any noticeable changes in the diffusion time of ATTO-488 (see Fig. 5(A) and (B)). The values of the effective diffusion time obtained from the fit of experimental data by G∞(t) are roughly the same as in pure water. This means that the molecular interactions in diluted surfactant solutions are too weak to affect the free diffusion of ATTO-488. Our previous work on the mobility of fluorescent dye TAMRA in low concentration solutions of hexaethylene glycol monododecyl ether (C12E6) is consistent with the present results.45 The diffusion coefficient of TAMRA also does not change in the C12E6 solution within the same range of surfactant concentrations, thus, the viscosity of the solution can be considered the same as that of water.
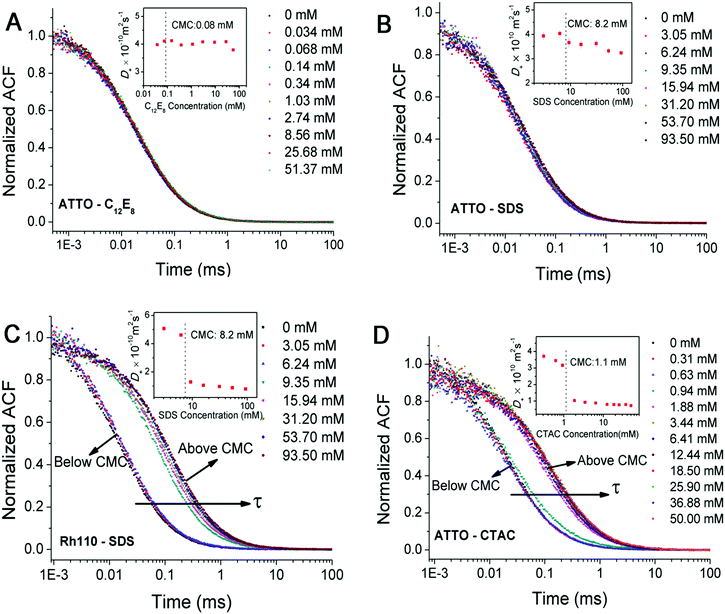 |
| | Fig. 5 (A–D) Normalized experimental autocorrelation curves for ATTO-488 and rhodamine 110 in the solutions of C12E8, SDS and CTAC of various surfactant concentrations, respectively. Inset: The effective diffusion coefficients (Deff) obtained from the fit of experimental curves by eqn (9). The overlapping sets of data points in (A) and (B) indicate that the diffusion time of ATTO-488 in the C12E8 and SDS solutions is independent of the surfactant concentration. In (C) and (D), a sudden increase in the diffusion time is observed in the Rh110–SDS and ATTO–CTAC systems at a concentration just above the CMC. The dyes are believed to bind to large micelles immediately once they encounter. | |
In contrast, the effective diffusion times for ATTO-488 in the CTAC solution and rhodamine 110 in the SDS solution experience a sudden increase when the surfactant concentration is just above the CMC (see Fig. 5(C) and (D)). The dyes are supposed to bind to micelles immediately and then move together in the solution all the time. Dosche's and Ghosh's groups also observed such an attachment of hydrophobic dyes to various surfactant micelles by using FCS.46,47
The formation of complexes between dyes and micelles depends mainly on hydrophobic effects and electrostatic forces. A higher affinity of the zwitterionic dye ATTO-488 to cationic CTAC micelles compared to anionic SDS and neutral C12E8 micelles can be attributed to the strong hydrophobic effect of the CTAC micelles which possess four additional alkyl groups in their chemical structure compared to SDS and C12E8 do. Rhodamine 110 can interact with SDS, CTAC and C12E8 micelles but its affinities decline due to the differences in molecular charges.
Kinetics of dye–micelle interactions
We categorize the dye–micelle interactions as: weak, intermediate or strong, according to the dependence of D+ on the surfactant concentration. For weak interactions, i.e., in the case of ATTO–C12E8 and ATTO–SDS systems, D+ changes marginally as a function of the surfactant concentration, which indicates the free diffusion of ATTO-488 in these solutions (inseted plot in Fig. 5(A) and (B)). For strong interactions, namely in Rh110–SDS and ATTO–CTAC systems, D+ drops sharply when the surfactant concentration exceeds the CMC (inset plots in Fig. 5(C) and (D)), which suggests that almost all dye molecules bind strongly to the micelles. For intermediate interactions, i.e., in the case of the Rh110–CTAC system, D+ decreases moderately when the surfactant concentration increases. In the absence of analytical theory, these three concentration-related behaviours of D+ in FCS were also reported by molecular dynamics simulation of a generic bead–spring polymer model and spherical tracers with no attraction (single diffusion), weak attraction (single slow diffusion), and strong attraction (double diffusion) to the polymer in the work of Vagias's group.27
Since FCS has showed a good accuracy in evaluating the equilibrium constant of the Rh110–CTAC system at relatively high concentrations of micelles, we use this method for other dye–micelle systems as well. As in the case of CTAC, the diffusion coefficients of SDS and C12E8 micelles are determined by DLS at 25 °C and they amount to 0.92 × 10−10 m2 s−1 and 0.34 ×10−10 m2 s−1, respectively (see Fig. S3 and S4 in the ESI† for details). These values are consistent with the published results.32,48 In Table 3, we show the values of K for the studied dye–micelle systems. In the case of strong interactions (Rh110–SDS and ATTO–CTAC systems), K is roughly one order of magnitude larger than that in the case of intermediate interactions. The reported value of K for the interaction between rhodamine 123 and C12E7 is (1.6 ± 0.2) × 104 M−1,25 close to the value for the Rh110–C12E8 system studied in this work.
Table 3 Equilibrium constants K, the association k+ and dissociation k− rate constants, and the association rate constants kdc for the diffusion-controlled reaction estimated using the Smoluchowski equation for the studied dye–micelle systems. K is fitted by eqn (11) and k± are obtained from formula (8) and eqn (6)
| |
K (×104 M−1) |
−ΔG (kJ mol−1) |
k
+ (×108 M−1 s−1) |
k
− (×103 s−1) |
k
dc (×1010 M−1 s−1) |
| Rh110–C12E8 |
2.32 ± 0.21 |
24.90 |
12.16 ± 6.07 |
52.4 ± 26.2 |
2.9 |
| Rh110–CTAC |
4.32 ± 0.32 |
26.44 |
2.14 ± 0.01 |
6.58 ± 0.06 |
1.50 |
| Rh110–SDS |
15.8 ± 0.09 |
29.66 |
0.51 ± 0.08 |
0.32 ± 0.05 |
1.36 |
| ATTO–CTAC |
46.7 ± 7.8 |
32.34 |
0.74 ± 0.01 |
0.16 ± 0.01 |
1.35 |
Next, we use the equilibrium constant obtained by FCS at high concentrations of micelles to determine the relaxation rates of the same dye–micelle system, which is achieved by fitting the FCS autocorrelation function in the low concentration regime. In other words, we apply the same procedure as in the case of the aforementioned Rh110–CTAC system. The fitted values of k+ and k− for intermediate and strong interactions are collected in Table 3 together with the diffusion-controlled association rate constant kdc. The reported values of the association rate constant for the dye–micelle interactions vary from 106 M−1 s−1 to the diffusion controlled limit, i.e., 1010 M−1 s−1.23,24,26 The values of k+ in our dye–micelle systems are roughly two orders of magnitude smaller than kdc (see Table 3). The latter can be estimated from the Smoluchowski equation:
where
Ddm and
Rdm are the sums of the diffusion coefficients and the hydrodynamic radii of the dyes and micelles, respectively, and
NA is Avogadro's constant.
We describe the dye–micelle reaction as a two-step process: the intermediate formation and complex formation
| | 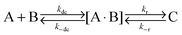 | (13) |
In the first step, one micelle and one dye molecule get into contact to form an intermediate [A·B] driven by free diffusion under the diffusion-controlled rate
kdc. Next, the dye–micelle complex is formed through hydrophobic and electrostatic interactions with a reaction rate
kr. The value of
kr can be simply estimated
via49| | 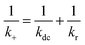 | (14) |
The calculated value of
kr is roughly equal to
k+ in our dye–micelle systems, indicating that complex formation is a reaction-controlled process.
The micelles behave as “soft cages” which hinder the entrance and exit of dyes when they collide driven by Brownian motion. The activation energies, labelled as ΔG+ and ΔG−, for the studied dye–micelle interactions are shown in Fig. 6. In thermodynamics we can only determine the Gibbs free energy difference but not the absolute values. The free energy differences (ΔG) between the bound and unbound states for various dye–micelle systems are calculated on the basis of the expression: ΔG = −RTlnK, and the values of the rate constants are shown in Table 3. The energy barriers of intermediate interactions are much lower than those of strong interactions. The stability of the dye–micelle complex is determined by its residence time expressed as τR = 1/k−.50 The values of the dissociation rate constant for strong interactions are at least one order of magnitude smaller than those for the intermediate interactions, suggesting longer times of the bound state.
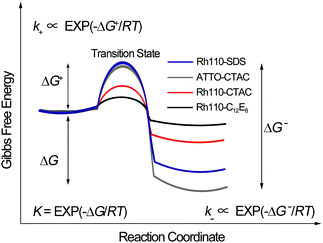 |
| | Fig. 6 A simple scheme of the changes in Gibbs free energy of the studied dye–micelle systems due to complex formation. Here, R is the gas constant and T denotes temperature. The equilibrium constant K depends on the free-energy difference (ΔG) of the bound and unbound states. The rate constants (k+, k−) depend on the free-energy differences (ΔG+, ΔG−) between the two states and the transition state. The arbitrary setting of the same initial state in Fig. 6 is to simplify the illustration of ΔG. | |
Triplet-state kinetics
The fraction of dye molecules in the triplet state, p, and the triplet-state lifetime, τt, are also fitted by formulas (8) and (9). Table 4 shows the fitted values of p and τt, which are close to the published results for rhodamine 110 (1.9 μs, 10%) and ATTO-488 (2.9 μs, 10%) in aqueous media, obtained by the method of total internal reflection fluorescence correlation spectroscopy (TIR-FCS).51
Table 4 Values of the triplet state parameters: the triplet-state lifetime τt and the fraction of dyes in the triplet state p fitted by (8) and (9), respectively
| |
τ
t
(μs) |
p
(%) |
τ
t
(μs) |
p
(%) |
|
Fitted by equation eqn (9).
Fitted by formula (8).
|
| Rh110–C12E8 |
2.7 ± 1.3 |
13.6 ± 4.0 |
3.3 ± 0.3 |
14.5 ± 2.1 |
| Rh110–CTAC |
3.9 ± 1.0 |
13.8 ± 3.9 |
2.3 ± 0.6 |
7.7 ± 0.8 |
| Rh110–SDS |
3.4 ± 1.6 |
9.7 ± 2.9 |
2.0 ± 0.6 |
4.0 ± 0.5 |
| ATTO–CTAC |
3.9 ± 1.7 |
13.6 ± 3.5 |
5.9 ± 1.4 |
8.3 ± 0.8 |
Conclusions
In this work, we use FCS to study the reaction kinetics of selected dye–micelle systems. The interactions between dyes and micelles are categorized into three types: weak, intermediate and strong, in terms of the equilibrium constant K. To fit the FCS experimental data, we apply a new approximate form of the FCS autocorrelation function, Ga(t), derived on the basis of Magde's formalism. Our results show that Ga(t) can be used to determine the association and dissociation rate constants by studying diluted micellar solutions, in which the relaxation rate can be reduced below the fast-reaction limit. Then, the rate constants follow from the dependence of the relaxation rate on the micelle concentration. On the other hand, FCS studies in the region of high micelle concentrations, in which the fast-reaction limit usually applies, provide information about K for the dye–micelle system. The values of K obtained by FCS are consistent with the results from an independent technique, i.e., TDA and with the data obtained from references. Our results show that the association rates of the dye–micelle interactions are much smaller than the diffusion-controlled rate. Therefore, a two-step reaction mechanism is used to explain the complex formation between dyes and micelles. Generally, our method can potentially facilitate kinetic studies of molecular interactions involving drugs, proteins and DNA.
Acknowledgements
This work was supported by a grant from the National Science Centre granted on the basis of decision number UMO-2012/07/B/ST4/01400 (Opus 4). This work was done using equipment from the NanoFun laboratories founded by POIG.02.02.00-00-025/09. We thank Dr Sylwester Gawinkowski and Maria Pszona for the measurements of quantum yields.
Notes and references
- A. Michelman-Ribeiro, D. Mazza, T. Rosales, T. J. Stasevich, H. Boukari, V. Rishi, C. Vinson, J. R. Knutson and J. G. McNally, Biophys. J., 2009, 97, 337–346 CrossRef CAS PubMed.
- R. N. Day and F. Schaufele, Mol. Endocrinol., 2005, 19, 1675–1686 CrossRef CAS PubMed.
- C. C. Govern, M. K. Paczosa, A. K. Chakraborty and E. S. Husebyb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8724–8729 CrossRef CAS PubMed.
- L. M. Berezhkovskiy, Expert Opin. Drug Metab. Toxicol., 2008, 4, 1479–1498 CrossRef CAS PubMed.
- M. Goyal, M. Rizzo, F. Schumacher and C. F. Wong, J. Med. Chem., 2009, 52, 5582–5585 CrossRef CAS PubMed.
- G. G. Mironov, V. Okhonin, S. I. Gorelsky and M. V. Berezovski, Anal. Chem., 2011, 83, 2364–2370 CrossRef CAS PubMed.
- A. S. M. Dyck, U. Kisiel and C. Bohne, J. Phys. Chem. B, 2003, 107, 11652–11659 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- W. Al-Soufi, B. Reija, M. Novo, S. Felekyan, R. Kuhnemuth and C. A. M. Seidel, J. Am. Chem. Soc., 2005, 127, 8775–8784 CrossRef CAS PubMed.
- W. Bujalowski, Chem. Rev., 2006, 106, 556–606 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- C. Perez-Arnaiz, N. Busto, J. M. Leal and B. Garcia, J. Phys. Chem. B, 2014, 118, 1288–1295 CrossRef CAS PubMed.
- M. Airoldi, G. Barone, G. Gennaro, A. M. Giuliani and M. Giustini, Biochemistry, 2014, 53, 2197–2207 CrossRef CAS PubMed.
- E. L. Elson and D. Magde, Biopolymers, 1974, 13, 1–27 CrossRef CAS.
- D. Magde, E. L. Elson and W. W. Webb, Biopolymers, 1974, 13, 29–61 CrossRef CAS PubMed.
- D. Magde, W. W. Webb and E. Elson, Phys. Rev. Lett., 1972, 29, 705–708 CrossRef CAS.
- R. Rigler, U. Mets, J. Widengren and P. Kask, Eur. Biophys. J., 1993, 22, 169–175 CrossRef CAS.
- H. Qian and E. L. Elson, Appl. Opt., 1991, 30, 1185–1195 CrossRef CAS PubMed.
- M. Eigen and R. Rigler, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5740–5747 CrossRef CAS.
- X. Z. Zhang, A. Poniewierski, S. Hou, K. Sozanski, A. Wisniewska, S. A. Wieczorek, T. Kalwarczyk, L. L. Sun and R. Holyst, Soft Matter, 2015, 11, 2512–2518 RSC.
- K. Koynov and H. J. Butt, Curr. Opin. Colloid Interface Sci., 2012, 17, 377–387 CrossRef CAS.
- D. Granadero, J. Bordello, M. J. Perez-Alvite, M. Novo and W. Al-Soufi, Int. J. Mol. Sci., 2010, 11, 173–188 CrossRef CAS PubMed.
- M. Novo, S. Felekyan, C. A. M. Seidel and W. Al-Soufi, J. Phys. Chem. B, 2007, 111, 3614–3624 CrossRef CAS PubMed.
- J. Bordello, M. Novo and W. Al-Soufi, J. Colloid Interface Sci., 2010, 345, 369–376 CrossRef CAS PubMed.
- S. Freire, J. Bordello, D. Granadero, W. Al-Soufi and M. Novo, Photochem. Photobiol. Sci., 2010, 9, 687–696 CAS.
- V. C. Reinsborough and J. F. Holzwarth, Can. J. Chem., 1986, 64, 955–959 CrossRef CAS.
- A. Vagias, R. Raccis, K. Koynov, U. Jonas, H. J. Butt, G. Fytas, P. Kosovan, O. Lenz and C. Holm, Phys. Rev. Lett., 2013, 111, 088301 CrossRef PubMed.
- E. Feitosa, W. Brown, K. Wang and P. C. A. Barreleiro, Macromolecules, 2002, 35, 201–207 CrossRef CAS.
- M. Zulauf, K. Weckstrom, J. B. Hayter, V. Degiorgio and M. Corti, J. Phys. Chem., 1985, 89, 3411–3417 CrossRef CAS.
- G. Olofsson, J. Phys. Chem., 1985, 89, 1473–1477 CrossRef CAS.
- E. Dutkiewicz and A. Jakubowska, Colloid Polym. Sci., 2002, 280, 1009–1014 CAS.
- L. T. Okano, F. H. Quina and O. A. El Seoud, Langmuir, 2000, 16, 3119–3123 CrossRef CAS.
- R. Hołyst, A. Poniewierski and X. Zhang, Soft Matter, 2016 Search PubMed , accompanying paper.
- A. Bielejewska, A. Bylina, K. Duszczyk, M. Fialkowski and R. Holyst, Anal. Chem., 2010, 82, 5463–5469 CrossRef CAS PubMed.
- A. Majcher, A. Lewandrowska, F. Herold, J. Stefanowicz, T. Slowinski, A. P. Mazurek, S. A. Wieczorek and R. Holyst, Anal. Chim. Acta, 2015, 855, 51–59 CrossRef CAS PubMed.
- A. Lewandrowska, A. Majcher, A. Ochab-Marcinek, M. Tabaka and R. Holyst, Anal. Chem., 2013, 85, 4051–4056 CrossRef CAS PubMed.
- P. O. Gendron, F. Avaltroni and K. J. Wilkinson, J. Fluoresc., 2008, 18, 1093–1101 CrossRef CAS PubMed.
- T. Dertinger, A. Loman, B. Ewers, C. B. Muller, B. Kramer and J. Enderlein, Opt. Express, 2008, 16, 14353–14368 CrossRef PubMed.
- O. Krichevsky and G. Bonnet, Rep. Prog. Phys., 2002, 65, 251–297 CrossRef CAS.
- H. Zettl, Y. Portnoy, M. Gottlieb and G. Krausch, J. Phys. Chem. B, 2005, 109, 13397–13401 CrossRef CAS PubMed.
- B. A. Scalettar, J. E. Hearst and M. P. Klein, Macromolecules, 1989, 22, 4550–4559 CrossRef CAS.
- J. Gapinski, J. Szymanski, A. Wilk, J. Kohlbrecher, A. Patkowski and R. Holyst, Langmuir, 2010, 26, 9304–9314 CrossRef CAS PubMed.
- H. U. Kim and K. H. Lim, Bull. Korean Chem. Soc., 2004, 25, 382–388 CrossRef CAS.
- R. Holyst, A. Bielejewska, J. Szymanski, A. Wilk, A. Patkowski, J. Gapinski, A. Zywocinski, T. Kalwarczyk, E. Kalwarczyk, M. Tabaka, N. Ziebacz and S. A. Wieczorek, Phys. Chem. Chem. Phys., 2009, 11, 9025–9032 RSC.
- J. Szymanski, A. Patkowski, A. Wilk, P. Garstecki and R. Holyst, J. Phys. Chem. B, 2006, 110, 25593–25597 CrossRef CAS PubMed.
- F. Luschtinetz and C. Dosche, J. Colloid Interface Sci., 2009, 338, 312–315 CrossRef CAS PubMed.
- S. Ghosh, U. Mandal, A. Adhikari and K. Bhattacharyya, Chem. – Asian J., 2009, 4, 948–954 CrossRef CAS PubMed.
- R. B. Dorshow, C. A. Bunton and D. F. Nicoli, J. Phys. Chem., 1983, 87, 1409–1416 CrossRef CAS.
- A. J. Elliot, D. R. Mccracken, G. V. Buxton and N. D. Wood, J. Chem. Soc., Faraday Trans., 1990, 86, 1539–1547 RSC.
- A. C. Pan, D. W. Borhani, R. O. Dror and D. E. Shaw, Drug Discovery Today, 2013, 18, 667–673 CrossRef CAS PubMed.
- H. Blom, A. Chmyrov, K. Hassler, L. M. Davis and J. Widengren, J. Phys. Chem. A, 2009, 113, 5554–5566 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm01791f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a