
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Relating side chain organization of PNIPAm with its conformation in aqueous methanol
Debashish
Mukherji
*a,
Manfred
Wagner
a,
Mark D.
Watson
ab,
Svenja
Winzen
a,
Tiago E.
de Oliveira
c,
Carlos M.
Marques
d and
Kurt
Kremer
*a
aMax-Planck Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: mukherji@mpip-mainz.mpg.de; kremer@mpip-mainz.mpg.de
bUniversity of Kentucky, Lexington, KY 40506-0055, USA
cUniversidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
dInstitut Charles Sadron, Université de Strasbourg, CNRS, Strasbourg, France
First published on 2nd September 2016
Abstract
Combining nuclear magnetic resonance (NMR), dynamic light scattering (DLS), and μs long all-atom simulations with two million particles, we establish a delicate correlation between increased side chain organization of PNIPAm and its collapse in aqueous methanol mixtures. We find that the preferential binding of methanol with PNIPAm side chains, bridging distal monomers along the polymer backbone, results in increased organization. Furthermore, methanol–PNIPAm preferential binding is dominated by hydrogen bonding. Our findings reveal that the collapse of PNIPAm is dominated by enthalpic interactions and that the standard poor solvent (entropic) effects play no major role.
1 Introduction
Polymer conformations in solvent mixtures often exhibit puzzling and paradoxical behavior. One such phenomenon is co-non-solvency that occurs when two competing (miscible) good solvents for a polymer are mixed together, as a result the same polymer collapses within intermediate solvent–cosolvent mixing ratios in bulk1–9 and near surfaces.10,11 One popular system that shows co-non-solvency is the conformational behavior of poly(N-isopropylacrylamide) (PNIPAm) in aqueous alcohol mixtures. Even when the phenomenon is usually associated with PNIPAm,1–4 the name co-non-solvency was first coined when polystyrene chains were dissolved in a mixture of cyclohexane and DMF solution.12 Interestingly, not only PNIPAm and polystyrene, rather a large number of polymers can exhibit the phenomenon of co-non-solvency.13–18 Furthermore, these systems present both LCST and UCST temperature effects. This suggests that the effect, unlike the common chemical believe, is not “only” restricted to polymers exhibiting LCST behavior and thus is independent of specific chemical details.The microscopic origin of the puzzling coil–globule–coil transition is a matter of intense debate. Here extensive experimental,1–5,9 theoretical6,8,19–21 and computer simulations4,7,8,22 studies have been performed. On the experimental side, studies have employed infrared spectra, light scattering and neutron scattering.1–5,9 In some cases, also in conjunction with analytical theory1,5 and parameter estimation for interaction strength.9 On the theory side, ever since the first theoretical work employing the Flory–Huggins theory,1 several analytical works have been proposed to explain co-non-solvency based on the cooperativity effect,6 particle based theory,8 Flory–Huggins lattice model at high polymer concentrations,20 and the off-lattice statistical model,21 to name a few. Furthermore, former simulations are mostly limited to a few studies.4,7,8 In this context, using a semi-grand canonical molecular dynamics approach, two of us have previously shown that PNIPAm has a significantly higher affinity (or preferential binding) towards methanol than towards water by a factor of ∼4kBT per monomer.7 This is further exemplified by potential of mean force (PMF) calculations, where a clear preferential interaction of methanol with PNIPAm was observed.23 This indicates that the polymer collapse in miscible good solvents, such as water and methanol for PNIPAm, is dictated by the relative (enthalpic) interaction strengths between methanol–PNIPAm and water–PNIPAm. Recently, it has been shown that the effect of co-non-solvency can also be observed in tertiary butyl alcohol in methanol–water mixtures, that is driven by enthalpic interactions.24 Furthermore, the enthalpically driven collapse of PNIPAm is against the common understanding of standard poor solvent collapse of LCST polymers, where solvent entropy gain plays a crucial role in polymer collapse. Furthermore, because a wide variety of polymers show the co-non-solvency phenomenon when dissolved in appropriate mixtures of solvents, a unified concept of co-non-solvency was proposed within a generic approach.8,19 Therefore, simple Lennard-Jones (LJ) interactions between monomer–(co)solvent are sufficient to explain the co-non-solvency effect at constant temperature8,19 and more effects,25,26 while ignoring all chemical details that often only contribute to a mere numerical prefactor. However, LJ representation of a (co)solvent bead may represent several methanol and/or water molecules.8
What makes the co-non-solvency of PNIPAm an interesting and puzzling effect is that concepts known from the conventional polymer science often are insufficient to describe the phenomenon. For example: (1) a polymer collapses when the solvent quality remains good or even gets increasingly better by the addition of better cosolvent,7,8 (2) standard poor solvent collapse or entropic effects are irrelevant, (3) the phenomenon is independent of any underlying LCST or UCST temperature behavior of polymers,13–19 and (4) is driven by large (local) concentration fluctuations of different solvent components near the polymer,19 making the mean-field description unsuitable. In this work, we revisit the phenomenon of co-non-solvency of PNIPAm in aqueous methanol mixtures by combining nuclear magnetic resonance (NMR), dynamic light scattering (DLS) and μs long, two million particles all-atom simulations. Note that we have used all-atom simulation instead of a semi-grand canonical setup.7 This is because in the case of a good solvent chain, the chain extension covers almost a full simulation domain consisting of ∼30 nm box boundary. To better correlate the different approaches, we match the polymer length Nl to be similar, in the unit of persistence length ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) p, which was chosen as Nl ∼ 100p. Our results provide experimental support for the claims presented in the four points mentioned above.
p, which was chosen as Nl ∼ 100p. Our results provide experimental support for the claims presented in the four points mentioned above.
The remainder of the paper is organized as follows: in Section 2 we briefly state the methodology for simulations, material synthesis and experimental measurements. Section 3 presents results and discussion. Finally we draw our conclusions in Section 4.
2 Materials, models and methods
2.1 Nuclear magnetic resonance measurements
The H-NMR experiments27,28 were measured with a 5 mm triple resonance TXI 1H/13C/15N probe equipped with a z-gradient on an 850 MHz Bruker AVANCE III system. For proton spectra, 128 transients were used with a 9.5 μs long 90° pulse and a 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 Hz spectral width together with a recycling delay of 5 s. The temperature was regulated at 298.3 K and calibrated with a standard 1H methanol NMR sample using the Topspin 3.1 software (Bruker). Temperature was controlled with a VTU (variable temperature unit) and an accuracy of ±0.1 K.
600 Hz spectral width together with a recycling delay of 5 s. The temperature was regulated at 298.3 K and calibrated with a standard 1H methanol NMR sample using the Topspin 3.1 software (Bruker). Temperature was controlled with a VTU (variable temperature unit) and an accuracy of ±0.1 K.
Diffusion Ordered NMR Spectroscopy (DOSY-NMR) experiments were performed with a gradient strength of 5.350 G mm−1 on a Bruker Avance-III 850 NMR Spectrometer. The gradient strength of probes was calibrated by using a sample of 2H2O/1H2O at a defined temperature and compared with the theoretical diffusion coefficient of 2H2O/1H2O (values taken from Bruker diffusion manual) at 298.3 K.
The diffusion delay time (Δ), Bruker term D2O was optimized for the TXI probe for 60 ms, while the gradient pulse length was kept at 1.6 ms. The optimization was realized by comparing the remaining intensity of the signals at 2% and 95% gradient strengths. The intensity loss of the echo was in the range of 90%. Using longer diffusion time causes a loss of signal intensity (from the echo) due to a short spin lattice relation time T1, which was determined with the inversion recovery method27 before diffusion measurements.
The diffusion measurements were done with a 2D DOSY sequence29 by incrementing in 16 linear steps from 2% to 100% with the TXI probe. The calculation of the diffusion value was automatically done with the mono exponential function:30
 | (1) |
The 2D sequence for diffusion measurement used double stimulated echo with three spoil gradients for convection compensation and with an eddy current delay of 5 ms for reduction31,32 (acronym Bruker pulse program: dstebpgp3s). The spin–spin relaxation times T2 were obtained via the CPMG method (Carr–Purcell–Meiboom–Gill)33,34 using eight different increments. In the experiment, the time between the inversion (180° pulse) and the read pulse (90° pulse) is incremented in eight steps and the quantitative analysis of the eight integrals (from the eight increments) exponentially fitted.
2.2 Size exclusion chromatography and light scattering
2.3 PNIPAm samples and synthesis
Three different PNIPAm samples were used for our experiments. These include a commercial PNIPAm from Sigma-Aldrich [cat # 535311-10G]. Using light scattering and size exclusion chromatography, we estimate absolute molecular weight Mw and poly dispersity (PDI) to be Mw (SLS) = 293 kDa, Mw (GPC) = 309 kDa, and PDI = 2.69. Nl value is estimated using the equation Nl = (Mw/PDI)/M0, where M0 is the NIPAm formula weight. This corresponds to Nl = 962 (sample PNIPAM-963). Furthermore, we have prepared two PNIPAm samples by RAFT polymerization giving chain lengths bracketing that corresponding to Nl ∼ 100p to better correlate all-atom simulations and experimental observations (details will be described in the next section).
Before starting the polymerization procedure, N-isopropyl acrylamide (NIPAm, Acros, 99%) was recrystallized three times in series by adding a benzene solution (∼30% NIPAm by weight) to an approximate 12-fold excess of pentane at room temperature. 4,4′-Azobis(4-cyanovaleric acid) (ACVA, Aldrich, ≥98%,), 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (BM1433, >95%, boron molecular) and all other materials were purchased from commercial sources and utilized as received.
:1:0.4 in a 25 mL Shlenk tube and pump/purged 3 times with vacuum/argon. DMF (anh) was added via syringe to give a 20% by weight NIPAm solution that was sparged with argon for 30 minutes while cooling in ice. The reaction was initiated by placing in a 60 °C bath, and aliquots were taken via syringe at time intervals to estimate conversion by NMR and molecular weight by GPC. After 10 hours (∼80% conversion by NMR), the solution was cooled in ice and the polymer precipitated 3 times by dripping into rapidly stirring ether (from MeOH for 2nd and 3rd cycle), and the resulting pale yellow solid dried under reduced pressure. 1H NMR (850 MHz, D2O) δ: 3.90 (br, 1H), 2.20–1.90 (br, overlapping, 1H), 1.80–1.34 (br, overlapping, 2H), 1.16 (br, 6H). Mw (GPC): 26.5 kDa, PDI = 1.13. Prior to precipitation Mw (GPC) = 18.6 kDa, PDI = 1.4.
Sample 2 (PNIPAM-288) with Nl = 288: conducted as above, but with NIPAM, BM1433, and ACVA in a molar ratio of 500:1:1 and reaction stopped at 60% conversion. 1H NMR identical to PNIPAM-207. Mw (GPC): 39.8 kDa, PDI = 1.22. Prior to precipitation Mw (GPC) = 30.2 kDa, PDI = 1.4.
2.4 All-atom simulations
We employ all atom molecular dynamics simulations using GROMACS package.35 We use the Gromos96 force field36 for methanol, the SPC/E water model37 and the force field parameters for PNIPAm are taken from ref. 4. The temperature is set to 298 K using velocity rescaling with a coupling constant 0.5 ps.38 The electrostatics are treated using Particle Mesh Ewald.39 The interaction cutoff is chosen as 1.0 nm. The time step for the simulations is set to 2 fs. Initial equilibration of every configuration is performed at ambient pressure for 20 ns, where the pressure coupling is done using a Berendsen barostat40 with a coupling time of 0.5 ps. The final configuration of constant pressure simulations, with equilibrated density, is used for canonical simulations.We choose a PNIPAm chain of length Nl = 256 that corresponds to ∼100p, with p being the persistence length of a PNIPAm chain. Note that an atactic PNIPAm chain has p ∼ 2–3 monomers. We choose five different methanol mole fractions xm in MD simulations. In Table 1 we present our system parameters. The production runs are performed for at least 1 μs long MD trajectory each. During the production run observables such as gyration radius Rg and structure factor S(q) are calculated. In Fig. 1 we present the time evolution of Rg for three different xm. It can be seen that the structure is rather stable over long simulation time scales.
| x m | N | N w | N m | L box (nm) | R g (nm) |
|---|---|---|---|---|---|
| 0.10 | 7.0 × 105 | 6.3 × 105 | 0.7 × 105 | 28.3 | 2.53 |
| 0.25 | 6.0 × 105 | 4.5 × 105 | 1.5 × 105 | 28.1 | 2.28 |
| 0.60 | 5.0 × 105 | 2.0 × 105 | 3.0 × 105 | 28.6 | 5.22 |
| 0.80 | 5.0 × 105 | 1.0 × 105 | 4.0 × 105 | 29.8 | 6.24 |
| 1.00 | 6.0 × 105 | 0 | 6.0 × 105 | 32.8 | 7.37 |
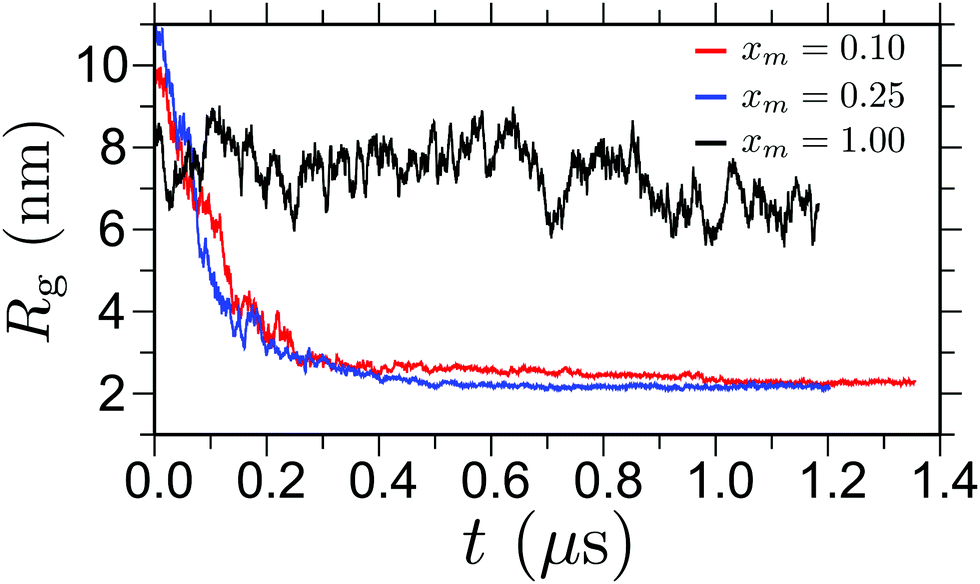 | ||
| Fig. 1 Time evolution of polymer gyration radii Rg for three different methanol mole fractions xm. The results are shown for a chain length Nl = 256 and at temperature T = 298 K. | ||
3 Results and discussions
3.1 PNIPAm conformation in aqueous methanol revisited
We first revisit PNIPAm conformation in water and methanol mixtures. In Fig. 2 we show Rg as a function of xm. We have also included the data from the earlier generic simulations corresponding to Nl = 100p.8 It can be appreciated that, by matching p between the all-atom and previous generic simulations, we obtain a very good (almost quantitative) agreement. Furthermore, Fig. 2 also suggests that 1σ ∼ 1 nm. This can be rationalized as follows: in our generic model p ∼ 1 monomer, whereas for a PNIPAm chain p ∼ 2.5–3 monomers or ∼1 nm. This leads to a mapping of 2.5–3 NIPAm monomers onto one generic bead. Since the size of a generic monomer is 1σ, thus our observation of 1σ ∼ 1 nm is a reasonably good estimate. To make comparison with the empirical data, we have estimated PNIPAm radii by both DOSY NMR and DLS measurements on three PNIPAm samples of varying sizes. For this purpose, we have used samples PNIPAm-207 or Nl ∼ 207 = 83p and PNIPAm-288 or Nl ∼ 288 = 115p. Fig. 2 contains the experimental data for PNIPAm-207 that show a similar trend as the all-atom and the generic simulations. A slight quantitative variation in Rg is because of the difference in Nl's between experiment and all-atom simulations, which varies by ∼15p. As the Rg for the smaller PNIPAm samples is below that can be accurately determined by SLS (i.e., Rg < 10 nm), we obtain hydrodynamic radii Rh from diffusion coefficient data (DLS and DOSY NMR) using the Stokes–Einstein–Sutherland relation.41,42Rh is then translated into Rg using the expressions Rg = 1.5Rh for coil conformations and Rg = (3/5)1/2Rh for globule.43,44 Note that we present experimental data of Rg for the well defined collapsed structures and expanded chain conformations. Near the transition region 0.25 < xm < 1.0, we do not present data because of the nontrivial relation between Rh and Rg.
 | ||
| Fig. 2 Gyration radius Rg as a function of methanol mole fraction xm. Results are shown for Rg obtained from all-atom simulations for a chain of length Nl = 256 ∼ 100p and experimental measurements for Nl ∼ 207 ∼ 83p (sample PNIPAm-207). For the pure water (xm = 0.0) and pure methanol (xm = 1.0) we use the data obtained from dynamic light scattering (DLS). For the collapsed structure at xm = 0.1 and 0.25, we present the data calculated using nuclear magnetic resonance (NMR). For comparison, we also include data from our previous generic simulations8 with Nl ∼ 100p and an analytical expression presented in eqn (2). Arrows are the indicative of the corresponding y-axis of the corresponding data set. | ||
Having shown the results for the collapse–swelling–collapse transition, we now want to briefly explain the origin of this reentrant transition. In this context, it was previously shown that the initial collapse at lower xm values are due to preferential binding of methanol molecules with the PNIPAm chain.7 Therefore, when a small amount of methanol molecules is added, these molecules try to bind to more than one monomer acting as sticky contacts between distal monomers along the polymer backbone. This tendency leads to the formation of segmental loops along the backbone initiating the process of polymer collapse. When the xm exceeds a certain concentration, so that the system can overcome the solvent translational entropy, the polymer re-opens after complete decoration of the polymer with methanol molecules. In this case, conformational entropy contributes to a logarithmic correction19 and, therefore, only leads to a weak effect on describing the overall phenomenon, which is otherwise dominated by enthalpy. The methanol molecules forming enthalpically driven sticky contacts between distal monomer units were termed as bridging cosolvent and their fractions defined as ϕB.8 The analytical expression for ϕB19 can be converted into gyration radius using an argument using the formulation19 based on,43,44
 | (2) |
![[scr V, script letter V]](https://www.rsc.org/images/entities/char_e149.gif) is the magnitude of the negative excluded volume −||, which can be estimated from the simulations and analytical theory.8 Note that ϕB gives the direct measure of using the relation = 100ϕB(xc). Furthermore, an analytical expression can be derived for ϕB using a Langmuir like adsorption isotherm taking into account the competitive displacement of both solvent and cosolvent.8 In Fig. 2 we also include Rg estimated using eqn (2). It can be seen that the experimental, all-atom simulation and generic simulation data can be well described by the analytical theory. Suggesting a reasonable correlation between the phenomena of co-non-solvency and the bridging scenario proposed earlier.8
is the magnitude of the negative excluded volume −||, which can be estimated from the simulations and analytical theory.8 Note that ϕB gives the direct measure of using the relation = 100ϕB(xc). Furthermore, an analytical expression can be derived for ϕB using a Langmuir like adsorption isotherm taking into account the competitive displacement of both solvent and cosolvent.8 In Fig. 2 we also include Rg estimated using eqn (2). It can be seen that the experimental, all-atom simulation and generic simulation data can be well described by the analytical theory. Suggesting a reasonable correlation between the phenomena of co-non-solvency and the bridging scenario proposed earlier.8
Fig. 3 shows a scaling plot of Rg obtained from all-atom simulations and from NMR and DLS measurements of the three polymer samples included in this work. For comparison we have also included data taken from the published literature.3,7 It can be appreciated that the data from different Nl obtained from different methods falls within the universal scaling law, further showing a nice quantitative agreement between different methods.
 | ||
| Fig. 3 Gyration radius Rg as a function of chain length Nl. Results are shown for coil (at xm = 1.0) and globule (at xm = 0.1) conformations. Data for Rg are obtained from different experiments, simulations and also from published work from the literature, as described in the legend. Symbols are respective data and the lines are power law fits shown in the legend. For coil conformation Rg ∝ Nl3/5 and for globular conformations Rg ∝ Nl1/3. | ||
3.2 Good solvent collapse
One of the most intriguing features of this phenomenon is that PNIPAm collapses in good solvent. This makes the polymer conformation completely decoupled from the thermodynamic solvent quality (see chemical potential data in ref. 7). Therefore, we expect the chains to maintain a self avoiding walk statistics between the bridging points, i.e. an exponent of ν = 3/5. A quantity that best describes the polymer conformation is the static structure factor S(q), which usually requires very long simulation trajectories and also long chain lengths. In this context, thus far all-atom simulations are mostly limited to oligomer systems (consisting of Nl = 20–40 to 8–16p). In this work, we calculate S(q) for a collapsed chain at xm = 0.1 over last 0.5 μs long all-atom MD run for Nl = 256 ∼ 100p. In Fig. 4 we show S(q), which shows a cross over from an approximate q−5/3 scaling between 4 nm−1 ≤ q ≤ 10 nm−1 (or corresponding length scale of 1.6 nm ≥ ≥ 0.62 nm) to a q−4 scaling for q ≤ 4 nm−1. This suggests that – while a PNIPAm chain remains globally collapsed, it consists of rather large good solvent blobs with typical sizes of ∼ 1.3 nm.
 | ||
| Fig. 4 Static structure factor S(q) of a collapsed PNIPAm structure at xm = 0.1. For the calculations of S(q) we only consider the alkane backbone of a PNIPAm chain. | ||
3.3 Folded PNIPAm structure and side chain organization
We have used solution NMR to monitor side chain dynamics of PNIPAm as a function of xm (predeuterated MeOD in D2O). Typical 1H NMR spectra are presented in Fig. 5. All the signals decrease in area and broaden with increasing xm between 7.5% ≤ xm ≤ 37.6% and then again increases when xm ≥ 37.6%. The decrease in area and broadening of the signals indicate slowing dynamics, which can be reflected in spin–spin relaxation times T2, as also reported by others as a means to map out polymer collapse.48 The T2 values for the Ha signal plotted in Fig. 6, as a function of xm, give the expected clear signature of the coil–globule–coil scenario as a result of the side chains becoming less mobile when the polymer is in the collapsed state. | ||
| Fig. 5 Left panel shows schematic representation of a monomer of PNIPAm. In NMR experiments we identify the rigidity of Ha and Hb indicated in this schematic. Right panel presents nuclear magnetic resonance (NMR) spectra highlighting Ha and Hb hydrogens as indicated in the left panel. Results are shown for different methanol mole fractions xm, starting from pure water xm = 0.0 (or 0%) to pure methanol xm = 1.0 (or 100%). The signal around 3.75 ppm corresponds to Ha and Hb peak appears around 1.10 ppm. Note that for clear representation of the data, we have aligned the peak positions of Ha. We have used Sigma Aldrich sample of PNIPAm-962. | ||
 | ||
| Fig. 6 In-plane relaxation time T2 of Ha hydrogen as a function of methanol mole fractions xm. The data is obtained by integrating the intensity peak around 3.75 ppm in Fig. 5. | ||
The data from the NMR experiments also suggests that the side group rigidity of a PNIPAm chain is because of the fact that a PNIPAm collapses in a way that the inner core of the folded structure is occupied by the side groups. MD simulations also support this claim for a collapsed structure. In this context, as described earlier, when there is preferential binding of PNIPAm with methanol, it is expected to observe methanol encapsulation within a collapsed structure of PNIPAm. Therefore, it is important to monitor if the side chain rich inner core also has methanol molecules sitting in between. For this purpose, we have performed a separate set of NMR experiments to identify preferential binding of the methanol molecules with PNIPAm chains.
3.4 Preferential adsorption of methanol on PNIPAm and solvent intake
To experimentally observe the preferential interaction of the PNIPAm with methanol, we have used a concept where preferential PNIPAm–methanol binding7 can lead to local aggregation of methanol near the polymer structure that leads to depletion away from the polymer, see a schematic in Fig. 7(a). For this purpose we have prepared a NMR tube separated into two compartments by a membrane filter, see Fig. 7(b). Both compartments contain 15% aqueous methanol, and 1.188 g PNIPAM was added to the upper compartment. This essentially becomes an osmosis experiment, with quantitative 1H-NMR spectra collected to monitor changes in solvent composition in the lower compartment, from which solvent uptake by the PNIPAM can be estimated. For all different time dependent proton spectra, the same phase correction, baseline and integration parameters (integral width for the proton and methyl group of methanol) were applied. | ||
| Fig. 7 Part (a) presents a schematic representation of the concept where local aggregation of cosolvents near a polymer lead to depletion away from the macromolecular structure. Part (b) shows a schematic showing the 5 mm tube with external reference capillary, a sealing function and a paper filter in the center that separates PNIPAM from bulk aqueous methanol mixture at the bottom. Part (c) presents the amount of excess methanol molecules encapsulated by the PNIPAm collapsed sample as measured from the depletion of methanol in the lower part of the NMR tube. Here 3% of methanol is with respect to the initial 15% of the methanol content. | ||
In Fig. 7(c) we present the amount of depleted methanol in the lower panel of the NMR tube in Fig. 7(b) that is engulfed by the PNIPAm sample in the upper panel of the NMR tube. The measurements were conducted over sixteen days. It can be seen that the methanol increases by ∼3% in the polymer system within the first 3–4 days. Beyond 4 days, solvent intake data shows a plateau suggesting no evaporation of methanol from the airtight experimental setup. Note that a rather large polymer concentration is needed in the upper panel of Fig. 7(b) to observe any significant solvent intake. The solvent intake, as observed in our NMR experiments, further supports our earlier claim that preferential adsorption of the methanol with PNIPAm drives the polymer collapse.7 This scenario can be further validated by looking into the potential of mean force (PMF) between PNIPAm–methanol and PNIPAm–water calculated using the umbrella sampling.23,45,46 In Fig. 8 we show PMF between different PNIPAm–(co)solvent pairs, which show a clear signature of preferential enthalpic interactions between PNIPAm–methanol. Furthermore, in a given polymer, the energy density within the solvation volume is dictated not only the interaction energy, but also by the sizes of the solvent (methanol and water in this case). Therefore, it should still be mentioned that the enthalpic interactions (or bridging) are usually not given by a single methanol molecule, rather a few collectively lead to sticky contacts.8
 | ||
| Fig. 8 Potential of mean force v(r) showing NIPAm–methanol and NIPAm–water interaction strengths for two different pressures. Simulations are performed at a temperature of 298 K and the data is taken from ref. 23. | ||
The window of PNIPAm collapse, or the LCST behavior, is strongly dependent on the temperature of the systems.1,2 Here, it is important to mention that – just because a polymer exhibits an LCST behavior, it does not suggest that the polymer collapse at a constant T should also be driven enhanced solvent entropy by the addition of methanol in water. Our arguments are based on the claim that the interaction asymmetry between PNIPAm–methanol and PNIPAm–water dictate PNIPAm collapse in aqueous methanol mixture.8 The smaller the asymmetry, the narrower the window of polymer collapse.19 To elucidate that a PNIPAm collapses because of interaction asymmetry, we have calculated PMF between NIPAm–water and NIPAm–methanol for T = 278 K. The data is shown in Fig. 9. As expected, at a reduced T the asymmetry in interactions also reduces, making the background binary fluid homogeneous for the polymer. This can not facilitate a polymer to collapse in a binary mixture. Interestingly previous experiments1,2 have shown that the PNIPAm remains in coil state for T = 278 K, thus showing a nice correlation capturing the temperature effects between the earlier experiments1,2 and our simulations.
 | ||
| Fig. 9 Same as Fig. 8. But for a temperature T = 278 K. | ||
3.5 Mechanism of polymer collapse
Lastly we want to comment on the possible mechanism of cosolvent bridging leading to PNIPAm collapse in aqueous methanol mixtures. In this context, it is already presented in the previous section and consistent with previous work,7 that there is a preferential binding of PNIPAm with methanol. To further illustrate the bridging scenario driven by preferential adsorption, we calculate the number of hydrogen bonds nH-bond/Nl between a methanol molecules and a NIPAm monomer using all-atom simulations. In Fig. 10(a) we show nH-bond/Nl as a function of xm. In the range 0.10 ≤ xm ≤ 0.25, NIPAm shows a strong tendency of hydrogen bonding with the methanol molecules that is evident from the maximum deviation from the expected linear behavior.4 Without attempting to describe any specific geometry or arrangement of H-bond donor and acceptors, we can assume the –OH end of the methanol points towards the PNIPAm amide linkage, such that the CH3 of methanol now forms part of the local solvent accessible surface previously defined by the amide group. The sticky contacts could then be formed between these CH3 groups and the isopropyl group of a distal NIPAm unit (see Fig. 10(b)), or between multiple CH3 groups of bound methanol molecules (see Fig. 10(c)). Note that in Fig. 10(b) and (c), for simplicity of representation, we only highlight hydrogen bonds between hydrogen of methanol and the oxygen of NIPAm amide groups that we expect to be most dominant. However, it should be mentioned that there might be several more scenarios. For example, there is a possibilities of bonding between methanol oxygen and hydrogen of the amide group and methanol hydrogen with nitrogen of the amide groups. Furthermore, it should also be mentioned that a single CH3 interaction with isopropyl group is ∼kBT, which may not sound as a large enough interaction strength. However, these methanol mediated sticky contacts are each likely facilitated by a few methanol molecules making the sticky contact attraction strength of the order of several ∼kBT and not only by a single methanol molecule, as simplified in the schematics shown in Fig. 10(b) and (c). Here we want to emphasize that the solvation properties are intimately linked to the energy density within the solvation volume. Hence, not only dictated by the individual interaction strengths, but also related to number of (co)solvent particles within the solvation volume. Therefore, if one can reduces the energy density within the solvation volume, such that the solvent–cosolvent interaction contrast also reduces, one should expect to see narrower window of collapse. Indeed, it had been experimentally observed that for larger alcohols (such as ethanol or propanol) window of collapse reduces by ∼30–40% in comparison to methanol.47 | ||
| Fig. 10 Part (a) shows number of hydrogen bonds nH-bond/Nl between a methanol molecule and a monomer of PNIPAm as a function of methanol mole fraction xm. The dashed red line is the linear extrapolation. Data is shown for all-atom simulations of chain length Nl = 256 and for the temperature T = 298 K. Parts (b) and (c) present schematic of two possible scenarios of bridging methanol molecules, which we expect to be most relevant. | ||
4 Conclusions
We have revisited the co-non-solvency of PNIPAm in aqueous methanol mixtures. For this purpose, we have combined nuclear magnetic resonance and dynamics light scattering experiments with the all-atom molecular dynamics simulations, complementing our earlier studies of generic simulations8 and analytical theory.19 These findings strongly support that the initial collapse at lower methanol concentration is due to the methanol–PNIPAm enthalpic (bridging) effects and the reopening at larger methanol concentrations is entropic. Furthermore, preferential PNIPAm–methanol binding leads to increased organization of the PNIPAm side chains, which is intimately linked to the global conformational behavior of PNIPAm in aqueous methanol mixtures.While we study a specific system of PNIPAm in aqueous methanol mixture, our proposed mechanism of PNIPAm collapse at lower methanol concentrations provide a natural explanation to other phenomena, such as the initial collapse of PNIPAm in aqueous urea mixtures.48,49 Where the collapse was proposed is initiated by hydrogen bonded bridging of urea molecules between two NIPAm monomers that are far along the polymer backbone.
It should also be mentioned that when dealing with polymer physics and/or the thermodynamics of polymer solutions, two key considerations, out of several, are absolutely needed to make any reasonable comparison to experimental data: (1) a polymer should be studied and not an oligomer and (2) time scale of simulations compared to the polymer relaxation time. In this context, our simulations of μs long trajectory of a Nl = 100p chain consisting of two million particles is the largest all-atom simulations performed on this PNIPAm based systems. A reasonably good agreement between all-atom simulations and experiments complementing earlier generic simulations, suggests that the co-non-solvency phenomenon is indeed driven by enthalpy.8
Acknowledgements
D. M. thanks Burkhard Dünweg, Martin Müser, Kostas Ch. Daoulas, Paulo A. Netz, Robinson Cortes-Huerto, and Torsten Stühn for countless stimulating discussions, Jens-Uwe Sommer for useful discussions during the SOMATAI-2016 meeting in Crete that motivated us to calculate Fig. 9 and Adam Moule for useful discussion regarding the methanol mediated sticky contacts and NMR experiments. We thank Robert Graf for stimulating discussions regarding NMR technique. Furthermore, we gratefully acknowledge the assistance of Christine Rosenauer with the DLS and GPC measurements. We thank Stefan Spang for preparing the NMR tube for the second set of NMR experiments. M. D. W., T. E. O., and C. M. M. acknowledges hospitality at the Max-Planck Institut für Polymerforschung, where this work was initiated and performed. We thank Robinson Cortes-Huerto, Takahiro Okhuma, and Torsten Stühn for critical reading of the manuscript.References
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415–2416 CrossRef CAS.
- G. Zhang and C. Wu, Phys. Rev. Lett., 2001, 86, 822–825 CrossRef CAS PubMed.
- J. Walter, J. Sehrt, J. Vrabec and H. J. Hasse, J. Phys. Chem. B, 2012, 116, 5251–5259 CrossRef CAS PubMed.
- H. Kojima, F. Tanaka, C. Scherzinger and W. Richtering, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1100–1111 CrossRef CAS.
- F. Tanaka, T. Koga and F. M. Winnik, Phys. Rev. Lett., 2008, 101, 028302 CrossRef PubMed.
- D. Mukherji and K. Kremer, Macromolecules, 2013, 46, 9158–9163 CrossRef CAS.
- D. Mukherji, C. M. Marques and K. Kremer, Nat. Commun., 2014, 5, 4882 CrossRef PubMed.
- K. Kyriakos, M. Philipp, C.-H. Lin, M. Dyakonova, N. Vishnevetskaya, I. Grillo, A. Zaccone, A. Miasnikova, A. Laschewsky, P. Mller-Buschbaum and C. M. Papadakis, Macromol. Rapid Commun., 2016, 37, 420–425 CrossRef CAS PubMed.
- M. Richter, M. Hunnenmörder and R. von Klitzing, Colloid Polym. Sci., 2014, 292, 2439–2452 CAS.
- Y. Yu, M. Cirelli, B. D. Kieviet, E. S. Kooij, G. J. Vancso and S. de Beer, Polymer, 2016 DOI:10.1016/j.polymer.2016.08.029.
- B. A. Wolf and M. M. Willms, Makromol. Chem., 1978, 179, 2265–2277 CrossRef CAS.
- A. Hiroki, Y. Maekawa, M. Yoshida, K. Kubota and R. Katakai, Polymer, 2001, 42, 1863–1867 CrossRef CAS.
- R. Lund, L. Willner, J. Stellbrink, A. Radulescu and D. Richter, Macromolecules, 2004, 37, 9984–9993 CrossRef CAS.
- B. E. Read, Trans. Faraday Soc., 1960, 56, 382–390 RSC.
- M. Ohkura, T. Kanaya and K. Kaji, Polymer, 1992, 33, 3686–3690 CrossRef CAS.
- Y. Kiritoshi and K. Ishihara, J. Biomater. Sci., Polym. Ed., 2002, 13, 213–224 CrossRef CAS PubMed.
- Y. Kiritoshi and K. Ishihara, Sci. Technol. Adv. Mater., 2003, 4, 93–98 CrossRef CAS.
- D. Mukherji, C. M. Marques, T. Stuehn and K. Kremer, J. Chem. Phys., 2015, 142, 114903 CrossRef PubMed.
- J. Dudowicz, K. F. Freed and J. F. Douglas, J. Chem. Phys., 2015, 143, 131101 CrossRef PubMed.
- Y. A. Budkov, A. L. Kolesnikov, N. N. Kalikin and M. G. Kiselev, EPL, 2016, 114, 46004 CrossRef.
- F. R. Rupero, T. Hajari and N. van der Vegt, J. Phys. Chem. B, 2015, 119, 15780–15788 CrossRef PubMed.
- T. E. deOliveira, P. A. Netz, D. Mukherji and K. Kremer, Soft Matter, 2015, 11, 8599–8604 RSC.
- K. Mochizuki, S. R. Pattenaude and D. Ben-Amotz, J. Am. Chem. Soc., 2016, 138, 9045–9048 CrossRef CAS PubMed.
- J. Heyda, A. Muzdalo and J. Dzubiella, Macromolecules, 2013, 46, 1231–1238 CrossRef CAS.
- S. de Beer, E. Kutnyanszky, P. M. Schön, G. J. Vancso and M. H. Müser, Nat. Commun., 2014, 5, 3781 Search PubMed.
- A. Jerschow and N. Mueller, J. Magn. Reson., Ser. A, 1996, 123, 222–225 CrossRef.
- A. Jerschow and N. Mueller, J. Magn. Reson., Ser. A, 1997, 125, 372–375 CrossRef CAS.
- M. Holz and H. Weingartner, J. Magn. Reson., 1991, 92, 115 CAS.
- J. E. Tanner, J. Chem. Phys., 1970, 50, 2523 CrossRef.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1992, 114, 3139–3141 CrossRef CAS.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- W. F. van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott and I. G. Tironi, Gromos43a1, Hochschulverlag AG an der ETH Zurich, 1996, pp. 1–1402 Search PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. A. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- W. Sutherland, Philos. Mag., 1905, 9, 781 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549 CrossRef.
- P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, London, 1979 Search PubMed.
- J. Des Cloizeaux and G. Jannink, Polymers in Solution: Their Modelling and Structure, Clarendon Press, Oxford, 1990 Search PubMed.
- M. Sprik and G. Ciccoti, J. Chem. Phys., 1998, 109, 7737 CrossRef CAS.
- J. Kahlen, L. Salimi, M. Sulpizi, C. Peter and D. Donadio, J. Phys. Chem. B, 2014, 118, 3960 CrossRef CAS PubMed.
- R. O. P. Costa and R. F. S. Freitas, Polymer, 2002, 43, 5879–5885 CrossRef CAS.
- J. Wang, B. Liu, G. Ru, J. Bai and J. Feng, Macromolecules, 2016, 49, 234–243 CrossRef CAS.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |