
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the syneresis of an OPV functionalised dipeptide hydrogel†
Ana M.
Castilla
a,
Matthew
Wallace
a,
Laura L. E.
Mears
a,
Emily R.
Draper
a,
James
Doutch
b,
Sarah
Rogers
b and
Dave J.
Adams
a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
bISIS Pulsed Neutron Source, Rutherford Appleton Laboratory, Didcot, UK
First published on 19th August 2016
Abstract
We describe a new dipeptide hydrogel based on an oligophenylene vinylene core. After gelation, the initial network evolves, expelling solvent and resulting in syneresis. We describe this process and the effects in the bulk properties of the material.
Introduction
Low molecular weight gels (LMWGs), assembled from small molecular building blocks, are emerging as useful soft materials.1 The tuneable and stimuli-responsive nature of supramolecular gels, as opposed to traditional polymeric gels, makes them appealing for applications such as sensors, in optoelectronics, tissue engineering or drug delivery.1,2 Following the application of a trigger (e.g. a pH change, the addition of a salt, a temperature change), a LMWG in solution undergoes hierarchical self-assembly. First, mono-dimensional structures (fibres) are formed that then entangle into a 3D network, trapping the solvent to form a gel.3 Significant work has been devoted to study the self-assembly processes that lead to gelation and how they relate to the bulk properties.3d Understanding and control of these processes would allow us to design materials with desired properties.Syneresis is the phenomenon by which a gel undergoes macroscopic contraction, expelling some of the solvent that was initially immobilised in the gel network. This ‘gel shrinking’ has been mostly studied in polymer-based gels for biomedical applications such as the controlled release of bioactive molecules.4 Only a very few studies report the shrinking process of supramolecular gels.5 The shrinking/swelling processes of a glycosylated amino acetate gelator were found to be thermal and pH-responsive,5a while these processes for an amphiphilic dendron gelator could be triggered with metal ions.5c We have reported a few peptide-based LMWG which exhibit syneresis6 however, no detailed study of the process has been reported to date. This volume phase transition is however an important process to control when developing materials; gel syneresis can be problematic for many applications where gel integrity is important, but controlled shrinkage can be exploited in the development of drug delivery systems or sensors.5 In this contribution, we discuss and study the syneresis of a peptide LMWG based on an OPV (oligophenylene vinylene) core (1, Fig. 1). The use of this π-conjugated OPV core is interesting for applications in organic electronics due to its known properties as an organic semiconductor.7 We have prepared several such gelators but only a small number of them exhibit syneresis; most either form stable gels or precipitates. It is rare to be able to compare two related molecules that show syneresis, albeit on different time scales. We provide a detailed study of the gel formation and subsequent contraction by different techniques providing insights into the origin of the gel syneresis.
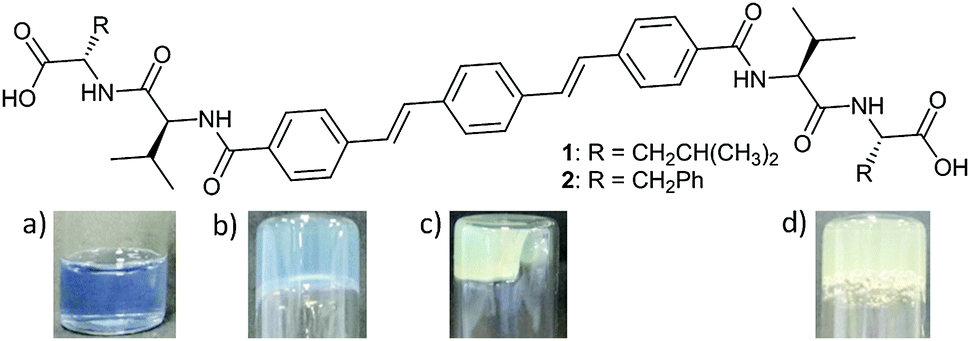 | ||
| Fig. 1 Top: Structures of OPV-based LMWGs 1 and 2. Bottom: Photos of (a) the initial solution of 1 (5 mg mL−1, pH = 10) and the gel of 1 (b) 6 hours and (c) 24 hours after the addition of GdL (3 mg mL−1) to the initial solution. (d) Hydrogel formed 24 hours after the addition of GdL (5 mg mL−1) to a basic solution of 2 (5 mg mL−1, pH = 10). NB. Samples shown in (a)–(c) contain bromophenol blue as a pH indicator. | ||
Results and discussion
OPV-based LMWGs 1 and 2 were prepared through amide coupling between OPV-3 diacyl chloride8 and the trifluoracetate salts of L-valyl-L-leucine methyl ester or L-valyl-L-phenylalanine methyl ester respectively, followed by cleavage of the methyl groups with lithium hydroxide (see ESI† for full details). A series of OPVs functionalised with L-alanine, L-valine, L-phenylalanine and glycine were also prepared (Scheme S1, ESI†). All these OPV-based compounds, except for the glycine derivative, dissolved in water upon deprotonation of the carboxylic groups with two equivalents of NaOH, resulting in solutions with pH ≈ 10–11. The glycine derivative forms instead a white suspension. LMWG 1 forms a free-flowing solution at high pH with a viscosity similar to water (Fig. S13, ESI†); NMR measurements (see Discussion below) indicate that 1 behaves in solution as a single molecule in equilibrium with large aggregates. Viscosity measurements of solutions of 2 at high pH, however, show them to be shear-thinning (Fig. S13, ESI†), suggesting 2 forms worm-like micelles at these conditions.9 The existence of large aggregates of 2 at high pH is also supported by NMR measurements (see below and Fig. S39 and S40, ESI†). Consistently, small angle neutron scattering (SANS) backs up the assignment of 2 forming worm-like micelles at high pH; the scattering from a solution of 1 at high pH is however clearly inconsistent with the formation of worm-like micelles (Section 4, ESI†).A pH-trigger was used to test the ability of these OPV-based LMWG to form hydrogels. To lower the pH, we added glucono-δ-lactone (GdL), which hydrolyses slowly to gluconic acid, lowering the pH controllably as we have previously reported.10 Addition of GdL (6 mg mL−1) to a basic aqueous solution of 1 (5 mg mL−1, pH = 10) resulted in a self-supporting gel showing significant syneresis. The gel shrank in a three-dimensional fashion maintaining the shape of the container (Fig. 1c). The fluid exuded over 72 hours after the addition of GdL was measured to be 60% of the initial solution volume. UV-Vis spectroscopy confirmed that no gelator was present in the exuded solution and hence is incorporated entirely in the shrunken gel (Fig. S14, ESI†). Attempting to quantify the syneresis with time during the shrinking is delicate, as one tends to damage the gel when only low volumes of water have been exuded. This is because the gel shrinks uniformly, not just downwards, making the removal of the water very difficult.
Gelator 2 was also observed to form hydrogels by this pH-triggered method (Fig. 1d). However, in this case the gel formed remained stable for 4 days, only undergoing syneresis over 5 days (Fig. S15, ESI†), and to a lesser extent than the gel formed using 1. Due to the small degree of syneresis in this case, absolute quantification was not attempted. All of the OPVs functionalised with single amino acids did not form gels by this pH-triggered method, instead forming precipitates. This difference in behavior is unsurprising; it has been shown previously that closely related molecules have very different gelation ability.11
We have previously shown that using the slow hydrolysis of GdL to gluconic acid in water to adjust the pH of aqueous solutions of peptide-based gelators allows for the self-assembly process to be followed by different techniques.12 Here, the pH changes accompanying the gelation of a solution of 1 after the addition of different amounts of GdL were visually followed by adding bromophenol blue as a pH indicator (blue above pH 4.6 and yellow below pH 3.0). When using 3 mg mL−1 of GdL to trigger gelation of a solution of 1 (5 mg mL−1, pH = 10), a weak gel forms within 6 hours and at a pH above 4.6 (Fig. 1b). Note, a lower amount of GdL was used here to slow down the rate of pH change. This gel showed no contraction for at least 2 hours but syneresis after 24 hours (Fig. 1c) when the colour of the sample had also switched to yellow, and the amount of syneresis increased over time (Fig. S16, ESI†).
The formation of the gel network was studied by monitoring simultaneously the evolution of rheological properties and changes in pH after addition of GdL (3 mg mL−1) to a solution of gelator 1 (5 mg mL−1, pH 10, Fig. 2). For these measurements, the experiment exposes the sample to constant oscillation. Our previous data on such gels12 has shown that the oscillation has little effect on the final gel properties. As for related LMWGs, the gel network develops in a two-stage process.12 The storage and loss moduli (G′ and G′′ respectively) start to increase at pH ≈ 7, indicating that self-assembly starts just before the apparent pKa of 1 (Fig. 3).
 | ||
| Fig. 2 Graph showing the evolution of the gel network after the addition of GdL (3 mg mL−1) to a basic solution of 1 (5 mg mL−1, pH = 10). Filled and empty circles represent storage and loss moduli respectively, and triangles the pH. The inset photographs show the solution and the gel before and after syneresis (note, these samples contain bromophenol blue as a pH indicator). | ||
 | ||
| Fig. 3 (a) Changes in pH with time of an aqueous solution of 1 (5 mg mL−1, initial pH = 10) after addition of GdL (3 mg mL−1). (b) Titration curve of an aqueous solution of 1 (5 mg mL−1, initial pH = 10) with HCl 0.1 M plotted in linear scale (left) and in logarithmic scale (right). Red dotted lines in (a) represent the region buffered by 1. Red lines in (b) denote the apparent pKas found for 1. | ||
The gel network has already formed at pH 6.7, when the mechanical properties reach a plateau where G′ > G′′. The subsequent strengthening of the gel, as the 3D network entangles and the pH decreases, is tracked by the increase of G′ and G′′ until reaching a second plateau where the mechanical properties of the gel stabilises. At longer times, a sudden decrease in storage and loss moduli was observed. This drop in mechanical properties after the gel is formed has previously been described for gels undergoing syneresis.6b The contraction of the gel, with concomitant expulsion of liquid, results in a weaker contact between the measuring plate and the sample hence, a decrease in the apparent mechanical properties measured. This occurs when the pH of the system was measured to be ca. 4.2, in agreement with the visual study. The self-assembly of gelator 2 followed the same two-stage process with no drop of the mechanical properties after reaching the second plateau (Fig. S18, ESI†). As mentioned earlier, gel 2 only started to shrink five days after the addition of GdL, which is after the rheological measurements were stopped.
Further time sweeps for different concentrations of 1 (3, 5 and 10 mg mL−1) and initial pH (7, 10 and 12) of the pre-gelation solution allowed us to determine the dependence of the time at which gel 1 undergoes syneresis with the starting pH and concentration of the pre-gelation solution (Fig. 4; note due to the much slower and less dramatic syneresis, similar data was not collected for 2). We took the slippage on the measuring plates as the time at which the gel contraction starts (Fig. S19–S21, ESI†). Fig. 4 clearly shows that the gel undergoes faster syneresis at lower concentrations of 1 and when starting from solutions at lower pH. Furthermore, in all cases gel contraction was observed to start when the system reaches a pH value within the interval of 3.5–4.5, rather than always at the same exact value. Consistently, the gelation kinetics, correlating with kinetics of pH change, is faster at higher temperatures (Fig. 5) and thus results in faster syneresis, yet gel contraction does not start always at the same pH. These observations suggest that syneresis is not simply an effect of pH, but likely a result of the self-assembly conditions.3d
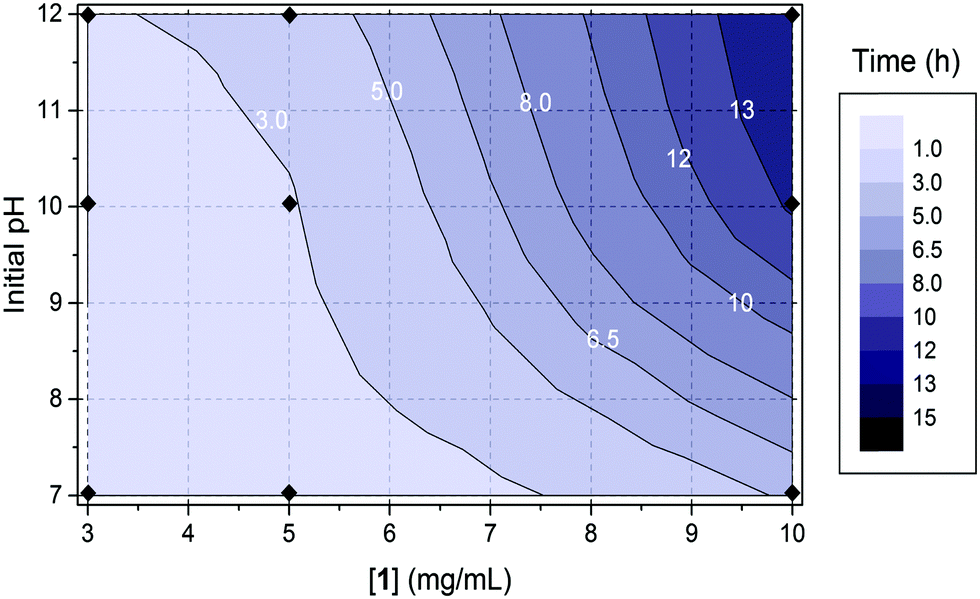 | ||
| Fig. 4 The dependence of the time at which gel 1 undergoes syneresis with the starting pH and concentration of the pre-gelation solution. The concentration of GdL used in all cases was 6 mg mL−1. The data points are indicated with black diamonds in the diagram: for 3 mg mL−1, t = 1.4 h (pH = 7), 2.3 h (pH = 10) and 2.8 h (pH = 12); for 5 mg mL−1, t = 1.7 h (pH = 7), 2.8 h (pH = 10) and 4.0 h (pH = 12); for 10 mg mL−1, t = 5.2 h (pH = 7), 13.4 h (pH = 10) and 14.9 h (pH = 12). | ||
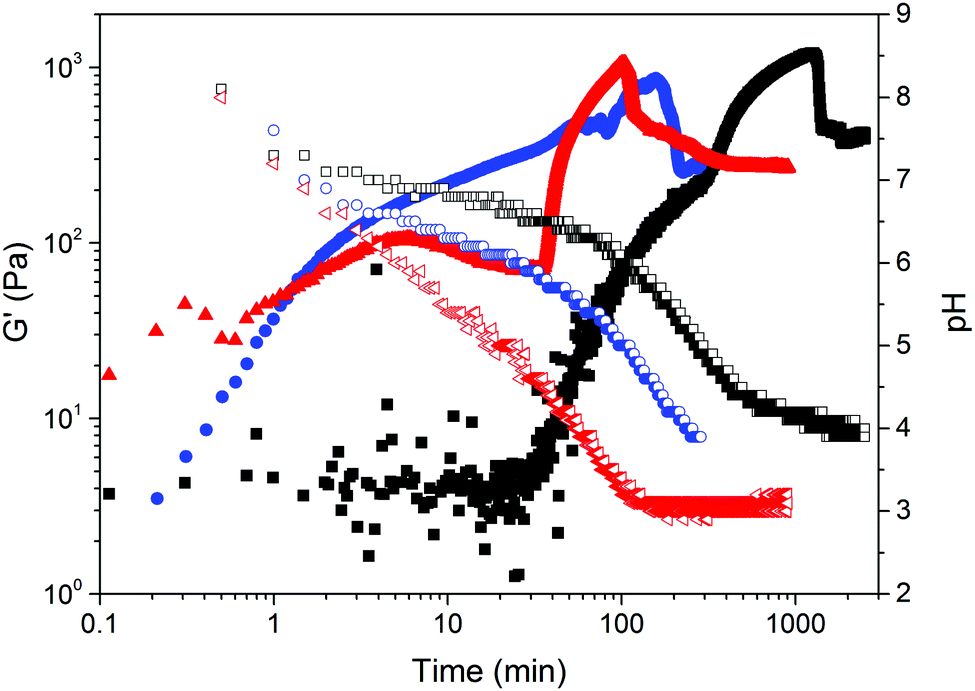 | ||
| Fig. 5 Evolution of pH (empty symbols) and G′ (full symbols) with time after the addition of 6 mg mL−1 of GdL to a solution of gelator 1 (5 mg mL−1, pH = 10) at different temperatures: 9 °C (black data), 25 °C (blue data) and 36 °C (red data). Rheology measurements were carried out at a strain of 0.5% and a frequency of 10 rad s−1. | ||
We further probed the self-assembly of 1 and the gel syneresis using solution-state NMR. The evolution of the charge and hydrophobicity of the fibres throughout the self-assembly process can be followed by tracking changes in the relative affinities of molecular probes (23Na+ and dioxane in this work, dissolved in the bulk solution of the gel) for the surface of the gel fibres.6b Measurements of 23Na NMR relaxation rates report on the mobility of the 23Na+ ions and thus provide a simple way to gauge the interaction of these ions with large charged structures such as gel fibres.6b,13 Similarly, the 2H linewidth of a deuterated solvent probe, such as dioxane-d8, reports on its molecular mobility and thus, the strength of its interaction with the hydrophobic gel fibres.14
Fig. 6a shows the longitudinal (T1) and transverse (T2) relaxation times of 23Na+ at different times since the addition of 4 mg mL−1 GdL to a solution of 1 (5 mg mL−1, pH 10). At pH 10 (t = 0), the 23Na+T1 and T2 are much shorter than the ca. 56 ms expected for a solution of small molecules,6b while 1 exhibits broad 1H NMR resonances and short T2 relaxation times (Fig. S38, ESI†).15 Together, these observations indicate that 1 is in equilibrium with self-assembled aggregates at pH 10. Upon addition of GdL, the 23Na+T2 initially decreases, indicative of the formation of larger structures that still bear a significant negative charge, before gradually increasing with time as the pH falls and this charge is presumably lost. The fitted linewidths of dioxane-d8 (0.05 vol%) as a function of time are plotted on Fig. 6b. In contrast to the 23Na+ relaxation measurements, the linewidth of dioxane-d8 is narrow in the absence of GdL, being comparable to the 0.6 Hz measured in a 0.05 vol% solution of dioxane-d8 in H2O, but as the pH falls and large structures begin to form, the linewidth increases indicating a significant interaction of the organic solvent with the now hydrophobic gel fibres. Upon syneresis of the gel at about 600 minutes, a sharp peak attributable to dioxane in the exuded fluid becomes apparent, superimposed on the broad peak of dioxane in the gel. The narrow component has a width comparable to the width at t = 0, while the broad component continues to broaden as the gel contracts. Time-lapse photography of a parallel sample confirms that the emergence of the sharp peak and the visible contraction of the gel coincide (Fig. S35, ESI†). The 2H resonance of HDO broadens only slightly over time, with the effect of the contraction of the gel being a lot less pronounced than on the dioxane resonance (Fig. S37, ESI†), indicating a stronger interaction of the organic solvent, and thus increased hydrophobicity of the fibres, with time. Overall, the NMR data suggest that, in line with our previous work,6b the syneresis of the gel can be linked to a loss of negative charge from the fibres and an increase in their hydrophobicity. Consistently, the FT-IR spectrum of the shrunken gel shows a more pronounced shoulder at 1716 cm−1 (Fig. S22, ESI†), assignable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of COOH groups, when compared to the non-contracted gel. Gelator 2 behaves similarly to 1 regarding loss of charge and increasing hydrophobicity of the gel fibres during gel formation (Fig. S39, ESI†) however, 2 does not show syneresis. Thus, these results support our previous inference that syneresis is not simply an effect of pH, and also indicate it is not related solely with charge reduction on the gel fibres.
O stretch of COOH groups, when compared to the non-contracted gel. Gelator 2 behaves similarly to 1 regarding loss of charge and increasing hydrophobicity of the gel fibres during gel formation (Fig. S39, ESI†) however, 2 does not show syneresis. Thus, these results support our previous inference that syneresis is not simply an effect of pH, and also indicate it is not related solely with charge reduction on the gel fibres.
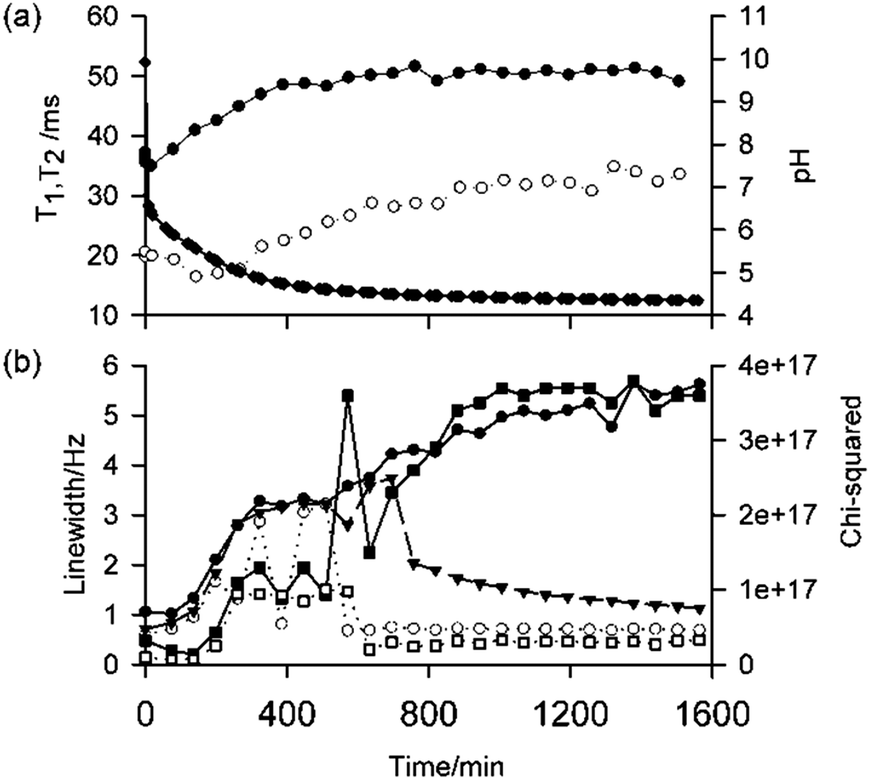 | ||
| Fig. 6 (a) Plots of 23Na+T1 (black circle) and T2 (white circle) relaxation times as a function of time following the addition of 4 mg mL−1 GdL to a solution of 1 at pH 10, along with pH measurements (black diamond). The lines are there to guide the eye. (b) Plot of fitted 2H linewidths of dioxane-d8 as a function of time: single Lorentzian fit (black triangle), wider component of double Lorentzian fit (black circle) and narrow component (white circle). The quality of fits are indicated by their chi-squared values (smaller value represents better fit): single component fit (black square) and double component (white square). Linewidths are the widths of the fitted Lorentzian peaks at half height. The data shown in (a) and (b) were acquired on the same sample. The time points at t = 0 were recorded prior to the addition of GdL. | ||
To explain the syneresis, we next examined the differences in the gel networks of 1 before and after contraction. We first studied the differences in mechanical properties. Both gel networks show typical behaviour of a LMWG, where G′ is an order of magnitude above G′′ and essentially independent of frequency up to 100 rad s−1. Strain sweeps show that the breakage point of the gel network is at approximately 16% before and after contracting, although at low strains G′ and G′′ for the non-syneresed gel show a weak strain dependence. The main difference is the increased stiffness of the contracted gel. The G′ (900 Pa) for the syneresed gel is twice the value of the non-contracted gel (440 Pa). This suggests a denser gel network for the contracted gel formed upon expulsion of water. This is consistent with an effective increase in the concentration from 5 mg mL−1 to 12.5 mg mL−1, assuming that 60% of the initial volume has been exuded by syneresis. Interestingly, we also observed an increase in the stiffness of syneresed gels formed with increasing amounts of GdL, which underwent faster gelation (Fig. S24, ESI†).
The primary fibre dimensions and the network within the hydrogels were probed in situ using SANS. The parameters used to model fit SANS data (Fig. S42 and Table S1 in the ESI†) of the gel formed by 1 indicate that the primary fibre structure is similar in the initial gel and once it has syneresed, with the radius (fitted as 2.1 ± 0.2 nm) and Kuhn length (proportional to the persistence length) of the segment only changing slightly, from 6.5 ± 0.5 to 5.0 ± 0.5 nm. There is a more distinct change over longer length scales (smaller Q), indicated by the increase of 11 nm in the fitted contour length of the fibre and in the network structure. The increase in the power law component implies an increase in segregation within the network as water is expelled. This implies an increase in domains of more densely packed fibres. We believe that this is consistent with our data above, where we have an intermediate state between a homogenous network and a fully segregated system, since we still have a gel (albeit more concentrated) and an aqueous phase (which does not contain the gelator). SANS data for the gel of 2 (Fig. S42 and Table S2, ESI†) indicate that the primary fibre radius is elliptical in cross-section, hence on average slightly larger (Rmin = 2.0 ± 0.1 nm and axis ratio 5.1 ± 0.5) and the Kuhn length longer at 16 ± 4 nm, as expected from the chemical structures. The gel network of 2 appears to be similar in homogeneity to the initially formed gel of 1, with a similar power law exponent.
Confocal microscopy was used to examine the morphology of the gel before and after contraction. This technique enables the study of wet samples and thus to directly probe the gels in their native state. This allows for overcoming the effect of syneresis during gel drying in sample preparation required by most microscopy techniques. Nile blue, which interacts with the gel fibres (Fig. S26, ESI†),16 was added to the pre-gelation solution to allow the fibres to be visualised. Representative confocal images are shown in Fig. S27 and S28 (ESI†). Images of the gel formed by 1 before contraction show very fine gel fibres, while the contracted gel shows a much denser network, supporting SANS data, where fibres cannot be distinguished. Images of the gel formed by 2 (Fig. S29, ESI†) showed a gel network with similar homogeneity to that of gel 1, but fibres of 2 are too thin to be visualised. SEM images are consistent with the confocal data (Fig. S30–S32, ESI†).
Conclusions
The contraction/swelling of supramolecular hydrogels has been previously ascribed to pH changes or electrostatic interactions between fibres,5a–c whereas the syneresis of a molecular organogel was demonstrated to be related with a structural change in the gel network.5d We have previously related syneresis with low hydrophobicity of the gelator.6a Indeed, 2 is slightly less hydrophobic than 1 (c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP = 5.03 for 1 and 4.73 for 2, as determined using an online prediction programme).17 However, there is often little link between hydrophobicity and gelation ability (within a certain range). We have shown that for Fmoc-dipeptides,6a the gelators that are less hydrophobic tend to undergo syneresis, which is the opposite of the effect seen here, again highlighting that gelation ability is difficult to predict. This current study indicates that the contraction of the gel formed by 1 is not an effect of pH or negative charges on the surface of the gel fibres. In addition, no structural changes have been observed to occur in the gel network upon syneresis, only an evolution to a more densely packed network. We thus hypothesise that the gel syneresis is related to the 3D arrangement of the self-assembled fibres in the gel network. We infer this arrangement favours closer interactions between fibres in the gel of 1 than in the gel of 2. This thus implies that syneresis will be strongly dependent on the self-assembly process, as opposed to simply the molecular structure.3d
logP = 5.03 for 1 and 4.73 for 2, as determined using an online prediction programme).17 However, there is often little link between hydrophobicity and gelation ability (within a certain range). We have shown that for Fmoc-dipeptides,6a the gelators that are less hydrophobic tend to undergo syneresis, which is the opposite of the effect seen here, again highlighting that gelation ability is difficult to predict. This current study indicates that the contraction of the gel formed by 1 is not an effect of pH or negative charges on the surface of the gel fibres. In addition, no structural changes have been observed to occur in the gel network upon syneresis, only an evolution to a more densely packed network. We thus hypothesise that the gel syneresis is related to the 3D arrangement of the self-assembled fibres in the gel network. We infer this arrangement favours closer interactions between fibres in the gel of 1 than in the gel of 2. This thus implies that syneresis will be strongly dependent on the self-assembly process, as opposed to simply the molecular structure.3d
Experimental
Preparation of LMWG solutions
To a vial containing a pre-weighed amount of gelator was added 0.1 M NaOH aq solution (2 equiv.) and H2O to a concentration of 5 mg mL−1, unless otherwise stated. This solution was vigorously stirred until complete dissolution of the gelator. The pH of the solution was 10–11.Hydrogel formation
The high pH solution of gelator was added to a pre-weighed amount of GdL (3 mg mL−1 of GdL for gel 1 and 5 mg mL−1 for 2, unless otherwise specified). The mixture was manually swirled and then left to stand still for the selected time at room temperature.For pH change photographs a few drops (20–50 μL) of bromophenol blue indicator solution were added to the gelator solution before GdL was added.
Preparation of hydrogels in plastic moulds
The mould was prepared by cutting the top off a 20 mL (2 cm diameter) plastic syringe. The selected volume of gelator solution was added to a vial containing a pre-weighted amount of GdL, the mixture was swirled and immediately transferred into the plastic syringe, standing vertically on its plunger that was secured to the table with Blu-Tack. This was covered with Parafilm and left to stand still for the selected time at room temperature. The gel was removed by carefully pushing the plunger. It was sufficiently stiff to be transferred onto the bottom plate of the rheometer.Rheological measurements
Dynamic rheological and viscosity measurements were performed using an Anton Paar Physica MCR101 or MCR301 rheometer. A parallel plates system was used to perform frequency, strain and time sweeps and a cone and plate system to perform viscosity measurements. For frequency and strain tests, 1.5–2 mL of the gels were prepared in plastic moulds as described above. For time sweeps, 450 μL of gel was prepared on the plate and, once the top plate was lowered, mineral oil was placed around the plate to prevent solution from drying out. For viscosity measurements samples were prepared at high pH as described above, and 2.1 mL of solutions were transferred onto the plate for measurement. All experiments were performed at 25 °C.pH Measurements
A calibrated FC200 pH probe (HANNA instruments) with a 6 mm × 10 mm conical tip was used for pH measurements. The stated accuracy of the pH measurements is ±0.1. The apparent pKa of the gelators were determined via titration of the gelator solutions (5 mg mL−1), prepared as described above, with a 0.1 M HCl solution. pH measurements were recorded after each addition of HCl until a stable value was reached. To prevent the gel from forming, the solutions were stirred continuously. Alternatively, to monitor the pH changes during the gelation process, 2 mL of a solution of gelator, prepared as described above, was added to a pre-weighted amount of GdL. After swirling to ensure dissolution of the GdL, the sample was placed in a circulating water bath at 25 °C and the pH measured every 0.5 minutes. In this case the sample was not stirred.Confocal microscopy
A Zeiss LSM510 on a Zeiss Observer Z1 (Zeiss, Jena, Germany) was used for imaging. The gel samples were prepared at a concentration of gelator of 5 mg mL−1 containing Nile blue (40 μL mL−1 of a 0.1 wt% solution) in CELLview Culture dishes, (35 mm diameter) and were excited at 633 nm and detected with a Zeiss Meta detector. A spectral filter of 650–710 nm was used to obtain the Nile blue emission. Data were captured using Zeiss Zen software (Zeiss, Jena, Germany) and analysed using Zeiss LSM image browser (version 4.2.0.121). Either a ×20 or ×100 lens was used.SEM Imaging
SEM images were obtained using a Hitachi S-4800 FE-SEM at 3 keV. Gel was deposited onto glass cover slips that were fixed onto aluminium 15 mm SEM stubs with carbon tabs and left to dry for 24 hours. Samples were not gold coated but measured at 1.5 kV in deceleration mode.UV-Vis absorption measurements
Solution UV-Vis absorption data was measured using a Thermo Scientific Nanodrop 2000/2000c spectrophotometer. The spectrophotometer was used in cuvette mode. Samples were prepared in PMMA plastic cuvettes with a path length of 1.0 cm.UV-Vis absorption data of the gels were obtained using a Shimadzu UV-2550 UV-Vis spectrophotometer running the UV Probe software, version 2.34. Spectra were measured with scan speed set to medium and using a slit width of 5.0 nm in absorption mode. UV-Vis of the gels were measured in 0.1 mm path length quartz demountable cuvettes. 100 μL of pre-gelation solution containing GdL was transferred to the cuvette while still liquid. The cuvette was sealed with Parafilm and the sample allowed to gel overnight before the spectrum was recorded.
Fluorescence measurements
Emission spectra were recorded using a PerkinElmer Luminescence spectrometer LS55 with a scan rate of 100 nm min−1 and at 25 °C. Solutions for fluorescence measurements were prepared as described above, diluted as specified in each experiment below and transferred into a PMMA fluorescence cuvette with a path length of 1 cm. To monitor gelation a pre-weighed amount of GdL was added to the gelator solution, prepared as described above, in a PMMA cuvette. The top was covered with Parafilm and allowed to gel overnight while spectra were recorded periodically.Fourier transformed infrared (FTIR) spectroscopy
IR spectra were collected on a Bruker FTIR at 2 cm−1 resolution by averaging over 64 scans. Hydrogels were prepared as described above. The hydrogels were loaded onto a CaF2 window, and another CaF2 window was placed on the top of the gels. Each spectrum was background subtracted. The peak intensities were obtained by fitting the spectra with PEAKFIT software using Gaussian functions. The fitting coefficients were all above 0.98.Acknowledgements
AMC, LLEM, and ERD thank the EPRSC for funding. DJA thanks the EPSRC for a Fellowship (EP/L021978/1). MW thanks the EPSRC for funding and Unilever for a Case Award. The NMR spectrometers used for this work were funded by the EPSRC (EP/K039687/1 and EP/C005643/1). Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beamtime allocation (RB1520075) from the STFC. We thank Steve King for additional help with the SANS work. This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547.References
-
(a) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed
; (b) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed
-
(a) Q. Lin, T.-T. Lu, X. Zhu, B. Sun, Q.-P. Yang, T.-B. Wei and Y.-M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC
-
(a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed
-
(a) H.-J. Schneider and R. M. Strongin, Acc. Chem. Res., 2009, 42, 1489–1500 CrossRef CAS PubMed
-
(a) F. Xie, L. Qin and M. Liu, Chem. Commun., 2016, 52, 930–933 RSC
-
(a) D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC
- H. A. M. Ardoña and J. D. Tovar, Bioconjugate Chem., 2015, 26, 2290–2302 CrossRef PubMed
- G. S. Vadehra, B. D. Wall, S. R. Diegelmann and J. D. Tovar, Chem. Commun., 2010, 46, 3947–3949 RSC
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC
-
(a) D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC
-
(a) M. L. Muro-Small, J. Chen and A. J. McNeil, Langmuir, 2011, 27, 13248–13253 CrossRef CAS PubMed
-
(a) L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed
-
(a)
M. Raue, A. Bernet, M. Küppers, S. Stapf, H. W. Schmidt, B. Blümich and T. Mang, in Intelligent Hydrogels, ed. G. Sadowski and W. Richtering, Springer International Publishing, 2013, vol. 140, ch. 4, pp. 45–51 Search PubMed
-
(a) E. Wikberg, T. Sparrman, C. Viklund, T. Jonsson and K. Irgum, J. Chromatogr. A, 2011, 1218, 6630–6638 CrossRef CAS PubMed
- B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed
- J. Raeburn, L. Chen, S. Awhida, R. C. Deller, M. Vatish, M. I. Gibson and D. J. Adams, Soft Matter, 2015, 11, 3706–3713 RSC
- http://www.molinspiration.com .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm01194b |
| This journal is © The Royal Society of Chemistry 2016 |