
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-network hydrogels improve pH-switchable adhesion†
Latifah
Alfhaid
a,
William D.
Seddon
b,
Nicholas H.
Williams
 b and
Mark
Geoghegan
*a
b and
Mark
Geoghegan
*a
aDepartment of Physics and Astronomy, Hounsfield Road, University of Sheffield, Sheffield S3 7RH, UK. E-mail: mark.geoghegan@sheffield.ac.uk
bDepartment of Chemistry, Brook Hill, University of Sheffield, Sheffield S3 7HF, UK
First published on 10th May 2016
Abstract
For environmentally-switchable adhesive systems to be reused repeatedly, the adhesive strength must not deteriorate after each adhesion cycle. An important criterion to achieve this goal is that the integrity of the interface must be retained after each adhesion cycle. Furthermore, in order to have practical benefits, reversing the adhesion must be a relatively rapid process. Here, a double-network hydrogel of poly(methacrylic acid) and poly[oligo(ethylene glycol)methyl ether methacrylate] is shown to undergo adhesive failure during pH-switchable adhesion with a grafted (brush) layer of polycationic poly[2-(diethyl amino)ethyl methacrylate], and can be reused at least seven times. The surfaces are attached at pH 6 and detached at pH 1. A single-network hydrogel of poly(methacrylic acid), also exhibits pH-switchable adhesion with poly[2-(diethyl amino)ethyl methacrylate] but cohesive failure leads to an accumulation of the hydrogel on the brush surface and the hydrogel can only be reused at different parts of that surface. Even without an environmental stimulus (i.e. attaching and detaching at pH 6), the double-network hydrogel can be used up to three times at the same point on the brush surface. The single-network hydrogel cannot be reused under such circumstances. Finally, the time taken for the reuse of the double-network hydrogel is relatively rapid, taking no more than an hour to reverse the adhesion.
Introduction
Switchable adhesion refers to the ability of a material or couple to change adhesion properties in situ by some environmental (but not mechanical, electrical, or magnetic) stimulus, be it pH,1–5 temperature,4,6–8 the ionic strength of the surrounding medium,9 or light.10 In most of these cases, a polymer conformational transition instigates the reversible adhesion. An environmental switchable adhesive must be shown to bond and debond in situ as those environmental conditions are changed. For an environmental pH-switchable adhesive, this would generally require making the adhesive interaction in water, and, whilst the two components are in contact, this can be reversed by changing the pH by adding an acid, base, or salt. At some value of pH or ionic strength, the adhesion will fail and the two components will detach. The importance of developing a robust and durable switchable adhesive system is related to the numerous applications of technology based upon the phenomenon, such as recycling4 and wound dressing.10An environmentally-switchable adhesive will also debond under the conditions at which adhesion is strong if sufficient mechanical strain is applied. Such a process generally results in the cohesive failure of one or both of the components,3,7 with a deleterious effect on subsequent adhesive behaviour. Designing a system immune from cohesive failure has important advantages because it may undergo a number of bonding–debonding cycles, with commensurate benefits on its commercial appeal, so a test of repeatability under conditions of adhesion is of considerable interest. Of course, some switchable adhesive systems are only designed to be used once,7,10 and this capability is unimportant for these systems.
Grafted polymer layers (known as brushes) have a number of applications such as in colloidal stabilization,11 compatibilization between organic and inorganic materials,12–14 control of cell attachment and detachment,6,15 and lubrication.16 Previous work has shown that the adhesion between two thin grafted oppositely charged polymer layers exhibits a relatively clean surface after detaching the two components with the structural integrity of the brush layers remaining good, although there was a continuous decrease in strength between the two components after three attachment–detachment cycles.9 Again using two grafted polymer layers, the reproducibility of the adhesion could be improved by using a polyzwitterion,8 rather than polyelectrolytes.
Hydrogels are networks of water-soluble polymers, the physical properties of which are controlled by their complex interconnected structure.17 They are generally better adapted for conformal contact with larger and rougher substrates than brushes, but their greater volume implies a longer equilibration time and thus a longer time for the adhesion to be reversed. Nevertheless, in the interaction of a hydrogel with a brush, it is sufficient that only the gel is adapted for conformal contact. Using a neutral (polyacrylamide-based) hydrogel and a polyanionic brush no damage was reported after debonding, but the time taken to reverse the adhesion was on the order of hours,5 as was also the case for the earlier work on oppositely charged polyelectrolytes.1,2
Double-network (DN) hydrogels are networks of a heavily crosslinked polyelectrolyte reinforced with a second network of a more lightly crosslinked hydrophilic polymer. The second network imparts much greater fracture resistance on the first, which is generally very brittle.18,19 This gives these double networks great potential in load-bearing technologies, and may be particularly important in areas of medical science. These networks may have useful properties, such as low friction20 and good biocompatibility.21 However, they are a new class of material so their properties are still being understood. Although DN hydrogels are a form of interpenetrating network which requires that the two components be chemically discrete, it may be that some of their properties are due to bonds between the different networks.22
Here, a double-network hydrogel of poly(methacrylic acid) (PMAA) and poly[oligo(ethylene glycol)methyl ether methacrylate] (POEGMA) is compared with a single-network (SN) PMAA hydrogel to assess its potential for switchable adhesion. This is done by testing environmental pH-switchable adhesion repeatedly on the same system, through the adhesion at pH 6 and retraction at pH 1. Re-adhesion occurs at different points on the brush surface. It is noted that DN hydrogels are stronger and more resistant to cohesive failure than SN hydrogels, although SN hydrogels do demonstrate good switchable adhesion performance. Data are also presented showing the repeatability of adhesion at pH 6, where the two components are attached and detached from the same point of the surface at the same pH. Besides demonstrating the applicability of the relatively new technological development of DN hydrogels to adhesion, the results more specifically demonstrate a vastly improved repeatability both for environmentally switchable adhesion and for repeatable adhesion when compared with the SN hydrogel equivalents. The results therefore indicate that, for excellent reversible adhesion properties involving hydrogels, the use of double networks is to be preferred.
For clarity, environmental pH-switchable adhesion is referred hereon as switchable adhesion, and the experiments performed at the same point on the surface at only pH 6 are denoted as tests of repeatable adhesion.
Experimental
Preparation of the single-network hydrogel
The PMAA (SN) hydrogel was prepared using free radical polymerization with water (100 mL, 5.5 mol), methacrylic acid (MAA; 98% Aldrich; 20 mL, 0.23 mol), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AMPA; 98% Aldrich; 0.03 g, 0.1 mmol), and N,N′-methylenebisacrylamide (MBA; 99% Aldrich; 0.06 g, 0.4 mmol). The resulting gel was placed in a mould in an oven at 80 °C for 2 h, and was then cut into hemispherical pieces and kept under DI water for at least three days before using them in adhesion experiments. The water was changed at least twice over this time.Preparation of the double-network hydrogel
The PMAA-POEGMA (DN) hydrogel was synthesized by first making the PMAA hydrogel and then immersing this hydrogel inside a solution of oligo(ethylene glycol)methyl ether methacrylate (OEGMA) which was also polymerized. A more densely crosslinked PMAA hydrogel from that used for the SN hydrogels was required. The ratio of water to MAA was kept the same, although a different initiator was used. The need for a more densely crosslinked PMAA gel in the DN hydrogel has been detailed elsewhere,18 where it is stated that the second network must be at a significantly greater concentration than the first network and must be loosely crosslinked with respect to the polyelectrolyte network.The first (PMAA) hydrogel was prepared using the same free radical polymerization method as for the SN hydrogel, but with different quantities: water (50 mL, 2.8 mol), MAA (98% Aldrich; 10 mL, 0.12 mol), potassium persulfate (KPS; 99% Aldrich; 0.13 g, 0.48 mmol), and MBA (0.94 g, 6 mmol). The mixture was placed inside an oven at 60 °C for 6 h, and the resultant gel was cut into hemispherical pieces and kept under DI water for 3 days. The water was changed at least twice over this time.
The PMAA gel was then immersed inside an OEGMA solution for 5 days. The OEGMA solution was made of water (60 mL, 3.3 mol), OEGMA with a number average molar mass of 950 Da (Aldrich; 12 g, 0.013 mol), KPS (0.04 g, 0.15 mmol), and MBA (0.02 g, 0.13 mmol). After this immersion, the gel was placed inside an oven at 60 °C for 6 h and then stored under DI water.
Polymer brush preparation
Poly[2-(diethyl amino)ethyl methacrylate] (PDEAEMA) brushes were grown from silicon substrates using surface-initiated atom-transfer radical polymerization (SI-ATRP). The silicon substrates used were bought from Prolog Semicor and had the following characteristics: diameter 50 mm, type dopant p-type boron, orientation (100) ± 1°, thickness 4000 ± 50 μm. The cleaned silicon substrates were immersed for 30 min in a 2% (v/v) solution of (3-aminopropyl)triethoxysilane (APTES; 98% Aldrich) in ethanol.23 The substrates were rinsed with ethanol, dried under nitrogen gas and then annealed for 30 min at 120 °C. The surface was functionalized with initiator by immersing the APTES-coated silicon substrates in a mixture of triethylamine (99% Aldrich; 0.41 mL, 3 mmol) and α-bromoisobutyryl bromide (98% Aldrich; 0.37 mL, 3 mmol) in dichloromethane (DCM) (99% Aldrich; 100 mL, 1.4 mol) for 30 min before being washed with ethanol and DCM and dried by a stream of nitrogen gas.24 For SI-ATRP,25 the following materials were used: 2-(diethyl amino)ethyl methacrylate (DEAEMA; 99% Aldrich; 10.84 mL, 54.0 mmol), 2,2′-bipyridine (bipy; 99% Aldrich; 0.39 g, 2.5 mmol), copper(I) bromide (CuBr; 98% Aldrich; 0.12 g 0.9 mmol), copper(II) bromide (CuBr2; 99% Aldrich; 0.06 g, 0.3 mmol), methanol (8 mL, 0.20 mol), and DI water (2 mL, 0.11 mol). The DEAEMA monomer, methanol and water were degassed separately inside round-bottom flasks for 30 min using nitrogen gas. The methanol and water were transferred into the monomer flask and then CuBr, CuBr2, and bipy were added. The solution was stirred and degassed with nitrogen for 30 min and then transferred into a glass container with the silicon substrate and left at room temperature for 24 h. Finally, the substrate was washed with methanol and ethanol, and then dried under a stream of nitrogen gas. The thickness of the dry brush layers was characterized using ellipsometry and found to be between 70 and 80 nm.Adhesion measurements
Measurements of the adhesion between PDEAEMA brushes and the single- and double-network hydrogels were made using a mechanical tester (Stable Micro Systems Texture Analyser TA.XTplus). The tester comprises a mechanical probe used to fix a hemispherical hydrogel inside its plastic jacket and a platform on which the brush substrate is placed. The mechanical probe brings the hydrogel into contact with the polymer brush surface with a force of 0.1 N. The gel was left in contact with the brush at this force for 2 min before being retracted at a speed of 50 mm min−1. For experiments whereby the pH was changed, the samples were allowed to equilibrate at the new pH before being retracted. The interface was illuminated and side-view images of the interface were taken using a camera. These measurements were performed at room temperature, 23 ± 2 °C.Results and discussion
Characterization of the hydrogels
The elastic modulus, K, of the gels was obtained by using the Hertz equation,26| a3 = PR/K, | (1) |
| pH | Modulus DN gel (MPa) | Swelling ratio DN gel | Modulus SN gel (MPa) | Swelling ratio SN gel |
|---|---|---|---|---|
| 1 | 1.18 ± 0.02 | 1.4 ± 0.1 | 0.42 ± 0.08 | 7.3 ± 0.3 |
| 5.8 | 1.09 ± 0.01 | 6.5 ± 0.6 | 0.30 ± 0.04 | 13.6 ± 0.4 |
Repeatable adhesion without environmental stimulus
Sample adhesion results for DN and SN hydrogels are shown in Fig. 1a, where a hemispherical hydrogel is retracted from a planar brush surface after being placed in contact for 2 min with a force of 0.1 N. It is immediately clear that the DN hydrogel shows significant adhesion at pH 6. The SN PMAA gel also adheres well to the PDEAEMA brush layer (with a dry thickness of 78 nm), but not to the same degree. At pH 1, there is no adhesion for either the single or double network, although the SN hydrogel does exhibit a long-range repulsive interaction over ∼1 mm, whereas the DN hydrogel has virtually no interaction with the brush. The long-range repulsive interaction between the uncharged SN hydrogel and the brush is likely to be a result of significant deformation in the SN hydrogel.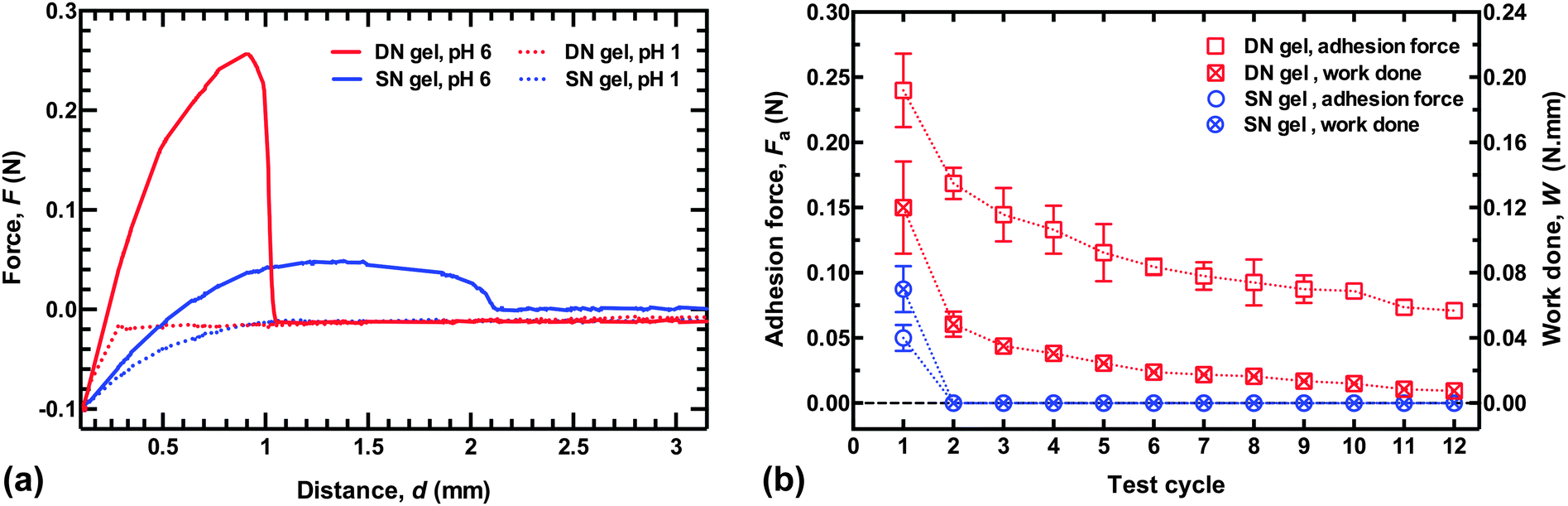 | ||
| Fig. 1 (a) Force as a function of retraction distance for hydrogel probes from a PDEAEMA brush surface. The hydrogels were placed in contact and a force of 0.1 N was applied 2 min before being retracted at a constant speed of 50 mm min−1. The zero distance corresponds to the point at which the load is applied. No adhesion force is observed at pH 1, but significant adhesion is seen at pH 6 for the DN hydrogel. (b) Repeated adhesion, where the abscissae represent the measurement number. The work done is the (positive) area under the force–distance curve. | ||
To test the quality of the repeated adhesion after debonding, force–distance curves were measured in deionized (DI) water. The probe was in contact with the brush for 2 min with a force of 0.1 N at pH 6, and then a retraction curve was measured at the same pH, before the probe was brought back to the same location (±1 mm). The probe was again left in contact with a force of 0.1 N for 2 min before being retracted at 50 mm min−1. The DN hydrogel could be retracted from the surface up to four times before the adhesion fell to below half the value of the first retraction (Fig. 1b). Even after twelve cycles, the adhesion force of the DN hydrogel was greater than that for the SN PMAA hydrogel after its first removal. The adhesion of the DN hydrogel remained repeatable over several cycles, while the adhesion of the SN hydrogel could not be repeated. The work done against attractive brush–gel interactions in retracting the gel from the brush layer was calculated from the area under the force–distance curve (for F > 0), and this decreased more rapidly with the number of cycles. Nevertheless, this was also substantially greater for the DN hydrogel than for the SN hydrogel. In real-world applications, for a given geometry the force required for debonding is likely to be a more useful parameter than the work done in separating the two components.
The reason that the SN hydrogel fails to achieve repeated adhesion is clear from optical microscopy images of the brush surfaces after the probe has been detached (Fig. 2). These show that the SN hydrogel had clearly undergone cohesive failure, leaving PMAA on the surface. This surface is now negatively charged, like the probe, and the two like-charged surfaces would therefore experience no adhesion. In contrast, the DN hydrogel left barely a mark on the surface, and signs of the contact event are not evident even after visualization with scanning force microscopy (Fig. 3).
 | ||
| Fig. 2 Optical microscope images of PDEAEMA brush layers after adhesion to the SN hydrogel (a) and the DN hydrogel (b). The scale is the same in both images and the bar represents 1 mm. Cohesive failure is clearly visible on the first brush resulted after detaching from the SN hydrogel at pH 6, whereas the DN hydrogel leaves no visible mark on the film. These images were taken after one test in DI water. | ||
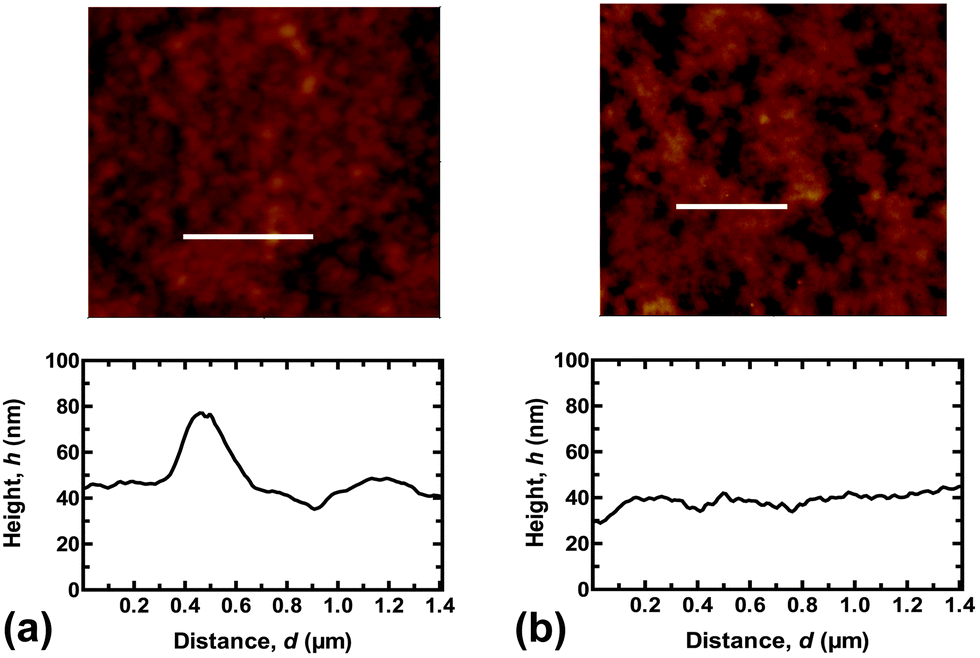 | ||
| Fig. 3 Scanning force microscopy images of the PDEAEMA brush surfaces (a) before and (b) after contact with the DN hydrogel. The surface morphology remains unaltered, but there is a small decrease in the root-mean-square roughness after the experiment from 9.8 to 5.6 nm. These images were taken after one test cycle at pH 6. The height profiles are taken from the white line on each image. | ||
Environmental pH-switchable adhesion
Environmentally-switchable adhesion between the polycationic brush layer and the two types of hydrogels was examined using a pH cycle. Adhesion between the two components was made at pH 6 by applying a force of 0.1 N for 2 min. The force was then removed, but the two components remained in contact. HCl was then carefully added to the solution with (slow) agitation by a magnetic stirrer until pH 1 was reached. The adhesion couple was left in contact for a further hour to equilibrate, still with no applied force; the gel was then retracted at 50 mm min−1, and the adhesion force measured. The system was washed with copious DI water to return it to pH 6 and the cycle was repeated. For these experiments the adhesion was measured at a different point on the sample after each cycle, i.e. the gel was brought into contact with a new area of brush surface that had not previously been in contact with the gel. Sample adhesion data and a summary of the results of this cycling are presented in Fig. 4. Here, the difference between the DN and SN hydrogels is considerably less; both gels were attached and detached from the brush surface seven times. After these seven cycles the gels were attached and detached in DI water at pH 6 to measure their final adhesion. Although the DN hydrogel had a pull-off force approximately twice that of the SN hydrogel (0.2 N), the work done in detaching both gels was similar.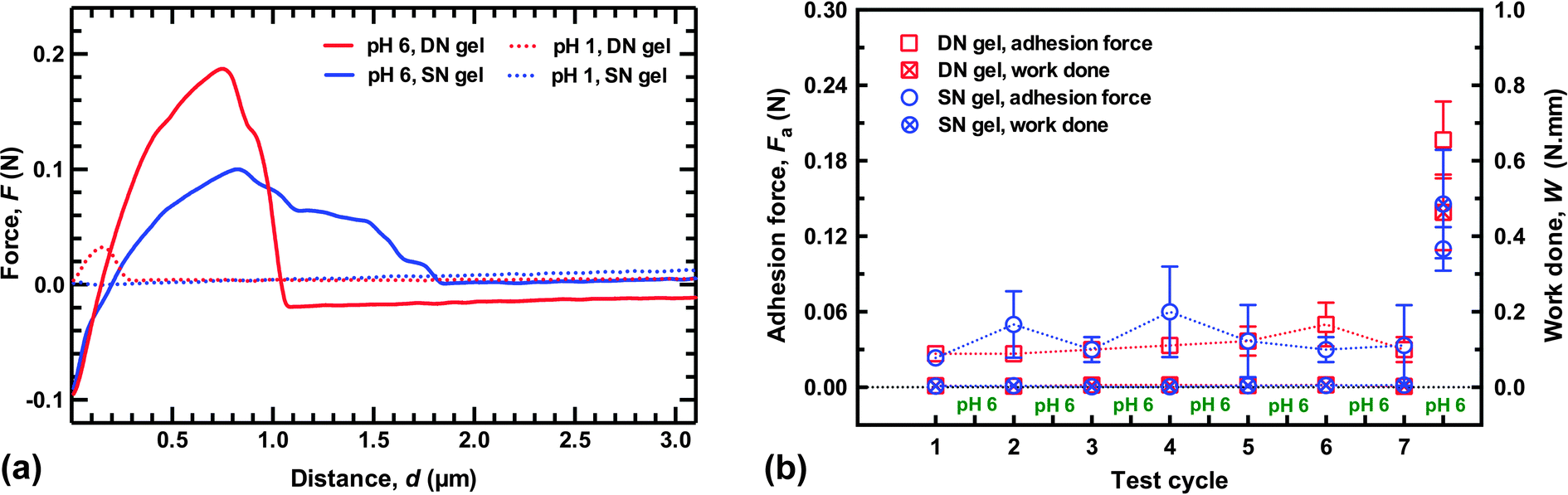 | ||
| Fig. 4 (a) Typical adhesion data for environmentally-switchable adhesion experiments. The pH 1 and pH 6 values are taken from the end of the experiment, i.e. the final removal of the probe at pH 1, and the ultimate detachment at pH 6. (b) Switchable adhesion results of the SN and DN hydrogels with the PDEAEMA brush layer (with thickness of 76 nm). Each datum represents an average of three different values from different points on the brush surface. The gel and brush were brought into contact underwater at pH 6 and removed at pH 1. Cycle 7.5 corresponds to their ultimate removal at pH 6 after the seventh cycle. The DN and SN hydrogels behave similarly during detachment at pH 1, although after the final detachment at pH 6 there is a difference in the maximum adhesion force of nearly a factor of two. | ||
It is worth noting that the DN hydrogel could be removed after only an hour without damaging the surface of the brush indicates that the DN hydrogel reduces considerably the time required to allow the adhesion to be reversed compared to the previous work,1,2 and also for a larger load than reported in those experiments. (It should be noted that in the earlier experiments, adhesive failure occurred without the external input used here, which should be considered in any comparison of times.) Although the DN hydrogels described here were detached after an hour, experiments performed after leaving the adhered couple in pH 1 solution for 20 minutes required a similar pull-off force and exhibited no damage when viewed under an optical microscope (Fig. 5). These results indicate that these DN hydrogels are appropriate for practical applications.
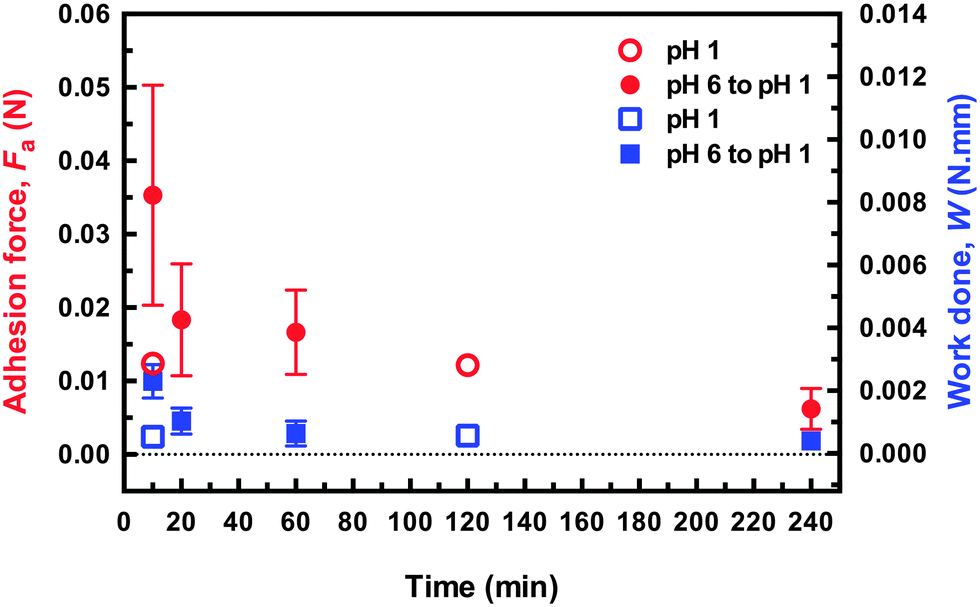 | ||
| Fig. 5 Adhesion force and work done in removing the DN hydrogel probe from the brush surface for different times after immersion in pH 1 solution for samples that were either brought into contact at pH 1 or pH 6. The applied force in both cases was 0.1 N, and was applied 2 min. The force–distance curves for these data are shown in the ESI.† | ||
Given the significant difference in performance of the two hydrogels without an environmental stimulus, it is worth asking why the switchable adhesion performance of the two networks is similar. From the data shown in Fig. 4a, it is clear that the DN hydrogel had very little interaction with the brush surface at pH 1 compared to the SN hydrogel. In fact, the DN hydrogel retains its shape during the attachment–detachment cycles whereas the SN hydrogel gradually deforms due to repetitive minor cohesive failures at the interface (Fig. 6). (For the experiments testing repeatability, the SN hydrogel did not experience a change in contact diameter because it was brought into contact with the area where it had previously undergone cohesive failure.) The increased contact area does not appear to create a stronger interface on subsequent attachments, which is likely to be due to a non-conformal surface left by a previous cohesive failure. During detachment the polyelectrolyte fragments into small regions dissipating energy, whilst the supporting neutral hydrogel supports a large deformation.21,27 As this occurs the system becomes softer and more malleable, allowing a gentle detachment from the surface. The SN hydrogel has no mechanism to dissipate stress around a weak point and so the gel fractures. Images of the fractured gel are shown as a function of cycle number in Fig. 6; in the same figure, images show that the DN hydrogel retains its hemispherical form. Experiments on brush–brush contact mechanics suggest that the interactions between oppositely charged polyelectrolytes are mainly due to electrostatic interactions;28 however in the present experiments the contact is not between a brush film and a brush-coated atomic force microscope tip, but rather a film and a macroscopic hydrogel. Furthermore a regime whereby electrostatic interactions dominate is likely to be restricted to relatively low pressures because excluded volume effects would otherwise be expected. PMAA hydrogels deswell when pH is decreased, and the resultant tension on the area of contact due to this deswelling will affect the debonding process. The DN hydrogel debonding may well be controlled by the POEGMA network dissipating energy around the contact region as the PMAA deswells. For the SN hydrogel, a pH-controlled contraction of the gel where it is in contact with the brush occurs, but without this additional mechanism for energy dissipation. Since the adhesive contacts between the gel and the brush are expected to be strong, the SN hydrogel undergoes cohesive failure initiated at the weakest points within it.
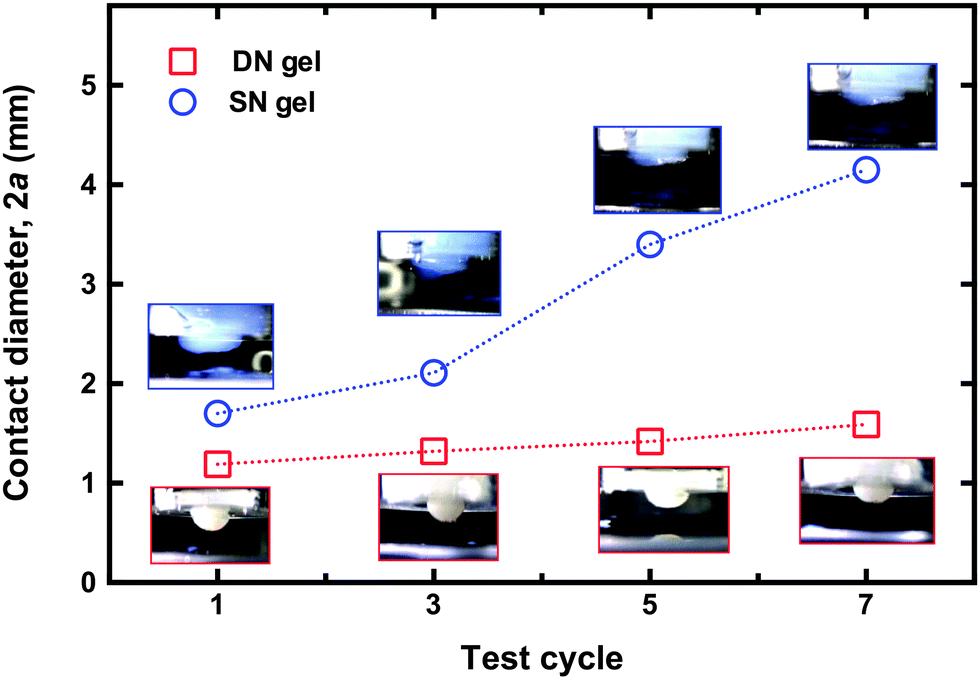 | ||
| Fig. 6 Contact diameter as measured from the flat part of each gel after removal from the brush at pH 1, an hour after the two components were brought into contact at pH 6. There is a marked increase in that for the SN hydrogel, whereas that for the DN hydrogel remains relatively constant. The insets show optical microscopy images for the SN gel after detaching it from the brush surface. Each image is shown above (SN hydrogel) or below (DN hydrogel) the corresponding datum. For scale purposes, the gel is housed in a white plastic jacket, which is 10 mm in diameter. The size of the interface due to cohesive failure is increasing with each detachment for the SN hydrogel, whilst the DN hydrogel is seen to retain its shape despite the numerous cycles. | ||
Because these environmental switchable adhesion measurements were performed on fresh regions of brush surface after each attachment–detachment cycle, the SN hydrogel remains viable. Were these experiments to have been performed on the same point, as was the case for the repeatable adhesion experiments, the SN hydrogel would not have been usable more than once. Furthermore, because the point of failure is not the interface between the brush and the gel, the mechanical properties of the gel are critical to the effectiveness of the adhesion couple.
Optimization of the hydrogels
These experiments provide different values for the adhesion of a hydrogel with an oppositely charged brush. It is likely that both the DN and SN hydrogels could be further optimized for these measurements, so no claim is made that the adhesion is the best that can be achieved. Factors influencing the properties of the hydrogels involve the degree of crosslinking and the water concentration during synthesis. Both of these influence the rigidity and swelling capacity of the hydrogel. The SN gel is softer than but otherwise similar to that used in previous work,1,2 and represents a good model for comparison. The balance between crosslink density and adhesion for PMAA is delicate; at large crosslink densities, it is possible that adhesive failure replaces cohesive failure as the mechanism of debonding of the gel from the brush, but the adhesion in these circumstances is rather weak.3 At smaller crosslink densities, it is possible that the adhesion may increase, although this would require unacceptable stretching of the gel during debonding for any realistic application, and cohesive failure would certainly result. The DN hydrogel was initially synthesized following a method described previously,29 but had to be adjusted because it was liable to stretch excessively under strain, with resulting cohesive failure of the DN hydrogel. (An example video and sample images of a less crosslinked DN hydrogel is included in the ESI;† the gel stretched without cavitation or fibrillation, and can even start to remove the brush from the silicon substrate.) The DN hydrogel used here is less soft than in the earlier work29 because an increased amount of crosslinker was used. As an aside, it is noted that POEGMA, which is a component of the DN hydrogels could be susceptible to hydrolysis over extended periods of time, which might limit the performance of the present system. However, similar esters are expected to have a half-life of about a day at pH 1, and to be even more stable at pH 6.30,31Conclusion
A DN hydrogel incorporating PMAA is shown to have strong adhesion to a PDEAEMA brush, displaying a resistance to cohesive failure that is absent from a comparable SN hydrogel. The POEGMA mesh of the PMAA network provides a resilience that allows the adhesion couple to be detached even at pH 6 (the pH at which it was adhered to the oppositely charged brush) and then reattached several times on the same place on the brush surface. The DN hydrogel remains intact when it is used for pH-dependent switchable adhesion after several adhesion cycles. In both cases the model SN hydrogel exhibits poorer performance. The SN hydrogel cannot be reused at the same point of contact after detaching at pH 6, due to extensive cohesive failure that resulted after the first retraction test, although it can be used for repeated pH-environmental switchable adhesion on a different area in the brush, where a rough and non-conformal contact caused by cohesive failure in the SN hydrogel is compensated by an increasing interfacial area to maintain performance. Here the increased area of the gel results from bringing the remaining SN gel into contact with fresh brush. Overall, it has been shown that because of their propensity for adhesive failure, the DN hydrogels are an effective means of controlling reversible and repeatable adhesion in pH-responsive systems.Acknowledgements
The Ministry of Education of the Kingdom of Saudi Arabia represented by Ha'il University is acknowledged for a PhD scholarship for LA. The EPSRC is acknowledged for a PhD scholarship for WDS. MG thanks Professor Costantino Creton (ESPCI ParisTech) for a useful discussion. LA thanks Dr Sina Naficy of the University of Wollongong for his advice concerning the synthesis of the double-network hydrogel.References
- R. La Spina, M. R. Tomlinson, L. Ruiz-Pérez, A. Chiche, S. Langridge and M. Geoghegan, Angew. Chem., Int. Ed., 2007, 46, 6460–6463 CrossRef CAS PubMed.
- R. La Spina, A. Chiche, M. R. Tomlinson and M. Geoghegan, Eur. Coat. J., 2011, 22–28 CAS.
- R. La Spina, PhD thesis, University of Sheffield, 2010.
- P. S. Shuttleworth, J. H. Clark, R. Mantle and N. Stansfield, Green Chem., 2010, 12, 798–803 RSC.
- G. Sudre, L. Olanier, Y. Tran, D. Hourdet and C. Creton, Soft Matter, 2012, 8, 8184–8193 RSC.
- J. Collett, A. Crawford, P. V. Hatton, M. Geoghegan and S. Rimmer, J. R. Soc., Interface, 2007, 4, 117–126 CrossRef PubMed.
- R. S. Gurney, D. Dupin, J. S. Nunes, K. Ouzineb, E. Siband, J. M. Asua, S. P. Armes and J. L. Keddie, ACS Appl. Mater. Interfaces, 2012, 4, 5442–5452 CAS.
- M. Kobayashi and A. Takahara, Polym. Chem., 2013, 4, 4987–4992 RSC.
- M. Kobayashi, M. Terada and A. Takahara, Soft Matter, 2011, 7, 5717–5722 RSC.
- C. G. Rolli, H. Nakayama, K. Yamaguchi, J. P. Spatz, R. Kemkemer and J. Nakanishi, Biomaterials, 2012, 33, 2409–2418 CrossRef CAS PubMed.
- E. P. K. Currie, W. Norde and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2003, 100–102, 205–265 CrossRef CAS PubMed.
- F. Brochard-Wyart, P. G. de Gennes, L. Léger, Y. Marciano and E. Raphael, J. Phys. Chem., 1994, 98, 9405–9410 CrossRef CAS.
- M. Geoghegan, C. J. Clarke, F. Boué, A. Menelle, T. Russ and D. G. Bucknall, Macromolecules, 1999, 32, 5106–5114 CrossRef CAS.
- K. P. O’Connor and T. C. B. McLeish, Macromolecules, 1993, 26, 7322–7325 CrossRef.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- T. Kreer, Soft Matter, 2016, 12, 3479–3501 RSC.
- W. Richtering and B. R. Saunders, Soft Matter, 2014, 10, 3695–3702 RSC.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- C. Creton and M. Ciccotti, Rep. Prog. Phys., 2016, 79, 046601 CrossRef PubMed.
- K. Arakaki, N. Kitamura, H. Fujiki, T. Kurokawa, M. Iwamoto, M. Ueno, F. Kanaya, Y. Osada, J. P. Gong and K. Yasuda, J. Biomed. Mater. Res., Part A, 2010, 93, 1160–1168 Search PubMed.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- T. Nakajima, H. Furukawa, Y. Tanaka, T. Kurokawa, Y. Osada and J. P. Gong, Macromolecules, 2009, 42, 2184–2189 CrossRef CAS.
- S. Alang Ahmad, A. Hucknall, A. Chilkoti and G. J. Leggett, Langmuir, 2010, 26, 9937–9942 CrossRef CAS PubMed.
- S. Alang Ahmad, G. J. Leggett, A. Hucknall and A. Chilkoti, Biointerphases, 2011, 6, 8–15 CrossRef PubMed.
- L. A. Fielding, S. Edmondson and S. P. Armes, J. Mater. Chem., 2011, 21, 11773–11780 RSC.
- H. Hertz, J. Reine Angew. Math., 1881, 92, 156–171 Search PubMed.
- T. Matsuda, T. Nakajima, Y. Fukuda, W. Hong, T. Sakai, T. Kurokawa, U. Chung and J. P. Gong, Macromolecules, 2016, 49, 1865–1872 CrossRef CAS.
- M. Raftari, Z. J. Zhang, S. R. Carter, G. J. Leggett and M. Geoghegan, Macromolecules, 2015, 48, 6272–6279 CrossRef CAS.
- S. Naficy, J. M. Razal, P. G. Whitten, G. G. Wallace and G. M. Spinks, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 423–430 CrossRef CAS.
- J. P. Guthrie, J. Am. Chem. Soc., 1973, 95, 6999–7003 CrossRef CAS.
- A. E. Rydholm, K. S. Anseth and C. N. Bowman, Acta Biomater., 2007, 3, 449–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the hydrogels, force–distance curves for the DN hydrogel for different times left at pH 1, and images showing a less-crosslinked DN hydrogel during detachment. A video of the weakly crosslinked DN hydrogel detaching the brush. See DOI: 10.1039/c6sm00656f |
| This journal is © The Royal Society of Chemistry 2016 |