
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics simulations of uniaxial deformation of thermoplastic polyimides†
V. M.
Nazarychev
a,
A. V.
Lyulin
b,
S. V.
Larin
a,
A. A.
Gurtovenko
ac,
J. M.
Kenny
ad and
S. V.
Lyulin
*ac
aInstitute of Macromolecular Compounds, Russian Academy of Sciences, Bol'shoi pr. 31 (V.O.), St. Petersburg, 199004, Russia. E-mail: s.v.lyulin@gmail.com
bTheory of Polymers and Soft Matter Group, Technische Universiteit Eindhoven, PO Box 513, 5600 MB Eindhoven, The Netherlands
cFaculty of Physics, St. Petersburg State University, Ul'yanovskaya str. 1, Petrodvorets, St. Petersburg, 198504, Russia
dMaterials Science and Technology Centre, University of Perugia, Loc. Pentima, 4, 05100 Terni, Italy
First published on 16th March 2016
Abstract
The results of atomistic molecular-dynamics simulations of mechanical properties of heterocyclic polymer subjected to uniaxial deformation are reported. A new amorphous thermoplastic polyimide R–BAPO with a repeat unit consisting of dianhydride 1,3-bis-(3′,4,-dicarboxyphenoxy)diphenyl (dianhydride R) and diamine 4,4′-bis-(4′′-aminophenoxy)diphenyloxide (diamine BAPO) was chosen for the simulations. Our primary goal was to establish the impact of various factors (sample preparation method, molecular mass, and cooling and deformation rates) on the elasticity modulus. In particular, we found that the elasticity modulus was only slightly affected by the degree of equilibration, the molecular mass and the size of the simulation box. This is most likely due to the fact that the main contribution to the elasticity modulus is from processes on scales smaller than the entanglement length. Essentially, our simulations reproduce the logarithmic dependence of the elasticity modulus on cooling and deformation rates, which is normally observed in experiments. With the use of the temperature dependence analysis of the elasticity modulus we determined the flow temperature of R–BAPO to be 580 K in line with the experimental data available. Furthermore, we found that the application of high external pressure to the polymer sample during uniaxial deformation can improve the mechanical properties of the polyimide. Overall, the results of our simulations clearly demonstrate that atomistic molecular-dynamics simulations represent a powerful and accurate tool for studying the mechanical properties of heterocyclic polymers and can therefore be useful for the virtual design of new materials, thereby supporting cost-effective synthesis and experimental research.
Introduction
Polyimides (PI) are heat-resistant polymers that are widely used in aircraft, airspace and mechanical engineering as all-purpose materials that can provide efficient performance under extreme environmental conditions and long-lasting load without significant degradation of their properties.1,2 Scientific interest in heat-resistant polymers, in general, and in aromatic polyimides, in particular, is also due to the fact that incorporation of some “hinge” groups (heteroatoms of oxygen, sulfur, sulfone group, etc.) into the repeat unit of a polymer chain can successfully be used to synthesize new thermoplastic polymers.1,2 Such polyimides and polyimide-based materials show both good mechanical properties3–12 and substantial resistance to high temperatures, chemicals and increased radiation levels.13However, due to the complex chemical structure of the diamine and dianhydride fragments of PI repeat units, the relationship between the chemical composition of the thermoplastic polyimides and their mechanical properties remains unclear.13 Furthermore, due to their complex chemical structure, the thermoplastic polyimides may show relatively low local translational and rotational mobilities of polymer chain fragments, in contrast to those observed for commodity polymers, such as polyethylene or polystyrene. Overall, these features can lead to specific changes in the relaxation patterns of different fragments of repeat units of thermoplastic polyimides subjected to an external deformation field. They can also significantly affect the correlation between their mechanical properties and temperature, pressure and other external factors, as compared to the corresponding correlations seen for polymers of simpler chemical structure.
Therefore, the synthesis of new polyimides requires large-scale experimental research, including an analysis of the effects of temperature and external pressure. Unfortunately, such experimental studies are quite resource-intensive.1,2 That is why computer simulation techniques used along with fully-atomistic computational models, are regarded as efficient and much cheaper methods for the comparative study of mechanical properties of new thermoplastic polyimides.
As far as the computer simulations of mechanical properties of polymers are concerned, it is still not clear how to correctly account for the influence of the thermal history (cooling rate14–16) and the load application rate (deformation rate17–29). Our study will focus exactly on these two issues, namely the sensitivity of the elasticity modulus to the cooling and deformation rates employed in simulations. This will allow us to determine the optimal values of the cooling and deformation rates to be used in simulations in order to appropriately model the thermomechanics of thermoplastic polyimides.
The next section of the paper focuses on the polymer materials in question and the simulation details, as well as on the uniaxial deformation strategy. The effects of the degree of equilibration, cooling and deformation rates, the molecular mass and the size of a periodic simulation cell on the elasticity modulus are studied in the subsequent sections of the paper. Finally, the influence of the temperature and external pressure on mechanical properties of the thermoplastic polyimides is reported and discussed.
Materials
Thermoplastic polyimide R–BAPO
The thermal properties of tetranuclear aromatic thermoplastic polyimides30–32 based on a dianhydride 1,3-bis-(3′,4,-dicarboxyphenoxy)diphenyl (dianhydride R) were investigated in our previous studies,33–36 while their mechanical properties were not studied computationally so far. To explore their mechanical properties we chose the polyimide R–BAPO36,37 (based on dianhydride R and a diamine 4,4′-bis-(4′′-aminophenoxy)diphenyloxide (diamine BAPO)) which was recently synthesized in the Institute of Macromolecular Compounds, Russian Academy of Sciences, see Fig. 1. | ||
| Fig. 1 Chemical structure of a repeat unit of the thermoplastic PI R–BAPO. | ||
Comparing three previously considered thermoplastic polyimides (R–BAPS, R–BAPB and R–BAPO),33–36 the choice of PI R–BAPO for studying the mechanical properties can mainly be justified by the following two reasons. First, in contrast to the partially crystallizing PI R–BAPB, the polyimide R–BAPO is fully amorphous, which allows us to rule out completely the effects related to the crystallization processes and investigate the mechanical properties of a fully amorphous polymer sample. Second, PI R–BAPO is characterized by a negligible anisotropy in the distribution of partial charges. This is in drastic contrast to PI R–BAPS, whose repeat unit contains a highly polar sulfone group that creates additional computational issues.
Methods
Simulation protocol
The simulation details related to the preparation of the equilibrated configuration can be found in our previous publications on the subject.33–36 Shortly, to generate the initial configuration, 27 polymeric chains with the degree of polymerization Np = 8 in the “polymer gas” state were compressed. After the compression step the pressure was reduced to 1 atm. To relieve the remaining residual stress, a cyclic annealing procedure was performed within the temperature range from 600 K to 300 K. The annealing was followed by equilibration for 2 μs at T = 600 K. Standard checks36 were used to prove that the equilibrium state has been reached. After that, a 1 μs production simulation was performed; instantaneous configurations of the system being saved every 100 ns. The selected 11 instantaneous states were used as initial states for the cooling procedure.The polymer samples were then cooled down to temperature of 290 K. In order to examine the influence of the simulated cooling rate γc on the mechanical properties, a cooling procedure was applied with five different values of γc, spanning the range from 1.5 × 1010 K min−1 to 1.5 × 1014 K min−1.
In order to reduce the required simulation time, we do not take into account the electrostatic interactions (EI)35 in the present study. Such an assumption is justified due to the rather small contribution of EI energy of PI R–BAPO to the total potential energy (∼25%),36 electrostatic interactions do modify the mechanical properties of PI R–BAPO but do not change the qualitative conclusions of the present study. Similar simulations without explicit EI are widely used in studies that dealt with polyimide mechanical properties;38–40 the possible influence of EI will be discussed in our future publication.
After the cooling step, each of the prepared 11 samples has been deformed with a stretching rate γd that was varied in the range from 10−1 nm ps−1 to 10−5 nm ps−1. For a periodic simulation cell of length of ∼5.8 nm, the corresponding deformation rate amounted to 1.8 × (106–1010) s−1. The simulations have been performed using the molecular-dynamics (MD) package Gromacs 4.5.641,42 and the force field Gromos53a5.43 All simulations required ∼1.6 million processor hours and were performed using 64 cores on Lomonosov (Intel Xeon X5570 CPUs) and Chebyshev (Intel Xeon E5472 CPUs) supercomputers at Moscow State University.
Simulation of mechanical properties
As mentioned above, the polymer samples were first cooled down to T = 290 K. This temperature is substantially lower than the glass-transition value of Tg ∼ 460–470 K36 measured in simulations of similar polyimides without EI. After cooling, the samples were subjected to uniaxial deformation following the deformation protocol proposed elsewhere for calculating the stress–strain dependences.44 Prior to the deformation, the LINCS algorithm45 that keeps the length of chemical bonds constant was disabled and the chemical bonds were described with the use of the harmonic potential from the same force field. After that, the simulated systems were additionally equilibrated for 400 ps. The LINCS algorithm had to be disabled due to the observed instability of simulations on a multi-processor system upon high-speed deformation. The density change after the LINCS deactivation did not exceed 1.5%.The uniaxial deformation changes the periodic cell size at a constant rate, along the positive direction of one of the reference axes – X, Y or Z –44 so that the isotropic Berendsen barostat46 with a time constant τp = 0.5 ps was replaced with the anisotropic Berendsen barostat with τp = 1 ps. In the direction of the applied deformation the samples remained incompressible, i.e. the compressibility of the system in this direction was set to zero. In the transverse direction the system compressibility was set to 4.5 × 10−10 Pa−1. Therefore, upon stretching a simulation cell elongates in the direction of deformation and compresses in the directions transversal to deformation in response to the external pressure (1 bar), see Fig. 2.
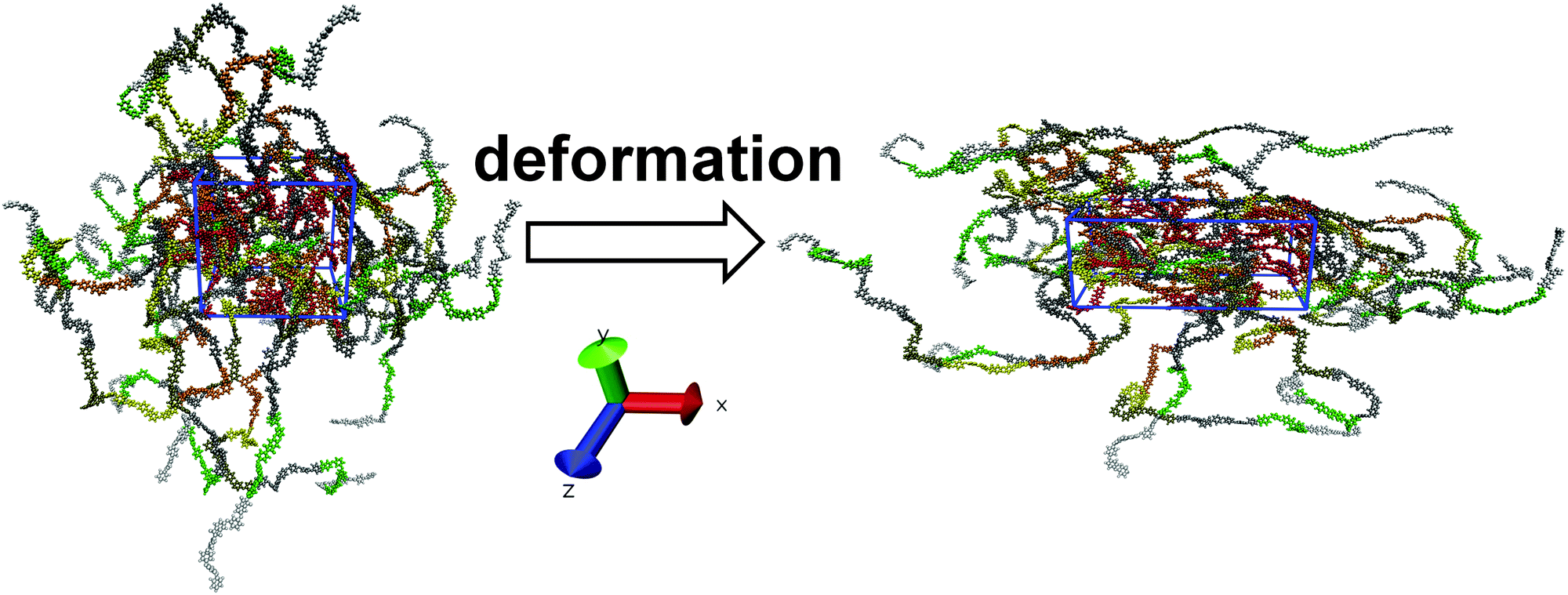 | ||
| Fig. 2 PI R–BAPO sample configuration before (left) and after (right) uniaxial deformation along the X axis. The periodic cell is highlighted in blue. | ||
During deformation the values of the pressure tensor Pi, i = x, y, z, and the simulation cell size Li in the stretching direction were saved each 1 ps. The characteristics obtained were converted to the dependence of the stress σ on the relative strain ε as47
| σ = −Pi, |
| ε = (Li − L0i)/L0i, | (1) |
| σ = Eε. | (2) |
Elastic properties can also be analyzed with the transversal dimensions fixed during the deformation.48 The value of the modulus determined in this way combines the Young's modulus E and the polymer bulk modulus, and differs from the elasticity modulus calculated using the present approach.
The yield peak σy (the onset of plastic deformation in the material) and the strain-hardening modulus Gh (the slope of the linear dependence σ(ε) in the post-yield deformation regime) were calculated as49–52
| σ = σy + Gh(λ2 − λ−1). | (3) |
Results
Influence of the degree of equilibration, cooling and deformation rates
Such behaviour of Young's modulus dependence on the degree of equilibration can be due to the preliminary annealing, where some equilibration of the samples in the glassy state has been done. This equilibration led to the rather weak dependence on the sample thermal history. Thus it might be sufficient for the accurate calculation of the mechanical properties in the present molecular-dynamics simulations.
Our findings also show that the modulus of elasticity for well-equilibrated polymer samples (Fig. S2 and Table S1, ESI†) hardly depends on their molecular mass (Fig. S3 and S4, ESI†). The dependence of mechanical properties on the size of a periodic simulation cell was examined as well, and no sufficient influence was observed (Table S2 and Fig. S5, ESI†). Such insensitivity of the elastic modulus is due to the fact that the modulus is mainly determined by the processes on the scales that do not exceed the entanglement length.
After switching off the LINCS algorithm, the samples obtained by cooling with five different rates γc from 1.5 × 1010 K min−1 to 1.5 × 1014 K min−1 were deformed at a stretching rate γd = 1.8 × 108 s−1, see Fig. 3a. The results show that the extracted values of the modulus E depend logarithmically on the cooling rate γc, see Fig. 3b.
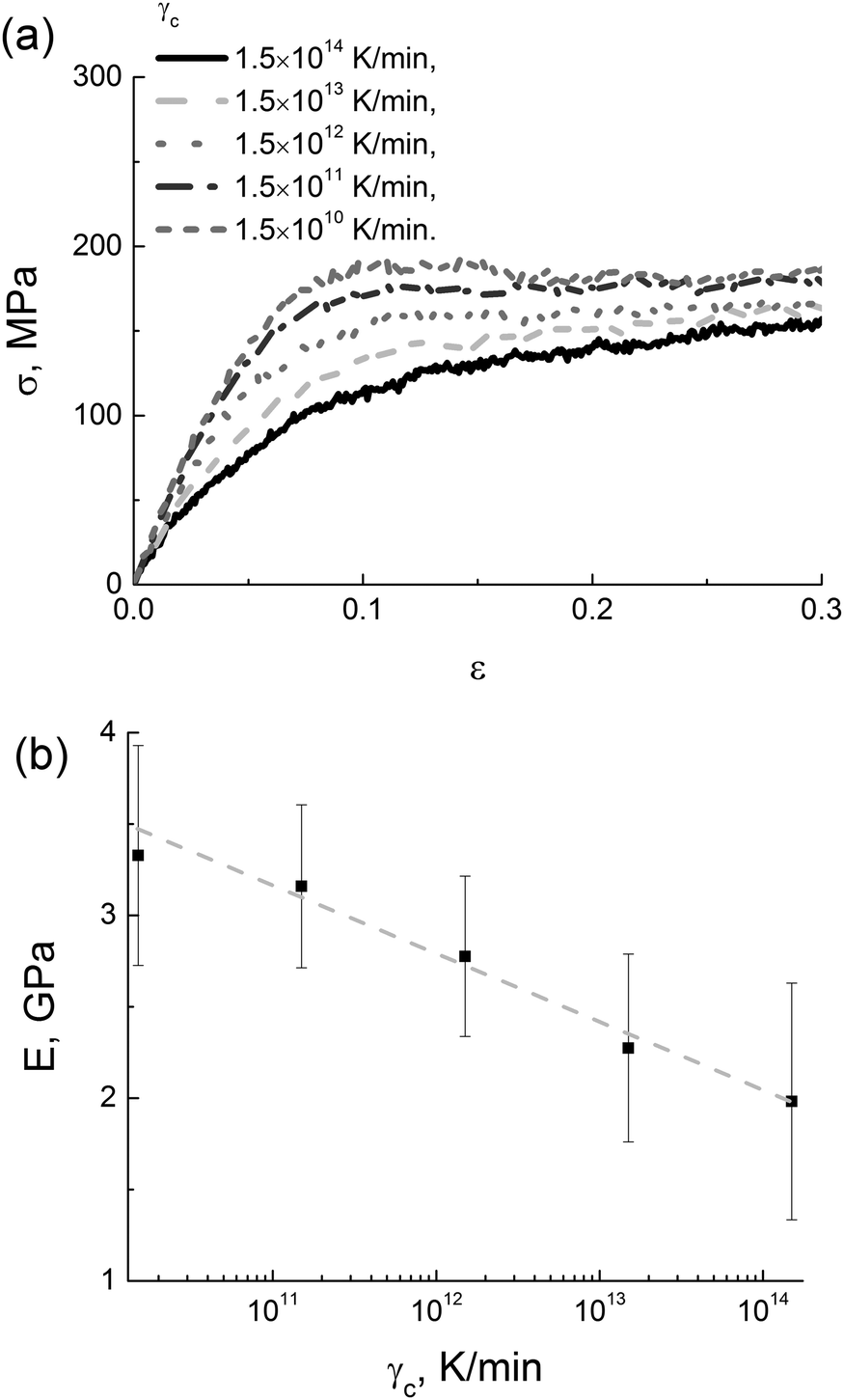 | ||
| Fig. 3 (a) Stress–strain dependences for different values of γc and (b) cooling-rate dependence of the elastic modulus in simulations; the logarithmic fit is shown by the dotted line. | ||
The logarithmic relationship between the elasticity modulus and the cooling rate is known from experimental studies on polyheteroarylenes.53 A similar dependence was also reported for a simple two-dimensional model of an amorphous glass.14 To the best of our knowledge, such a logarithmic dependence has never been observed in atomistic computer simulations of thermoplastic polyimides.
Such mechanical behavior of the PI samples with different thermal history is likely to be determined by both differences in the density at room temperature,49 see Fig. 4a, and different values of the typical intramolecular energy of excluded-volume interactions, see Fig. 4b.
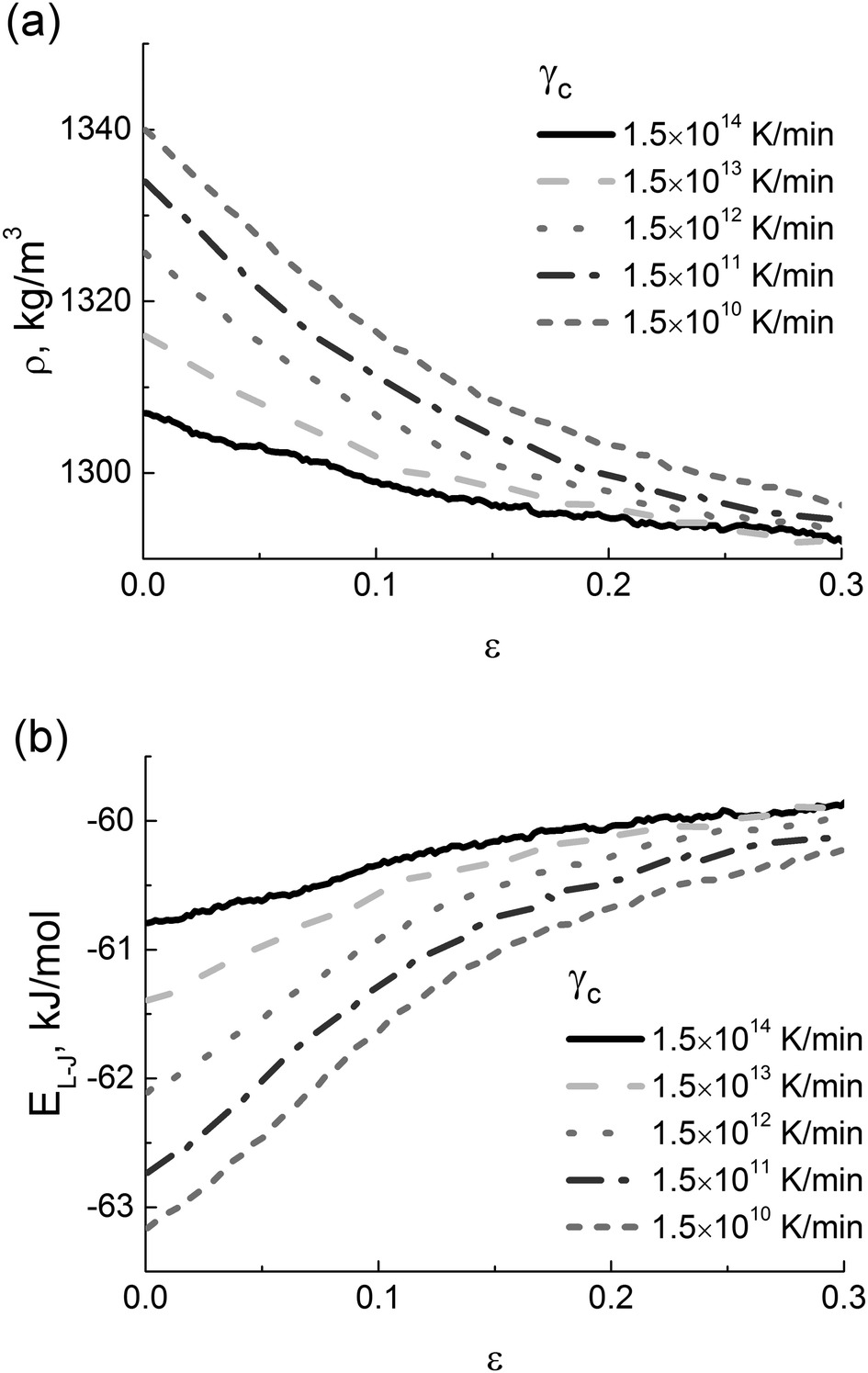 | ||
| Fig. 4 (a) Strain dependence of the PI density and (b) energy of excluded-volume interactions (EL-J) at various cooling rates γc. | ||
As the cooling rate γc decreases, the density of a sample in the glassy state systematically increases, leading to an increase in the number of contacts between polymer chains and a drop in the free volume. In the strain-hardening regime the dependences ρ(ε) that are calculated for various values of γc reach some constant density, see Fig. 4a. A similar behavior is observed for the strain dependence of the energy of excluded volume interactions, see Fig. 4b, where the saturation becomes clearly visible for deformations that exceed 20%. This is a manifestation of mechanical rejuvenation or (at least in part) of the removal of the thermal history upon deformation.54,55
 | ||
| Fig. 5 (a) PI R–BAPO stress–strain dependences for different deformation rates γd and (b) the modulus E(γd) as a function of the deformation rate γd; the logarithmic fit is shown by the dotted line. The filled square shows that the elasticity modulus clearly deviates from the logarithmic dependence for the deformation with the highest simulated rate, γd = 1.8 × 1010 s−1. | ||
The results obtained for PI R–BAPO confirm the logarithmic dependence E(γd) observed previously in coarse-grained simulations of polyethylene and polypropylene21,56 and also in experimental studies of various polymers and composite polymer-based materials.57,58
It is noteworthy that for the samples with very high deformation rates that exceed γd = 1.8 × 1010 s−1 (∼100 m s−1) and approach acoustic velocities, the dependence E(γd) clearly deviates from the logarithmic behavior, see Fig. 5b. Coarse-grained simulations of polyethylene demonstrated that the elasticity modulus also deviated from the logarithmic dependence21 at even higher rates (as high as 1011 s−1).
Some possible explanations for the observed non-logarithmic dependence of the elasticity modulus on the deformation rate can be linked to the presence of additional sub-Tg relaxation processes that are activated in polymer glasses under fast deformation. At high stretching rates the polymer internal structure undergoes substantial changes that are manifested in a rather rapid density drop, even in the linear viscoelastic regime (relative deformation of a few percent), see Fig. 6.
 | ||
| Fig. 6 The PI R–BAPO density ρ(ε) as a function of the relative strain for different values of γd. | ||
Thus, we can conclude that a correct choice of deformation-rate values in atomistic computer simulations is important for determining the mechanical properties of thermoplastic polyimides and for a subsequent comparison with experimental data. In the following the values of the deformation were taken below 1.8 × 109 s−1, i.e. well within the logarithmic regime of E(γd) dependence.
Influence of temperature and external pressure on mechanical properties
To get an insight into the performance range of thermoplastic PIs, it is essential to explore the impact of temperature and external pressure on their mechanical properties.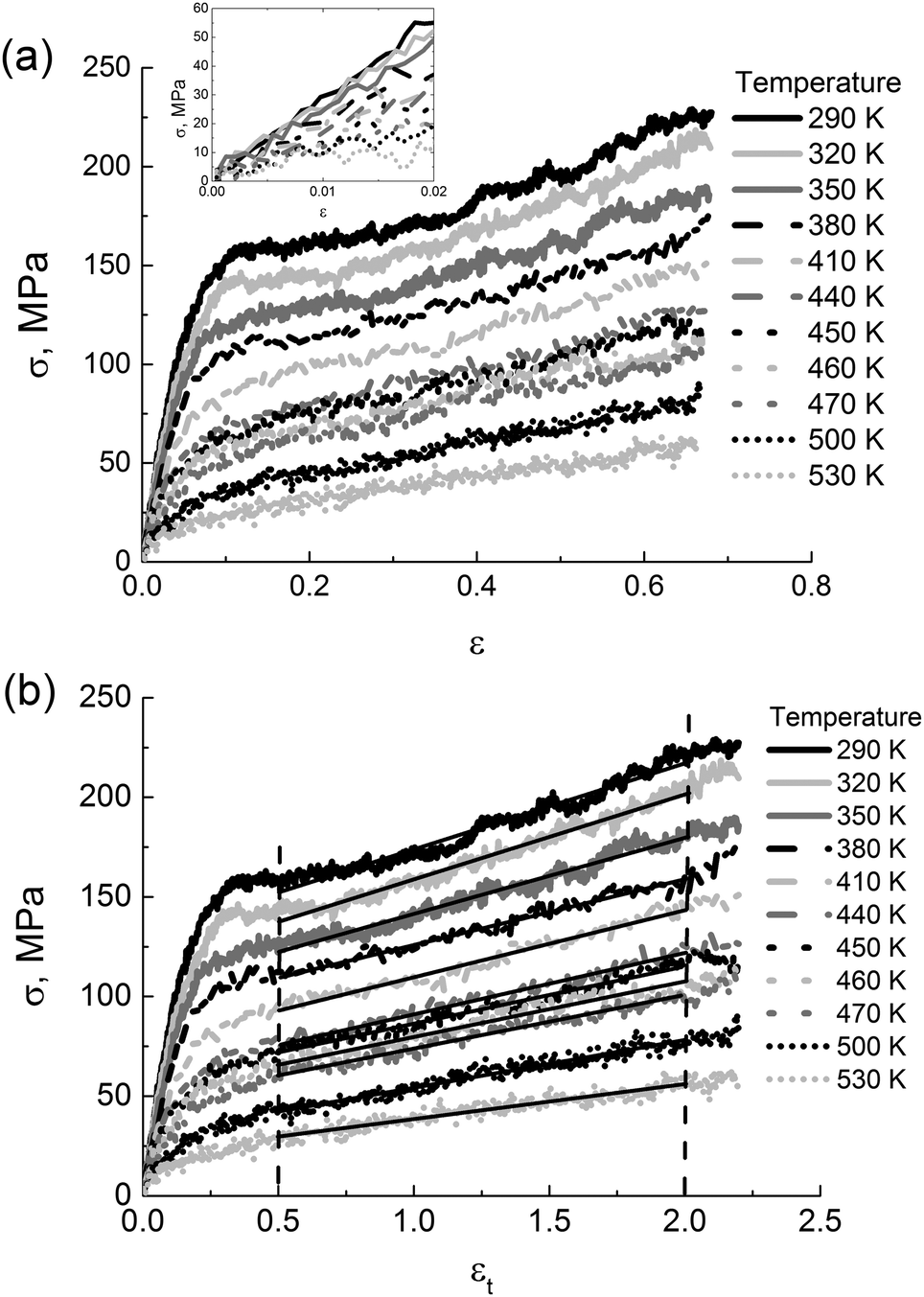 | ||
| Fig. 7 (a) The stress in PI R–BAPO samples as a function of the engineering strain ε at different temperatures. The inset shows the linear viscoelastic regime. (b) The stress in PI R–BAPO samples as a function of the strain εt at different temperatures. Solid black lines indicate the linear fits in the strain-hardening regime as highlighted by the vertical dotted lines, see eqn (3). | ||
The initial part of the σ(ε) dependence at different temperatures was used for calculating the temperature dependence E(T) of the elasticity modulus, see Fig. 8.
 | ||
| Fig. 8 The elasticity modulus as a function of temperature. A dashed grey line corresponds to the linear fit. Dashed arrows indicate Tbulk (E equals zero at Tbulk) and Tg of R–BAPO PI as estimated previously.36 | ||
The analysis of the data in Fig. 8 shows that the modulus drops almost linearly with temperature. Similar dependences E(T) were also found in other simulation studies,21,59 and they were also qualitatively confirmed by experimental data.60,61
When temperature approaches Tg from the low-temperature region, the value of E decreases by ∼45% as compared to the E value at room temperature. Extrapolation of the dependence E(T) to zero gives the flow temperature of Tbulk ∼ 580 K; this corresponds to the temperature at which the highly elastic response of the material to the applied load changes to a viscous flow. One can therefore conclude that the temperature T = 600 K that was selected for the equilibration stage corresponds to highly-mobile PI melt conditions.
The temperature range of the viscoelasticity for the majority of polymers is determined by their chemical structure. For thermoplastic PIs the highly-elastic state can be maintained in a rather broad temperature range from room temperature to temperatures that exceed by ∼100 K the glass transition temperature Tg = 470 K.36
We also analyzed the density–strain dependences at different temperatures, see Fig. 9.
 | ||
| Fig. 9 Density–strain dependences for PI R–BAPO at various temperatures. | ||
During deformation, the density decreases until a T-dependent constant value is reached at different strain values. The combined influence of temperature and deformation rate makes the sample plastic at lower values of ε when temperature increases. The state of viscous flow defines the material reaction to external pressure, and the polymer liquid becomes practically incompressible under such conditions. Note that we did not observe any significant increase in the density and the magnitude of (negative) excluded volume (LJ). The deformation above Tg has to be done with caution, due to the large fluctuations of these characteristics. To obtain reliable results, the number of simulated samples should be large enough to provide good statistics. The results obtained are in line with the simulation findings for mechanical properties of commodity polymers.44,59
With temperature the yield peak and the strain-hardening modulus decrease,62 see Fig. 10.
 | ||
| Fig. 10 Temperature dependence of the strain-hardening modulus. The inset shows the temperature dependence of the yield peak. The error bars are smaller than the symbol size. | ||
The analysis of the results presented in Fig. 10 shows that both the yield peak and the strain-hardening modulus decrease almost linearly as temperature increases. In the case of the yield peak dependence such behavior is in good agreement with other computer simulations.44 The theory of rubbery elasticity predicts that the strain-hardening modulus (Gh) should increase with temperature,
| Gh = kBTρ/Me | (4) |
 | ||
| Fig. 11 (a) The stress of PI R–BAPO samples under external pressure as a function of the engineering strain ε at T = 290 K. The inset shows the linear viscoelasticity regime. (b) The stress of PI R–BAPO samples under external pressure as a function of the strain εt at T = 290 K. Solid black lines are linear fits in the strain-hardening regime that is defined by the vertical dashed lines. | ||
The modulus increases linearly with external pressure P from 2.7 GPa (P = 0) to ∼3.7 GPa (P = 500 MPa), see Fig. 12.
 | ||
| Fig. 12 The PI R–BAPO elasticity modulus as a function of external pressure. | ||
The results obtained are qualitatively supported by experimental data for commodity polymers.63,64 To the best of our knowledge, the influence of the external pressure on mechanical properties of polyimides has not been reported yet.
The values of the yield peak σy and the strain-hardening modulus Gh also depend on the amount of the applied external pressure, see Fig. 13. With the increase of pressure both σy and Gh linearly increase. This correlation was established for the first time in simulations of atactic polystyrene under external pressure.49 Our findings in Fig. 13 are qualitatively supported by experimental data for other polymers.28,63,64 It is known that the ratio Gh/σy may act as an empirical relation that describes the strength of the polymer material49,50 (the so-called Considere criterion), which is often related to the neck formation as a result of the deformation when Gh/σy < 1/3. As Gh/σy exceeds 1/3, the material strength is supposed to increase. The dependence Gh/σy on external pressure at T = 290 K clearly distinguishes two regimes: Gh/σy is approximately equal to 1/3 under an external pressure of ≤200 MPa, and is above 1/3 under an external pressure of >200 MPa. Such behavior is likely to be associated with the substantial increase in the material strength under certain values of external pressure.
 | ||
| Fig. 13 (a) The PI R–BAPO strain-hardening modulus as a function of external pressure applied. The inset shows the Gh/σy as a function of the external pressure. The dashed line in the inset corresponds to Considere's criterion that determines the strength performance of the material. (b) The yield peak as a function of external pressure. | ||
Summarizing, the application of external pressure has a significant impact on practically all investigated mechanical properties, leading to a considerable increase in the elasticity modulus, the yield peak and the strain-hardening modulus. The strength of thermoplastic PI R–BAPO can be controlled by changing the external pressure; this property can be used to create new high-performance materials.
Conclusions
In this paper we employed atomic-scale molecular dynamics simulations to explore the mechanical properties of a novel thermoplastic polyimide R–BAPO under uniaxial deformation. The influence of various factors (cooling and deformation rates, external temperature and pressure) on the mechanical properties was extensively studied. It was demonstrated that the elasticity modulus is practically independent of the molecular mass, the size of the simulation box, and the degree of equilibration of the polymer sample. The simulations reported here successfully reproduce the well-known logarithmic dependence of the elasticity modulus on the cooling and deformation rates. It was established that the uniaxial stretching of PI samples with different thermal history leads to significant changes in the density and the energy of excluded-volume interactions during deformation. At the initial deformation stage, these values are determined by the material thermal history. Yet, as the deformation exceeds the yield strain, the dependences of both the density and the energy of excluded-volume interactions for the samples with different thermal history reach the same values; this effect can be interpreted as mechanical rejuvenation.The dependence of the elasticity modulus on the deformation in the acoustic regime (∼1010 s−1) deviates from the logarithmic dependence observed at much lower deformation rates (in both simulations and experiments). These findings most likely can be related to the thermally-activated nature of the polymer glass deformation due to the possible additional contributions of fast sub-Tg relaxation processes that are activated at high deformation rates.
Extrapolation of the T-dependence of the elasticity modulus to the zero modulus value was used to determine the temperature of transition from the highly elastic state to the viscous-flow state, Tbulk ∼ 580 K. Simulations showed the linear dependence of the yield peak and the strain hardening modulus on external pressure in agreement with previously obtained data.49 We also found an almost linear dependence of these characteristics on temperature. Our computational results also showed an increase in higher strength performance of R–BAPO with temperature (for temperatures below the glass transition temperature). Applying high external pressure (200 MPa and above) at a temperature of 290 K may lead to a further improvement of PI strength.
Overall, our study clearly demonstrates the ability of the state-of-the-art atomic-scale MD simulations to appropriately describe the mechanical properties of heterocyclic polymers. The developed protocols will be used in the nearest future for a comparative study of mechanical properties of composite materials based on these thermoplastic polyimides, which are reinforced with carbon nanofillers of various shapes and sizes.
Acknowledgements
This study was supported by the Russian Ministry of Education and Science within the State Contract No. 14.Z50.31.0002 (Megagrant). The simulations were performed using the computational resources of the Institute of Macromolecular Compounds, Russian Academy of Sciences, and the Chebyshev and Lomonosov supercomputers at Moscow State University.References
- M. I. Bessonov, M. M. Koton, V. V. Kudryavtsev and L. A. Laius, Polyimides: Thermally Stable Polymers, Plenum, New York, NY, 1987 Search PubMed.
- M. Ghosh, Polyimides: Fundamentals and Applications, CRC Press, 1996 Search PubMed.
- A. S. Argon and M. I. Bessonov, Philos. Mag., 1977, 35, 917–933 CrossRef CAS.
- J. W. Kang, K. Choi, W. H. Jo and S. L. Hsu, Polymer, 1998, 39, 7079–7087 CrossRef CAS.
- J. Huang, C. He, Y. Xiao, K. Y. Mya, J. Dai and Y. P. Siow, Polymer, 2003, 44, 4491–4499 CrossRef CAS.
- V. E. Smirnova, I. V. Gofman, E. M. Ivan’kova, A. L. Didenko, A. V. Krestinin, G. I. Zvereva, V. M. Svetlichnyi and V. E. Yudin, Polym. Sci., Ser. A, 2013, 55, 268–278 CrossRef CAS.
- I. V. Gofman, V. E. Yudin, O. Orell, J. Vuorinen, A. Y. Grigoriev and V. M. Svetlichnyi, J. Macromol. Sci., Part B: Phys., 2013, 52, 1848–1860 CrossRef CAS.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644–7655 CrossRef CAS PubMed.
- D. M. Delozier, K. A. Watson, J. G. Smith, T. C. Clancy and J. W. Connell, Macromolecules, 2006, 39, 1731–1739 CrossRef CAS.
- P. C. Irwin, Y. Cao, A. Bansal and L. S. Schadler, in 2003 Annual Report Conference on Electrical Insulation and Dielectric Phenomena, IEEE, 2003, pp. 120–123.
- L.-B. Zhang, J.-Q. Wang, H.-G. Wang, Y. Xu, Z.-F. Wang, Z.-P. Li, Y.-J. Mi and S.-R. Yang, Composites, Part A, 2012, 43, 1537–1545 CrossRef CAS.
- H. W. Ha, A. Choudhury, T. Kamal, D.-H. Kim and S.-Y. Park, ACS Appl. Mater. Interfaces, 2012, 4, 4623–4630 CAS.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- J. Ashwin, E. Bouchbinder and I. Procaccia, Phys. Rev. E, 2013, 87, 042310 CrossRef PubMed.
- Y. Shi and M. Falk, Phys. Rev. Lett., 2005, 95, 095502 CrossRef PubMed.
- K. Jordens, Polymer, 2000, 41, 7175–7192 CrossRef CAS.
- R. Hoy and M. Robbins, Phys. Rev. Lett., 2007, 99, 117801 CrossRef PubMed.
- L. C. A. van Breemen, T. A. P. Engels, E. T. J. Klompen, D. J. A. Senden and L. E. Govaert, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1757–1771 CrossRef CAS.
- J. Rottler and M. Robbins, Phys. Rev. E, 2003, 68, 011507 CrossRef PubMed.
- G. A. Medvedev, J. W. Kim and J. M. Caruthers, Polymer, 2013, 54, 6599–6607 CrossRef CAS.
- I. H. Sahputra and A. T. Echtermeyer, Modell. Simul. Mater. Sci. Eng., 2013, 21, 065016 CrossRef.
- E. Lerner and I. Procaccia, Phys. Rev. E, 2009, 80, 026128 CrossRef PubMed.
- J. Richeton, G. Schlatter, K. S. Vecchio, Y. Rémond and S. Ahzi, Polymer, 2005, 46, 8194–8201 CrossRef CAS.
- F. M. Capaldi, M. C. Boyce and G. C. Rutledge, Polymer, 2004, 45, 1391–1399 CrossRef CAS.
- A. D. Mulliken and M. C. Boyce, Int. J. Solids Struct., 2006, 43, 1331–1356 CrossRef CAS.
- E. M. Arruda, M. C. Boyce and R. Jayachandran, Mech. Mater., 1995, 19, 193–212 CrossRef.
- G. T. Gray, W. R. Blurnenthal, C. P. Trujillo and R. W. Carpenter, Mater. Res. Process. Sci., 1997, 7, 523–528 Search PubMed.
- R. W. Truss, P. L. Clarke, R. A. Duckett and I. M. Ward, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 191–209 CrossRef CAS.
- K. Cao, Y. Wang and Y. Wang, Mater. Des., 2012, 38, 53–58 CrossRef CAS.
- V. E. Yudin, V. M. Svetlichnyi, A. N. Shumakov, D. G. Letenko, A. Y. Feldman and G. Marom, Macromol. Rapid Commun., 2005, 26, 885–888 CrossRef CAS.
- V. E. Yudin, G. M. Divoux, J. U. Otaigbe and V. M. Svetlichnyi, Polymer, 2005, 46, 10866–10872 CrossRef CAS.
- V. E. Yudin, V. M. Svetlichnyi, A. N. Shumakov, R. Schechter, H. Harel and G. Marom, Composites, Part A, 2008, 39, 85–90 CrossRef.
- S. V. Lyulin, S. V. Larin, A. A. Gurtovenko, V. M. Nazarychev, S. G. Falkovich, V. E. Yudin, V. M. Svetlichnyi, I. V. Gofman and A. V. Lyulin, Soft Matter, 2014, 10, 1224–1232 RSC.
- S. V. Lyulin, A. A. Gurtovenko, S. V. Larin, V. M. Nazarychev and A. V. Lyulin, Macromolecules, 2013, 46, 6357–6363 CrossRef CAS.
- S. V. Lyulin, S. V. Larin, A. A. Gurtovenko, N. V. Lukasheva, V. E. Yudin, V. M. Svetlichnyi and A. V. Lyulin, Polym. Sci., Ser. A, 2012, 54, 631–643 CrossRef CAS.
- V. M. Nazarychev, S. V. Larin, A. V. Yakimansky, N. V. Lukasheva, A. A. Gurtovenko, I. V. Gofman, V. E. Yudin, V. M. Svetlichnyi, J. M. Kenny and S. V. Lyulin, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 912–923 CrossRef CAS.
- V. M. Nazarychev, S. V. Larin, N. V. Lukasheva, A. D. Glova and S. V. Lyulin, Polym. Sci., Ser. A, 2013, 55, 570–576 CrossRef CAS.
- D. Qi, J. Hinkley and G. He, Modell. Simul. Mater. Sci. Eng., 2005, 13, 493–507 CrossRef CAS.
- S.-H. Tzeng and J.-L. Tsai, J. Reinf. Plast. Compos., 2011, 30, 922–931 CrossRef CAS.
- J.-S. Gao, S.-C. Shiu and J.-L. Tsai, J. Compos. Mater., 2012, 47, 449–458 CrossRef.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, S. Uppsala and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- A. V. Lyulin, N. K. Balabaev, M. A. Mazo and M. A. J. Michels, Macromolecules, 2004, 37, 8785–8793 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- I. M. Ward and J. Sweeney, Mechanical Properties of Solid Polymers, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- C. Batistakis, M. A. J. Michels and A. V. Lyulin, Macromolecules, 2014, 47, 4690–4703 CrossRef CAS.
- B. Vorselaars, A. V. Lyulin and M. A. J. Michels, J. Chem. Phys., 2009, 130, 074905 CrossRef PubMed.
- R. N. Haward, Polymer, 1994, 35, 3858–3862 CrossRef CAS.
- R. S. Hoy, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 979–984 CrossRef CAS.
- D. J. A. Senden, J. A. W. Van Dommelen and L. E. Govaert, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1483–1494 CrossRef CAS.
- J. E. Lincoln, R. J. Morgan and E. E. Shin, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2947–2959 CrossRef CAS.
- A. V. Lyulin and M. A. J. Michels, Phys. Rev. Lett., 2007, 99, 085504 CrossRef PubMed.
- H. G. H. van Melick, L. E. Govaert and H. E. H. Meijer, Polymer, 2003, 44, 3579–3591 CrossRef CAS.
- H. Mae, M. Omiya and K. Kishimoto, Trans. Japan Soc. Comput. Methods Eng., 2008, 7, 6–11 Search PubMed.
- G. C. Jacob, J. M. Starbuck, J. F. Fellers, S. Simunovic and R. G. Boeman, J. Appl. Polym. Sci., 2004, 94, 296–301 CrossRef CAS.
- X. Li and A. F. Yee, Macromolecules, 2004, 37, 7231–7239 CrossRef CAS.
- D. Brown and J. H. R. Clarke, Macromolecules, 1991, 24, 2075–2082 CrossRef CAS.
- M. Kitagawa, J. Polym. Sci., Polym. Phys. Ed., 1977, 15, 1601–1611 CrossRef CAS.
- L. M. Nicholson, J. Mater. Sci., 2000, 5, 6111–6121 CrossRef.
- H. G. H. van Melick, L. E. Govaert and H. E. H. Meijer, Polymer, 2003, 44, 2493–2502 CrossRef CAS.
- A. W. Christiansen, E. Baer and S. V. Radcliffe, Philos. Mag., 1971, 24, 451–467 CrossRef CAS.
- S. Rabinowitz, I. M. Ward and J. S. C. Parry, J. Mater. Sci., 1970, 5, 29–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm00230g |
| This journal is © The Royal Society of Chemistry 2016 |