
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of heterocyclic capping groups on the self-assembly of a dipeptide hydrogel†
Adam D.
Martin
*a,
Jonathan P.
Wojciechowski
a,
Holly
Warren
b,
Marc
in het Panhuis
b and
Pall
Thordarson
*a
aSchool of Chemistry, The Australian Centre for Nanomedicine and the ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: adam.martin2@unsw.edu.au; p.thordarson@unsw.edu.au
bSoft Materials Group, School of Chemistry and ARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW 2522, Australia
First published on 22nd January 2016
Abstract
The mechanism and design rules associated with the self-assembly of short peptides into hydrogels is currently not well understood. In this work, four diphenylalanine-based peptides have been synthesised, bearing heterocyclic capping groups which have different degrees of hydrogen bonding potential and nitrogen substitution. For these four peptides, zeta potential and electrical impedance spectroscopy measurements were undertaken to monitor gelation, with the impedance data showing different gelation times for each peptide hydrogel. Through a combination of atomic force microscopy and rheological measurmeents, including dynamic strain and frequency sweeps, and thixotropic tests, the relationship between the mechanism of self-assembly in these hydrogels and their macroscopic behaviour can be established. It is observed that the degree of nitrogen substitution affects the self-assembly mechanisms of the hydrogels and as such, that there is an interplay between branching and bundling self-assembly pathways that are responsible for the final properties of each hydrogel.
Introduction
Short peptides are rapidly gaining popularity, due to their ease of manufacture through solid phase peptide synthesis (SPPS), the ability to select biorelevant amino acid sequences and potential uses in the fields of drug delivery, tissue engineering and cell scaffolding.1–10 Longer peptide sequences such as EAK-16 and RADA-16 are well known for their biocompatibility,11,12 and have been used to culture a variety of cells.13–15 In fact, RADA-16 is currently marketed under the name Puramatrix™ as a competitor to MatriGel for cell culture work.16 The advantages of using synthetic peptide hydrogels such as Puramatrix and the short peptides described herein is their chemical homogeneity relative to MatriGel, which is well-known for batch-to-batch variability.For longer peptides such as RADA-16 and EAK-16, complementary interactions between molecules are strong enough such that these peptides can form supramolecular gel networks, however shorter hydrogelators typically require the presence of additional aromatic groups in order to drive self-assembly into gel networks.17–19 This can be provided by aromatic amino acids such as phenylalanine, tyrosine or tryptophan, or through the use of an N-terminal capping group.20,21 Even with the presence of bulky aromatic groups, which may be expected to hinder the biological applicability of these short peptides, N-capped short peptides have been shown to be effective for culturing a variety of cell lines, including human dermal fibroblasts and Caco-2 cancer cells.22,23
The most commonly used N-terminal capping group is fluorenylmethyloxycarbonyl, or Fmoc, due to its use as an orthogonal protecting group in SPPS.24–28 More recently however, a variety of functional aromatic moieties have been appended to the N-terminus of short peptides, which in addition to inducing gelation in water, also impart some degree of functionality to the resulting hydrogels. The scope of groups used is varied, with photoactive spiropyrans being used to control the sol–gel transition, or stilbenes employed in order to create multicomponent hydrogels when mixed with an existing short peptide gelator.29,30 Functional groups which can be polymerised, such as carbazole, have been attached to the N-terminus of a peptide, as have groups designed to reinforce the hydrogen bonding within gel by providing addition sites for such interactions.31,32 In these studies above, the different capping groups employed result in the formation of gels which differ in their nanoscale structure. This manifests also as a difference in mechanical properties, which influences the potential applications of these materials.
In this work we have synthesised four different diphenylalanine based peptides 1–4 which bear heterocyclic capping groups with different degrees of nitrogen substitution and hydrogen bonding potential (Fig. 1). All four peptides form hydrogels, and we monitor gelation over a range of length scales using an array of characterisation techniques. In this way we examine the effect of the N-terminal capping group upon both the self-assembly of gelator molecules into hydrogels, and the final structure of the hydrogels which are formed. Indole-diphenylalanine, 1, has previously been synthesised and is known to form stiff hydrogels.33 Methylation of the indolic nitrogen, yielding N-methylindole-diphenylalanine, 2, removes the hydrogen bonding potential associated with the indole capping group. We then change the degree of nitrogen substitution on the five membered ring by adding an additional nitrogen whilst maintaining an available proton, giving benzimidazolone-diphenylalanine, 3. This hydrogen bonding potential is then removed in the final capping group used in this work, benzimidazole-diphenylalanine, 4. We monitor fibre charge during gelation through zeta potential and electrical impedance spectroscopy in order to investigate whether the presence of a free proton has an effect on the self-assembly of peptides 1–4. We then investigate the secondary structure of the gelators using circular dichroism and infrared spectroscopy. Finally, we examine the mechanical strength and recovery properties of these hydrogels using rheology, correlating the macroscopic hydrogel properties with data obtained from atomic force microscopy (AFM) of the peptide hydrogels at a range of different concentrations. In this way we are able to investigate the effect of nitrogen substitution and hydrogen bonding potential on the self-assembly and network structure of the four diphenylalanine peptides. This allows us to tune the resultant gel properties by carefully varying the nature of the heterocyclic capping group used. This is extremely important in the rational design of peptide hydrogels for applications such as tissue engineering or injectable drug delivery systems.
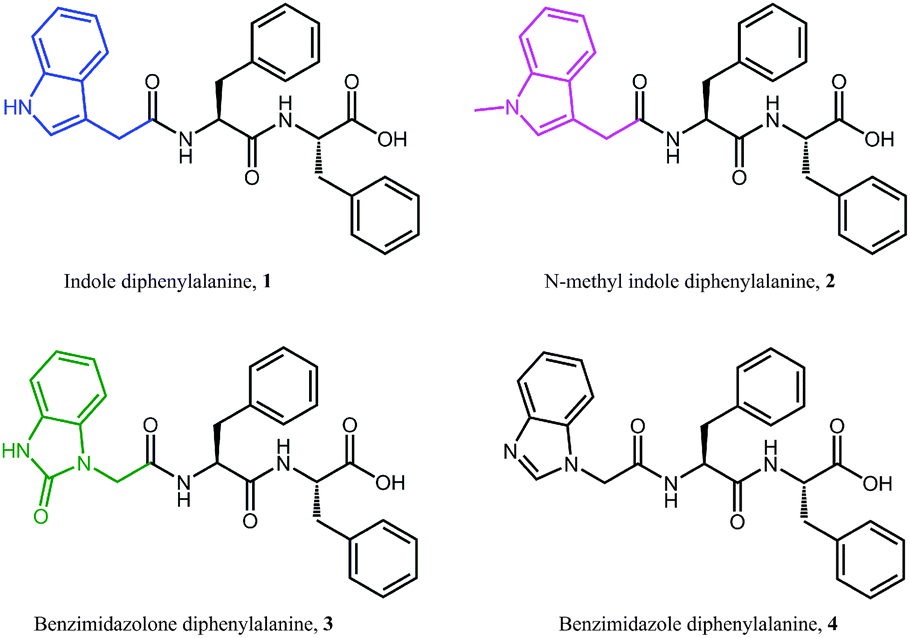 | ||
| Fig. 1 Molecular structure of the four dipeptides, each bearing different capping groups, used in this study. | ||
Results and discussion
Hydrogel formation
All four peptides formed hydrogels via a pH switch mechanism using glucono-delta-lactone (GdL), where the final pH of the gel is approximately 4–5.34 Attempts at gel formation through heat–cooling in phosphate buffered saline, or solvent switching using DMSO and either water or DMEM (Dulbecco’s Modified Eagle Medium) were unsuccessful. All peptides formed opaque hydrogels apart from 3, which formed a transparent hydrogel, which is desirable for future cell culture applications due to ease of imaging. The minimum gelation concentration for 1–4 was recorded as 0.3, 0.6, 0.02 and 0.1% (w/v), respectively. From this, it can be seen that methylation of the indolic proton in 2 has doubled the minimum gel concentration and that 3 forms hydrogels at extremely low concentrations, making this peptide a supergelator. Interestingly, the trends observed for the minimum gel concentrations of peptides 1–4 correspond to the trends in their clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values (1.85, 1.92, 1.08 and 0.72, respectively), where clogP is a measure of the calculated hydrophilicity of the peptide. From these results it seems as though a more hydrophilic peptide gives a lower minimum gel concentration, perhaps due to better solvation of the peptide monomer, thus allowing a greater self-assembly space to be explored.
P values (1.85, 1.92, 1.08 and 0.72, respectively), where clogP is a measure of the calculated hydrophilicity of the peptide. From these results it seems as though a more hydrophilic peptide gives a lower minimum gel concentration, perhaps due to better solvation of the peptide monomer, thus allowing a greater self-assembly space to be explored.
All peptides except for 2 give rise to a negative peak in their circular dichroism (CD) spectra at approximately 220 nm, which is typical of a β-sheet arrangement (Fig. 2a). The CD spectrum of 2 gives only weak signals, indicating a lack of secondary organisation. This is supported by the relatively sharp signal observed in the Amide I region of the ATR-IR spectrum for 2 (Fig. 2b), indicative of decreased hydrogen bonding relative to the other peptides.35 This indicates that methylation of the indolic proton in 2 has disrupted the peptides ability to form β-sheets, implying that in this instance the selection of capping group plays a role in the formation of β-sheets.
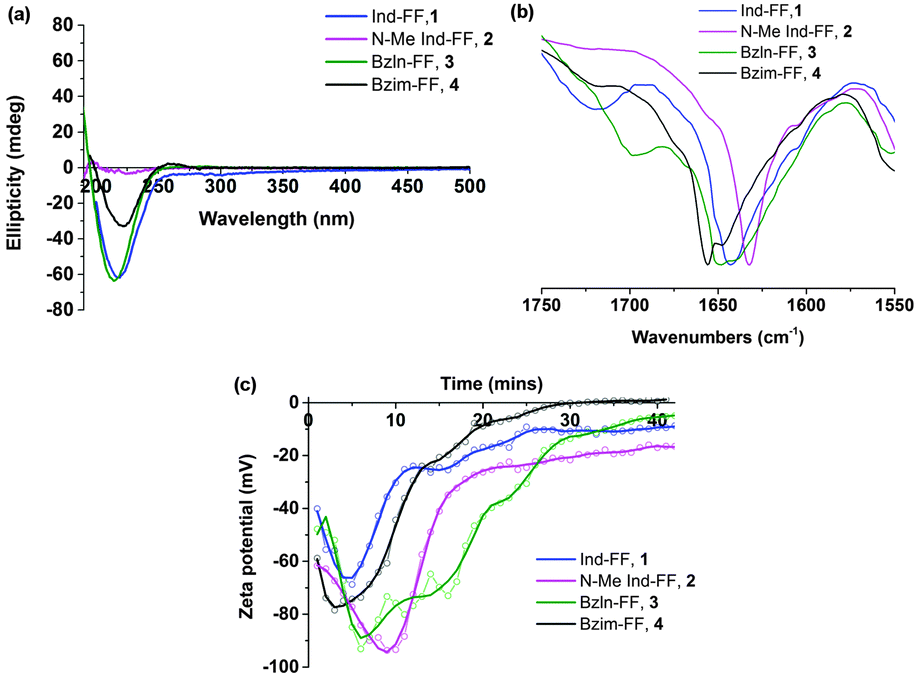 | ||
| Fig. 2 Circular dichroism (a), Amide I region of ATR-IR (b) and zeta potential measurements (c) of peptide gelators 1–4. For CD studies a 1% (w/v) hydrogel was diluted 1:8 with milliQ water. ATR-IR and zeta potential measurements were performed at 0.6% (w/v). The zeta potential graph has been smoothed using a Savitsky–Golay filter. | ||
Zeta potential measurements (Fig. 2c) were performed in order to study the gelation of peptides 1–4. In all cases, an initial decrease in zeta potential was observed before a gradual increase, accompanied by a decrease in ion mobility, which is expected during gelation. The initial decrease in zeta potential that was observed for all systems is intriguing, and suggests that upon GdL addition any existing supramolecular structures are broken up into monomers before gelation proceeds. It is also interesting to note that for 2, the zeta potential plateaus at −20 mV, suggesting that there is still some repulsion between fibres. This may help to explain the increased minimum gelation concentration and decreased stability for this peptide relative to the other peptides in this study.
Electrical impedance spectroscopy measurements have previously been shown to accurately indicate the onset of gelation for polymer gels,36–38 and gelation time can be calculated by fitting two linear regression lines to early and late stage impedance data and finding their intersection. Initially, the impedance of the gel solutions increases due to aggregation of the gel monomers and immobilisation of water. Upon gelation, the system reaches a steady state, most likely where the rate of fibre formation and dissolution is equal. This is seen in the impedance plot as a plateau.
For all of our self-assembled peptide hydrogels, we are able to reliably fit linear regression lines and thus find gelation times (Fig. 3). For indole-diphenylalanine, 1, and benzimidazolone-diphenylalanine, 3, we see a sharp change in gradient after 20 and 30 minutes respectively, indicating that gelation has occurred. For N-methylindole diphenylalanine, 2, and benzimidazole diphenylalanine, 4, the change in gradient is slightly less noticeable, but occurs after 18 and 12 minutes, respectively. This order of gelation is also seen in the zeta potential measurements, confirming that both electrical impedance and zeta potential are reliable, novel methods for monitoring the onset of gelation in short peptide hydrogels. For both 2 and 4, impedance continues to increase after 50 minutes, indicating that dynamic processes are still occurring. For 2, this is due to N-methylindole diphenylalanine being an unstable gel, which fits with other observations including zeta potential measurements (Fig. 2c) and visual observations of gel precipitation over time. For 4, this is due to slow crystallisation of the hydrogel over time.
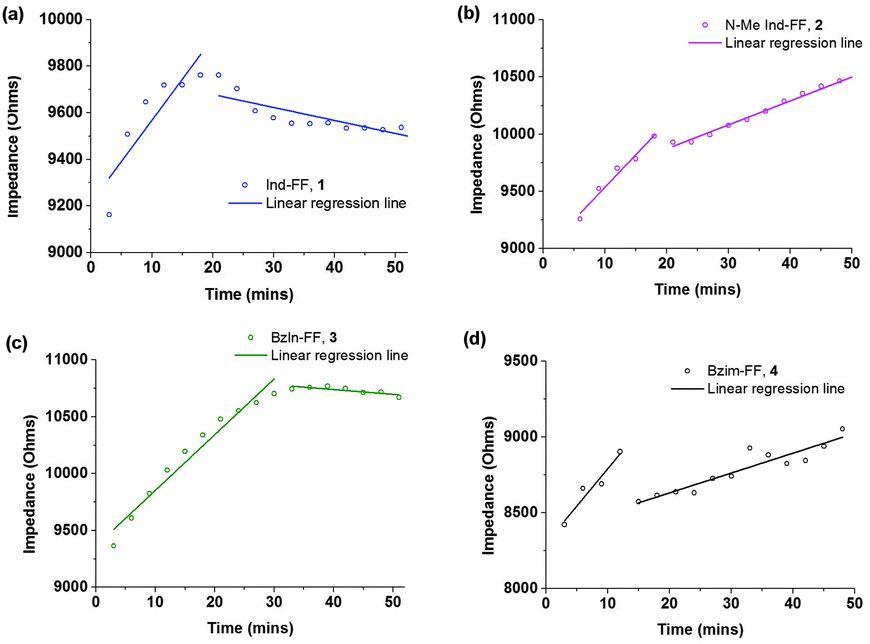 | ||
| Fig. 3 Time dependent impedance data for 1–4. Measurements were performed at 25 °C and impedance data was averaged over 0.1 to 100 kHz, where the impedance is independent of frequency. | ||
Mechanical behaviour of hydrogels
Each of the hydrogels 1–4 prepared were subjected to rheological testing. First, a strain sweep was performed in order to establish the linear viscoelastic region (LVE), where the storage modulus G′ and loss modulus G′′ are independent of the applied strain. In the strain sweep graphs (Fig. 4a–d) it can be seen that the LVE is similar for indole and N-methyl indole-diphenylalanine, with both materials displaying reversible network characteristics up to a strain of 1%. The LVE for benzimidazolone-diphenylalanine, 3, extends to 3% and for benzimidazole-diphenylalanine, 4, to 10%. It is interesting to note that the strain tolerance (for irreversible network deformation) is highest for the softer hydrogels, which have a higher degree of nitrogen substitution in their capping group. Further insight into this can be provided using atomic force microscopy to study the network structure, which will be discussed later in this work. The crossover point (where G′′ = G′) is where the network is starting to break and the material displaying fluid-like behaviour. This value varies from 60% for hydrogel 4 to as low as 3% for hydrogel 3.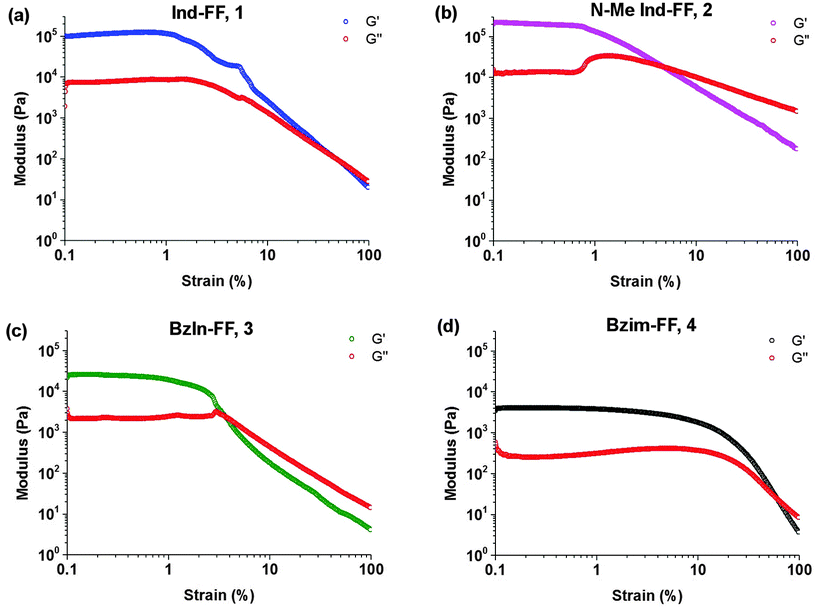 | ||
| Fig. 4 Strain sweeps performed on hydrogels 1–4. Gels were prepared at 0.6% (w/v) and storage (G′) and loss modulus (G′′) measurements performed at a constant frequency of 1 Hz and temperature of 25 °C. | ||
With the LVE now determined for each hydrogel, frequency sweeps were performed at a constant strain of 0.5% (within the LVE for all hydrogels), with all hydrogels displaying frequency independent behaviour over the range from 0.1 to 10 Hz. Frequency sweeps are often used to give an indication of the stiffness of the hydrogels (approximated by the storage modulus G′). From these measurements (Fig. 5a–d) it can be seen that the indole-based hydrogels 1 and 2 are quite stiff, with storage moduli above 105 Pa. The high stiffness of hydrogels of 1 have been reported previously,33 however the high storage modulus of hydrogels of 2 is surprising. This indicates that hydrogen bonding does not play a large role in determining the mechanical characteristics for these systems. Instead the degree of nitrogen substitution appears to play a crucial role for these peptides. The two benzimidazole-based hydrogels 3 and 4 also display similar stiffness values. That these hydrogels are an order of magnitude weaker than their indole-based counterparts, despite 1 and 3 both having a free hydrogen available for potential hydrogen bonding, is further evidence that gel strength is determined not determined by hydrogen bonding potential for these systems, but instead by the degree of nitrogen substitution on the heterocyclic capping group.
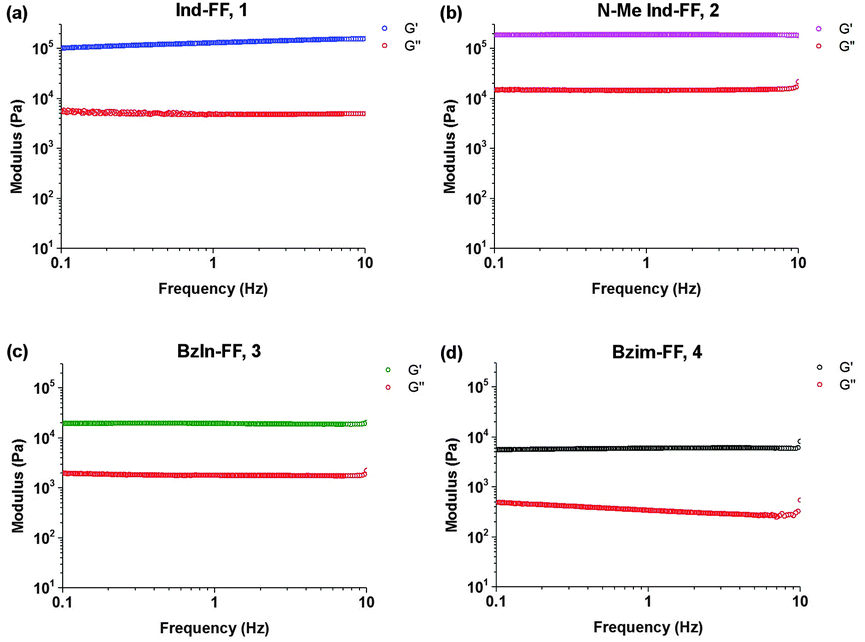 | ||
| Fig. 5 Frequency sweeps performed on hydrogels 1–4. Gels were prepared at 0.6% (w/v) and measurements performed at a constant strain of 0.5% and temperature of 25 °C. | ||
Thixotropy tests were performed in order to assess the ability of these hydrogels to recover from being subjected to high strain, similar to that applied when a hydrogel is injected through a syringe needle. In order to do this, we alternated between applying a small oscillatory strain (0.5%, low strain) and a large strain (100%, high strain) and monitored the resulting shear modulus behaviour of each hydrogel. Under high strain the gels exhibit a rapid decrease in G′ resulting in fluid-like behaviour (G′′ > G′). It can be seen that hydrogels 1 and 4 display rapid recovery of G′ after changing the applied strain from high to low. This recovery is most likely due to the presence of crystalline domains in the hydrogel structure, which is discussed in more detail below. It should be noted that even with this remarkable behaviour, gels of 1 do not recover back to their initial value of G′, and over a number of strain cycles the storage modulus G′ slightly decreases. This could indicate that there may be some structural changes within the hydrogel. For hydrogels of 4, this behaviour is more pronounced, with an associated increase in the loss modulus G′′. Despite this, the rapid recovery of both 1 and 4 back to almost their initial gel strength makes them prime candidates as injectable delivery vectors.
For 2 (Fig. 6b), we see that the gel is not reformed upon the application of an initial strain, instead G′ and G′′ hover close to zero. This is not surprising, given that 2 was observed to have a low degree of stability, based on circular dichroism, zeta potential, impedance and visual observations. It can be seen for 3 (Fig. 6c) that after the first application of a high strain, the hydrogel recovers relatively quickly (approximately 200 seconds), however to a stiffness that is below the original stiffness of the gel. Each subsequent breakage of the gel results in a slower recovery, which is evidenced by no steady state value of G′ being reached, even after 600 seconds. In addition to this longer recovery time, the modulus of the gel decreases with each application of strain.
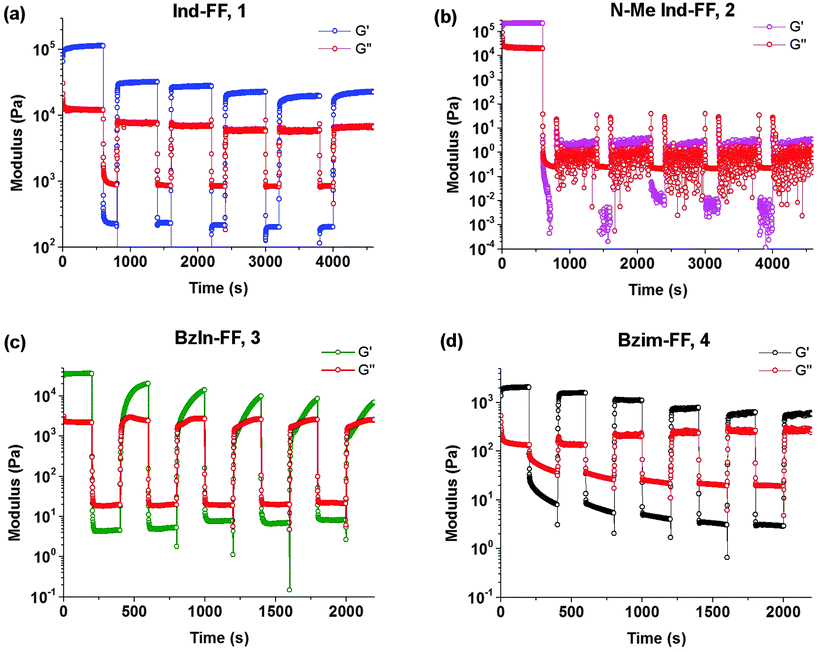 | ||
| Fig. 6 Thixotropy tests performed on hydrogels 1–4. After gelation, storage modulus (G′) and loss modulus (G′′) measurements were performed at a constant frequency (1 Hz) using low strain (0.5%, 600 seconds) followed by high strain (100%, 200 seconds). This protocol was repeated six times. Gels were prepared at 0.6% (w/v) and measurements performed at a constant temperature of 25 °C. | ||
Structure of hydrogel networks
Atomic force microscopy (AFM) was performed on hydrogels of 1–4 at different concentrations in order to understand of how their gel networks evolve and to establish a relationship between gel strength, fibre morphology and thixotropic behaviour. It has been previously reported that 1 forms large fibres that are composed of smaller, 2 nm fibres.33 These images were obtained at a lower concentration however, and it is known that aggregation effects can occur during sample preparation, despite efforts to reduce this happening. At a concentration of 0.5% (w/v), a dense fibrous network is observed, with fibre sizes of approximately 10 nm (Fig. 7B). Similar to previous AFM and TEM (transmission electron microscopy) results, these fibres are long and straight, exhibiting little branching behaviour. Increasing the concentration to 1% (w/v) gives a fibrous network which is denser still, and in addition to the long, straight fibres which are still present, minimal branching behaviour is observed (Fig. 7C). Furthermore, fibre size increases to 30–40 nm, suggesting that additional bundling is occurring. In addition to AFM, previous cryo-SEM (scanning electron microscopy) work on 1 has also shown the presence of crystalline domains in addition to fibres (Fig. S7, ESI†). It is proposed that these crystalline domains are critical to the recovery behaviour of this gel after the application of a large external strain, due to the ability of the strain to be dissipated within these domains, thus allowing rapid recovery.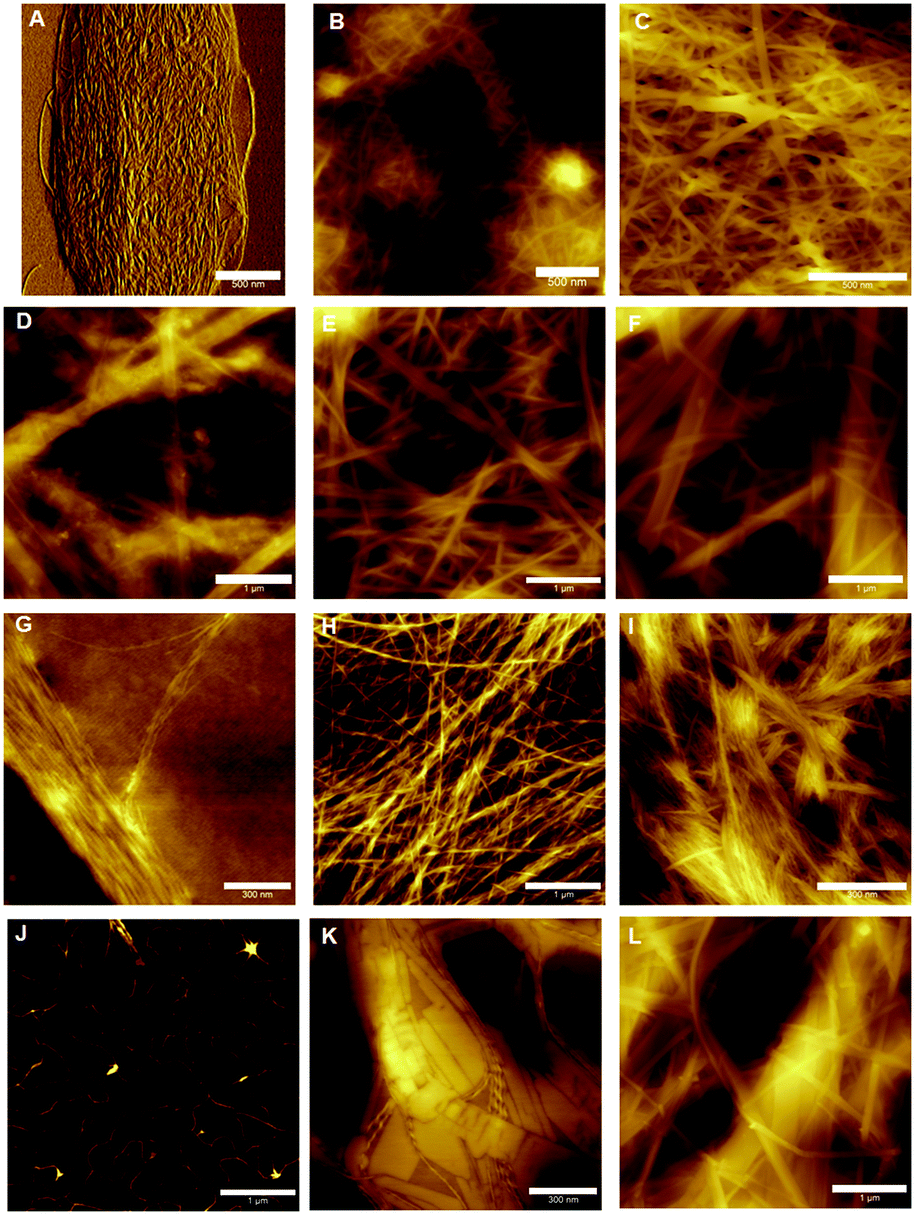 | ||
| Fig. 7 AFM images of the gel networks of 1–4 evolving with concentration. 1 at concentrations of 0.1, 0.5 and 1% (w/v) (A–C), 2 at concentrations of 0.1, 0.5 and 1% (w/v) (D–F), 3 at concentrations of 0.01, 0.02 and 0.1% (w/v) (G–I) and 4 at concentrations of 0.02, 0.1 and 0.2% (w/v) (J–L). All samples were spread coated onto a freshly cleaved mica substrate. Scale bars denote 500 nm (A–C), 1 μm (D–F, H, J and L) and 300 nm (G, I and K). | ||
For hydrogels of 2, at low concentrations the gel network is a mixture of large fibres (diameter > 100 nm), smaller fibres (diameter 10–70 nm) and amorphous precipitate (Fig. 7D). This is further evidence of the instability of hydrogels of 2 at low concentrations, and this lack of uniformity at low concentrations, coupled with minimal fibre branching, is likely the reason for the high minimum gel concentration of this peptide. While it is this fibre morphology that most likely gives the indole-based hydrogels of 1 and 2 their strength due to the lack of junction zones, which have been shown to be the most probable points of failure,39 it is also the reason that these gels are brittle, as an applied strain cannot be redistributed by this inflexible network, resulting in hydrogel failure. At 0.5% (w/v), a more dense fibrous network is formed (Fig. 7E), with a population of large (50–100 nm) and smaller (20–30 nm) fibres seen. At even higher concentrations the population of fibres becomes weighted more heavily in favour of the larger diameter fibres, as expected (Fig. 7F). At all concentrations, minimal branching behaviour is observed, which is consistent with observations for similarly strong hydrogels of 1.
The presence of precipitates indicates that fibre formation is not necessarily favoured, and that the fibres are likely a kinetically trapped intermediate. The application of a high strain as in thixotropy studies (Fig. 6b) results in these fibres being destroyed, and due to the repulsion between fibres (Fig. 2c) the gel network cannot reform.
Lower concentrations were employed when studying hydrogels of benzimidazolone-diphenylalanine, 3, owing to its lower minimum gelation concentration. At 0.01% (w/v), short nanofibrils uniformly 1 nm in diameter are observed (Fig. 7G). This suggests that these nanofibrils are molecular fibres, and the bundling together which is observed here is likely due to drying effects. Doubling the concentration to 0.02% (w/v) results in the formation of a fibrous network, with fibre sizes of 5–8 nm for individual fibres. The wrapping of individual fibres around each other to form twisted bundles is clearly visible, with the best example of this being the horizontal fibre in the top third of Fig. 7H. This entanglement of fibres leads to branching between fibres and this, coupled with the smaller fibre diameters, is responsible for the lower stiffness of the resultant hydrogel.40 Increasing the concentration further does not result in an increase in the size of the individual fibres, suggesting a favourable fibre diameter is reached at low concentrations, however strong lateral associations between fibres do occur, Fig. 7I. These associations may be the reason behind the slow recovery of hydrogels of 3 upon the application of a large external strain (Fig. 6c). The external strain disrupts these fibre bundles, which then are dispersed throughout the solution after cessation of the high strain. This would result in the lower strength gel network observed, with increased recovery times indicating these dispersed fibres slowly attempting to reorganise into their initial structure.
Hydrogels of benzimidazole-diphenylalanine, 4, show a coexistence of nanocrystals and fibres. At low concentrations (0.02% (w/v)), protofibres 1 nm in diameter are observed (Fig. 7J). Branching behaviour is already evident in these protofibres and it is interesting to note that both benzimidazole-based hydrogels 3 and 4 form these molecular fibres at low concentrations, compared to the indole-based hydrogels which form much larger, unbranched structures. This suggests a different method of fibre formation, most likely due to the different degree of nitrogen substitution in the capping groups of 1 and 2versus3 and 4. Increasing the concentration to 0.1% (w/v) results in the formation of both nanocrystals and helical fibres (Fig. 7K); giving a fascinating insight into the competing processes of gel formation and crystallisation. As with 1, any applied strain can potentially either be dispersed either through a rearrangement of the fibre and nanocrystal structure, or dissipated into the crystalline sections of the network, leading to the rapid recovery of hydrogels of 4 after an applied external strain. At higher concentrations (0.2% (w/v)) long, flat fibres are the dominant feature (Fig. 7L), with diameters of 10–30 nm.
It is clear from this work that there are a number of factors that determine the physical properties of a hydrogel network. These factors stem from the molecular structure of the gelator molecules, and manifest in the hierarchical self-assembly of these gelators into fibrous networks. For the peptides studied here, it would seem that the degree of nitrogen substitution controls the stiffness of the resultant hydrogel, due to the propensity of these networks to undergo bundling compared to branching. The degree of bundling in polymer hydrogels has previously been shown to affect their stiffness proportional to N3, where N is the number of fibres in a bundle.41 Conversely, higher levels of nitrogen substitution on the capping groups result in softer gels, and these networks exhibit a higher degree of branching behaviour. The self-assembly processes behind branching versus bundling in hydrogel networks is not yet well understood, however in this work we have shown that for these diphenylalanine peptide hydrogels, recovery from an applied strain is dependent on the presence of crystalline domains within the structure. Through characterisation across different length scales, we have shown that the nanoscale structure of these materials defines their macroscale properties. Such an approach is required in order both to understand the behaviour of these hydrogels and to rationally design such materials in future.
Conclusions
In conclusion, we have synthesised four diphenylalanine-based peptides 1–4, capped at their N-terminus with heterocycles that display different degrees of hydrogen bonding potential and nitrogen substitution. Using zeta potential and electrical impedance measurements we monitored and obtained good agreement for the gelation times of 1–4. Zeta potential measurements of 2, N-methyl indole-diphenylalanine, provided a key insight into the unstable nature of this hydrogel by showing that even upon gelation; the fibrous network is negatively charged. Despite this instability, hydrogels of 2 were shown to be just as stiff as hydrogels of indole-diphenylalanine, 1. Peptides displaying a greater degree of nitrogen substitution at their capping group, benzimidazolone and benzimidazole-diphenylalanine (3 and 4 respectively) formed softer networks.Atomic force microscopy studies revealed two different self-assembly mechanisms. Indole-based gelators were shown to bundle together, forming long, straight fibres whilst benzimidazole-based gelators underwent significant branching, giving softer networks. This work suggests that for these materials, network structure and thus mechanical properties are controlled by the degree of nitrogen substitution of the capping group. Finally, the thixotropic behaviour of these systems was found to depend on the presence of crystalline domains within the hydrogel network, as these crystalline domains can dissipate strain effectively, resulting in a rapid recovery from an applied external strain. The establishment of structure–property relationships within this family of diphenylalanine gelators has significant implications for the future applications of these materials as extracellular matrix mimics and injectable systems. In particular, it is envisaged that a greater understanding of design rules arising from this work will allow for the rational design of functional and hydrogels for biomedical applications.
Experimental section
All chemicals and solvents used were purchased from Sigma Aldrich and used as supplied. Indole-diphenylalanine has been synthesised as previously reported.33Synthesis of peptides and capping groups
N-Methyl indole-3-acetic acid was synthesised via methylation of the commercially available indole-3-acetic acid in good yield (77%) following a previously reported procedure.42 Benzimidazolone was synthesised in four steps starting from ortho-phenylenediamine and urea using a literature procedure to give benzimidazolone,43 which was then reacted with Boc anhydride and the mono-Boc derivative isolated. This compound was alkylated using tert-butyl bromoacetate and sodium hydride before a one-pot cleavage of both the Boc protecting group and tert-butyl ester yielded benzimidazolone acetic acid in a 15% overall yield across four steps. Full experimental details, including characterisation data, can be found in the ESI.†All peptides were synthesised using solid phase peptide synthesis, employing HOBt/HBTU as the coupling reagents. N-Methyl indoleacetic acid was coupled to the diphenylalanine peptide in this manner; however for benzimidazolone a DIC-based coupling method was required. The full SPPS procedure, including yields and characterisation can be found in the ESI.†
Circular dichroism
CD measurements were performed using a ChirascanPlus CD spectrometer, with data collected between wavelengths of 180–500 nm with a bandwidth of 1 nm, sample ratio of 0.1 s per point and step of 1 nm. In a typical experiment, 1% (w/v) hydrogels were prepared and diluted 1:8 (v/v) in water. Temperature was kept constant at 20 °C and all experiments were repeated three times and averaged into a single plot.
Attenuated total reflectance-infrared spectroscopy measurements
Fourier transform infrared spectroscopy (FTIR) measurements were made on a Perkin Elmer Spotlight 400 FT-IR spectrophotometer equipped with a diamond crystal attenuated total reflectance (ATR) accessory. Hydrogels were prepared at 1% (w/v) and pressed between the diamond crystal and substrate. All spectra were scanned 16 times over the range of 4000–650 cm−1.Rheology measurements
Rheological measurements were performed on an Anton Paar MCR 302 rheometer using a 25 mm stainless steel parallel plate geometry configuration and analysed using RheoPlus v3.61 software. Typical rheology measurements involved casting 550 μL of a 0.6% (w/v) sol onto one of the stainless steel plates, lowering the other plate to the measurement position, and allowing two hours for the gel to form via the pH switch method described above. A Peltier temperature control hood and solvent trap was used to reduce evaporation and maintain a temperature of 25 °C for frequency and amplitude sweeps. Frequency sweeps were performed with a log ramp frequency (0.01–10 Hz) and constant strain (0.5%). Amplitude sweeps were performed with constant frequency (1 Hz) and log ramp strain (0.1–100%). The rheology plots displayed are an average of at least three repeats for each point and error bars denote two standard deviations from the log-averaged mean.AFM measurements
Hydrogels were prepared using the pH switch method described above. Upon addition of GdL and before gelation, one drop of the hydrogel solutions was cast onto a freshly cleaved mica substrate, followed by spreading of the drop over the mica using a glass slide, with the excess liquid wicked away using capillary action. These samples were left to dry in air overnight. Imaging was undertaken on a Bruker Multimode 8 atomic force microscope in Scanasyst mode in air, whereby the imaging parameters are constantly optimised through the force curves that are collected; preventing damage of soft samples. Bruker Scanasyst-Air probes were used, with a spring constant of 0.4–0.8 N m−1 and a tip radius of 2 nm.Zeta potential measurements
The zeta potential of hydrogel solutions were measured using a Malvern Instruments Zetasizer NanoZS, equipped with a He–Ne laser beam with a wavelength of 633 nm and scattering angle of 173°. Measurements were performed in folded capillary cells (Malvern Instruments, DTS1070) using hydrogels as prepared above at a concentration of 0.6% (w/v).Electrical impedance spectroscopy
The impedance behaviour of liquids and hydrogels of 1–4 was assessed as follows. Solutions were poured into a plastic sample holder (acrylic 1 cm width, 1 cm height, 2.5 cm length) containing two pieces of reticulated vitreous carbon (RVC, ERG Aerospace, USA, 20 pores per inch). A custom-designed instrument was used to measure electrical impedance spectroscopy for frequencies between 100 Hz and 100 kHz.30 Briefly, an alternating current signal (1 V peak voltage) was applied using a waveform generator (Agilent U2761A) across a circuit consisting of a known resistor (10 kΩ) and the gel sample. Current was calculated across the known resistor and then used to measure the impedance by measuring the potential difference across the known resistor with an oscilloscope (Agilent U2701A).Acknowledgements
The authors would like to acknowledge financial support from the University of Wollongong and Australian Research Council (ARC) through its Centre of Excellence Programs to MihP (CE140100012) and PT (CE140100036), Discovery Project Grant to PT (DP130101512), ARC Future Fellowship to PT (FT120100101) and the Australian Government by PhD Scholarship to JPW. Mr R. D. Gately is thanked for technical assistance.References
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519 CrossRef CAS PubMed.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117 RSC.
- X. P. Yao, Y. Liu, J. Gao, L. Yang, D. Mao, C. Stefanitsch, Y. Li, J. Zhang, L. L. Ou, D. L. Kong, Q. Zhao and Z. J. Li, Biomaterials, 2015, 60, 130 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511 CrossRef CAS PubMed.
- C. J. Newcomb, S. Sur, J. H. Ortony, O.-S. Lee, J. B. Matson, J. Boekhoven, J. M. Yu, G. C. Schatz and S. I. Stupp, Nat. Commun., 2014, 5, 3321 Search PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- H. Shigemitsu and I. Hamachi, Chem. – Asian J., 2015, 10, 2026 CrossRef CAS PubMed.
- S. Zhang, C. Lockshin, R. Cook and A. Rich, Biopolymers, 1994, 34, 663 CrossRef CAS PubMed.
- H. Yokoi, T. Kinoshita and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414 CrossRef CAS PubMed.
- S. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385 CrossRef CAS PubMed.
- T. Y. Cheng, M. H. Chen, W. H. Chang, M. Y. Huang and T. W. Wang, Biomaterials, 2013, 34, 2005 CrossRef CAS PubMed.
- K. Hamada, M. Hirose, T. Yamashita and H. Ohgushil, J. Biomed. Mater. Res., Part A, 2008, 84A, 128 CrossRef CAS PubMed.
- P. Worthington, D. J. Pochan and S. A. Langhans, Front. Radiat. Oncol., 2015, 5, 92 Search PubMed.
- S. Hsu, Y. Lin, J. Chang, Y. Lin and H. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881 RSC.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024 RSC.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315 CrossRef CAS PubMed.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 6595 RSC.
- M. Zhou, A. M. Smith, A. K. Das, M. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523 CrossRef CAS PubMed.
- W. T. Truong, W. Su, D. Gloria, F. Braet and P. Thordarson, Biomater. Sci., 2015, 3, 298 RSC.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365 CrossRef CAS.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587 RSC.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447 CrossRef CAS PubMed.
- Z. J. Qiu, H. T. Yu, J. B. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342 RSC.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848 CrossRef CAS PubMed.
- A. D. Martin, A. B. Robinson and P. Thordarson, J. Mater. Chem. B, 2015, 3, 2277 RSC.
- P. S. Kubiak, S. Awhida, C. Hotchen, W. Deng, B. Alston, T. O. McDonald, D. J. Adams and P. J. Cameron, Chem. Commun., 2015, 51, 10427 RSC.
- A. D. Martin, A. B. Robinson, A. F. Mason, J. P. Wojciechowski and P. Thordarson, Chem. Commun., 2014, 50, 15541 RSC.
- D. J. Adams, M. F. Butler, W. J. Firth, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- S. Fleming, P. W. J. M. Frederix, I. R. Sasseli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510 CrossRef CAS PubMed.
- D. M. Kirchmajer, B. Steinhoff, H. Warren, R. Clark and M. in het Panhuis, Carbohydr. Res., 2014, 388, 125 CrossRef CAS PubMed.
- H. Warren and M. in het Panhuis, Synth. Met., 2015, 206, 61 CrossRef CAS.
- H. Warren, R. D. Gately, P. O’Brien, R. Gorkin and M. in het Panhuis, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 864 CrossRef CAS.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651 CrossRef CAS PubMed.
- P. J. H. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Scwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte and A. E. Rowan, Nature, 2013, 493, 651 CrossRef CAS PubMed.
- S. R. Kandukuri, L. Y. Jiao, A. B. Machotta and M. Oestreich, Adv. Synth. Catal., 2014, 356, 1597 CrossRef CAS.
- Z. Yang, J. Wang, L. Li, C. Ye and H. Liu, J. Heterocycl. Chem., 2009, 46, 788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures including additional synthetic procedures, NMR and microscopy. See DOI: 10.1039/c6sm00025h |
| This journal is © The Royal Society of Chemistry 2016 |