
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled fibrillar networks of a multifaceted chiral squaramide: supramolecular multistimuli-responsive alcogels†
Jana
Schiller
a,
Juan V.
Alegre-Requena
ab,
Eugenia
Marqués-López
b,
Raquel P.
Herrera
b,
Jordi
Casanovas
c,
Carlos
Alemán
d and
David
Díaz Díaz
*ae
aInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 941 9434121; Tel: +49 941 9434373
bLaboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain
cDepartament de Química, EPS, Universitat de Lleida, Jaume II 69, 25001 Lleida, Spain
dDepartament d'Enginyeria Química – ETSEIB and Center for Research in Nano-Engineering, Universitat Politècnica de Catalunya, Av. Diagonal 647, 08028 Barcelona, Spain
eIQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain
First published on 11th April 2016
Abstract
Chiral N,N′-disubstituted squaramide 1 has been found to undergo self-assembly in a variety of alcoholic solvents at low concentrations leading to the formation of novel nanostructured supramolecular alcogels. The gels responded to thermal, mechanical, optical and chemical stimuli. Solubility studies, gelation ability tests and computer modeling of a series of structurally related squaramides proved the existence of a unique combination of non-covalent molecular interactions and favorable hydrophobic/hydrophilic balance in 1 that drive the anisotropic growth of alcogel networks. The results have also revealed a remarkable effect of ultrasound on both the gelation kinetics and the properties of the alcogels.
Introduction
Squaramides are four-membered vinylogously conjugated diamides derived from squaric acid and possess a very high synthetic versatility.1 Overall, the field of squaramides has experienced an extraordinary growth since the pioneering study of Rawal and co-workers2 using these compounds as organocatalysts. Moreover, squaramides have been recognized as bioisosteres of ureas3 exhibiting appealing properties for diverse research areas beyond organocatalysis,4–6 such as biomedicine,1 synthesis7 and crystal engineering.8–12 One of the most important features of these scaffolds is their capacity for selectively binding to different hydrogen bond acceptors through their acidic NH groups. Remarkably, squaramides play a dual role as they are also able to act as good hydrogen bond acceptors, making them suitable as receptors for cation recognition. This property has been explored to find promising candidates for the development of novel drugs1,13,14 and in the field of organocatalysis as chiral hydrogen bond donor catalysts.4 However, despite the increasing number of squaramide-based compounds in the literature, the study of their self-assembly properties in solution for the synthesis of soft functional materials has remained virtually unexplored.15Within this context, hierarchical gel-based materials have received increasing attention over the last decades16–27 due to their unique architectures and potential for high-tech applications in numerous fields including, among others,28–32 the fabrication of sensors,33 liquid crystals,34 electrically conductive scaffolds,35,36 templates for the growth of cells and inorganic structures,37,38 chemical catalysis,39 as well as in cosmetic and food industries.16 In contrast to chemical gels40–42 that are based on covalent bonds, (e.g., cross-linked polymers), physical gels1,43–50 are typically made of low molecular weight compounds self-assembled through non-covalent interactions (e.g., hydrogen-bonding, van der Waals, charge-transfer, dipole–dipole, π–π stacking and coordination interactions), which usually provides reversible gel-to-sol phase transitions as response to environmental stimuli.51,52 Systems based on both types of connections are also known.53,54 The solid-like appearance and rheological properties of the gels result from the immobilization of the liquid (major component) into the interstices of a solid matrix (minor component) through capillary forces.51,55 The formation of the 3D-network with numerous junction zones56,57 is a consequence of the entanglement of 1D-polymeric strands of gelator molecules16 typically of nm diameters and μm lengths.58,59
Herein, we report the unprecedented self-assembly properties of a chiral squaramide in numerous alcoholic solvents leading to the formation of multistimuli-responsive supramolecular alcogels. The remarkable effect of ultrasound during the preparation of the gels as well as the plausible gelation mechanism supported by quantum mechanical calculations are also discussed. This work expands the applications of squaramides towards materials synthesis.
Experimental
(A) Synthesis and characterization of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 569, 1798, 1656, 1650, 1572, 1521, 1465, 1342, 1162, 1094, 814, 665, 575, 549, 419. MS (ESI+) 470.3 [M + Na]. HRMS (ESI+) calcd for C23H33N3NaO4S 470.2084; found 470.2066 [M + Na].
569, 1798, 1656, 1650, 1572, 1521, 1465, 1342, 1162, 1094, 814, 665, 575, 549, 419. MS (ESI+) 470.3 [M + Na]. HRMS (ESI+) calcd for C23H33N3NaO4S 470.2084; found 470.2066 [M + Na].
(B) Preparation and characterization of gel materials
FTIR spectra were recorded using an Agilent Carry 630 spectrophotometer (Universität Regensburg) spectrophotometer.
Morphological characterization of the bulk samples was carried out by field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM). (a) FESEM: images were obtained with a Carl Zeiss Merlin field emission scanning electron microscope (0.8 nm resolution) equipped with a digital camera (SAI, Universidad de Zaragoza) and operating at 3 kV and 158 pA. Sample preparation: xerogel specimens were prepared by freeze-drying. Prior to imaging, a 5 nm sized Pt film was sputtered (40 mA, 30 seconds) on the samples placed on carbon tape. (b) TEM: images were recorded using a JEOL-2000 FXII transmission electron microscope (0.28 nm resolution) equipped with a CCD Gatan 694 digital camera (SAI, Universidad de Zaragoza) and operating at 200 kV (accelerating voltage). Sample preparation: 10 μL of the gel suspension was allowed to adsorb onto carbon-coated grids (300 mesh, from Aname). The grid was placed over a piece of filter paper in order to absorb the excess of solvent. (c) AFM: imaging was performed on a Multimode 8 (Bruker) instrument (LMA-INA, Zaragoza) in tapping mode at 0.5 Hz scanning rate using freshly cleaved mica surface as substrate and a single crystal silicon tip (TAP150A, 0.01–0.025 Ω cm, Antimony doped Si) at 128–152 kHz drive frequency. Drive amplitude ranged from 300 to 400 mV. Sample preparation: 10 μL of the alcogel suspension (ca. 5-fold dilution in MeOH) was placed on the substrate and homogeneously dispersed to form a thin layer that was allowed to dry in air overnight before measurement.
The growth of crystals in the gel phase was monitored using a Wild Makroskop M420 optical microscope equipped with a Canon Power shot A640 digital camera (Universität Regensburg). A polarization filter was used to observe the birefringence of the gels under polarized light.
Temperature-dependent 1H-NMR studies were carried out on a 400 MHz Bruker Avance instrument (Universität Regensburg) equipped with a BVT 2000 heating system (Bruker BioSpin GmbH).
Powder X-ray diffraction (PXRD) patterns were collected on a STOE STADI P powder diffractometer (Universität Regensburg) (StartFragment Transmission mode, flat samples, Dectris MYTHEN 1k microstrip solid-state detector) with CuKα1 radiation operated at 40 kV and 40 mA. Conditions: (a) 0.015°, time 1200 s per step, 2 theta range 10–55° (short measurement); (b) StartFragment 0.015°, time 320 s per step, 2 theta range 2–90° (long measurement).
UV-vis spectra were recorded in a Varian Cary 50 Bio UV-visible spectrophotometer.
Critical gelation concentrations (CGC) were determined by adding solvent in several portions (0.1 mL each) into the vial until everything remained dissolved, precipitation and/or gelation took place upon heating–cooling (and/or ultrasound) treatment as described above. The initial concentration for gelation tests was 100 g L−1.
Gel-to-sol transition temperatures (Tgel) were determined by the inverse-flow method by placing the sealed vials containing the organogels into a thermoblock (Fig. S1, ESI†), which was heated up at the rate of 2 °C min−1. In this work, the temperature at which the gel started to break was defined as Tgel. The average values of at least two random experiments were given. These values were correlated with the first differential scanning calorimetry (DSC) endothermic transition of model using a DSC7 (Perkin Elmer) instrument (Universität Regensburg) at a scan rate of 2 °C min−1 under nitrogen atmosphere (gas flow rate = 20 mL min−1). For the measurements, an appropriate amount of gel was placed into a pre-weighted Al pan (Perkin-Elmer), which was sealed and weight on a six-decimal plate balance. The pans were weighted again after each measurement to check for possible leakage.
| Entry | Solventa | Phaseb | Optical appearance | CGCe (g L−1) | Gelation timef | T gel (°C) | Procedureh | Temporal stabilityk |
|---|---|---|---|---|---|---|---|---|
| a Solvent volume = 1 mL. Systematic IUPAC name is provided for each case. b Abbreviations: G = gel; U = undissolved gelator; C = crystals. c Approximately 10% of the total volume remained ungelled. d Cluster of white needles are visible within the fresh gel sample, which turned into a transparent gel after 12 days (vide infra). e Values calculated by the inverse-flow method. Estimated error ± 0.5–1 g L−1. f Abbreviations: h = hour; min = minute; d = day. Unless otherwise noted, estimated error ± 1 min (for gelation times >1 min). g Estimated error ± 2 °C. h The heating–ultrasound treatment for the preparation of gels are highlighted in bold font. i To form this gel it was necessary to predissolve 1 in 20 μL of DMSO. j The sample was pretreated by heating–ultrasound as explain in the Experimental section. k The gels were stable at least for the indicated period. Symbol “>” indicates that the stability could possibly be much longer although it was not monitored. Abbreviation: nd = not determined due to unreliable variables; cryst = cyrstallization. | ||||||||
| 1 | Methanol | G | Transparent | 7 | 24 ± 2 h | 45 | Heating–cooling | >6 months |
| 2 | 2-Methylpentan-1-ol | G + U | nd | nd | nd | nd | Heating–cooling | nd |
| 3 | Butan-2-ol | G | Transparent | 20 | 24 ± 6 h | nd | Heating–coolingi | >4 months |
| 4 | Ethanol | G | Translucent | 5.8 | 10 min | 46 | Heating–ultrasound | Cryst. after 18 h |
| 5 | 2-Methylpropan-2-ol | G | Translucent | 3 | 5 min | 57 | Heating–ultrasound | >1 month |
| 6 | Butane-1,4-diol | G | Translucent | 5.5 | 10 min | 47 | Heating–ultrasound | >1 week |
| 7 | 2-Methylpropan-1-ol | G | Translucent | 4 | 1 min | 67 | Heating–ultrasound | >1 week |
| 8 | Phenylmethanol | G | Opaque | 21 | 8 min | 42 | Heating–ultrasound | >2 months |
| 9 | Acetone | G | Translucent | 7.1 | 1 min | 54 | Heating–ultrasound | >1 week |
| 10 | 2-Methylbutan-2-ol | G | Translucent | 3.3 | 5 min | 70 | Heating–ultrasound | >2 weeks |
| 11 | Propan-1-ol | G + C | Opaqued | 5.2 | 2 h | 64 | Heating–cooling | >6 months |
| 12 | Propan-1-ol | G | Translucent | 5.2 | 5 min | 56 | Heating–ultrasound | >4 months |
| 13 | Pentane-1,5-diol | G | Opaque | 10 | 15 min | 47 | Heating–cooling | >2 months |
| 14 | Pentane-1,5-diol | G | Translucent | 6.1 | 12 min | 36 | Heating–ultrasound | >1 week |
| 15 | Butan-1-ol | G | Transparent | 8.4 | 3.5 ± 0.25 h | 39 | Heating–cooling | >1 month |
| 16 | Butan-1-ol | G | Transparent | 8.4 | 6 min | 36 | Heating–ultrasound | >1 month |
| 17 | Hexan-1-ol | G + C | Transparent | 9.1 | 9 min | 35 | Heating–coolingj | >1 week |
| 18 | Hexan-1-ol | G | Transparent | 7 | 1 min | 75 | Heating–ultrasound | >2 months |
| 19 | 3-Methylbutan-2-ol | Gc | Transparent | 5 | 20 ± 4 h | nd | Heating–coolingj | >1 month |
| 20 | 3-Methylbutan-2-ol | G | Translucent | 3.7 | 2 min | 54 | Heating–ultrasound | >2 months |
| 21 | Pentan-2-ol | G | Transparent | 5.5 | 11 ± 1 d | 81 | Heating–cooling | >1 month |
| 22 | Pentan-2-ol | G | Transparent | 5.0 | 1 min | 65 | Heating–ultrasound | >2 months |
| 23 | 2-Methylbutan-1-ol | G + C | Transparent | 4.7 | 2.3 ± 0.15 h | 65 | Heating–cooling | Cryst. during gelation |
| 24 | 2-Methylbutan-1-ol | G | Translucent | 3.9 | 2 min | 53 | Heating–ultrasound | >1.5 months |
(C) Quantum mechanical calculations
All calculations were performed using the M06L functional,63,64 which was combined with the 6-31+G(d,p) basis set65 (i.e., M06L/6-31+G(d,p) level) The M06L is a meta-hybrid generalized gradient approximation (GGA) functional with very good response under dispersion forces, improving one of the biggest deficiencies in density functional theory (DFT) methods. Thus, the M06L functional is able to describe the geometry and interaction energy of complexes stabilized by non-covalent interactions, including π–π stacking, with accuracy close to that of couple cluster calculations with both single and double substitutions.66 All geometry optimizations were performed in methanol (ε = 32.6), which was described through a simple Self Consistent Reaction Field (SCRF) method. More specifically, the Polarizable Continuum Model67,68 (PCM) was used in the framework of the M06L/6-31+G(d,p) level to represent bulk solvent effects.Complexes formed by two interacting molecules (dimers) were considered for model squaramides 1, 3, 7, 8 and 10. For this purpose, around 50 starting geometries were constructed for each dimer by varying the relative position between the two interacting molecules. Accordingly, the formation of different specific and non-specific intermolecular interactions, as well as different geometries for each of such interactions, were considered. All these starting arrangements were submitted to geometry optimization at the PCM-M06L/6-31+G(d,p) level. Interaction energies (ΔEi) were corrected with the basis set superposition error (BSSE) by means of the standard counterpoise (CP) method.69 Calculations were performed using the Gaussian 0970 computer package.
Results and discussion
Design and synthesis of squaramides
In the frame of our interdisciplinary research program focused on the development of new hydrogen bond based functional molecules, some of us have recently focused on the synthesis of new squaramide structures,60,71 due to their important ability for selectively binding through cooperative hydrogen bonds to different acceptors.1,4–7 During the synthesis of squaramide 1 (Scheme 1) we observed a clear tendency to increase the viscosity of the solvent (i.e., MeOH). Hence, we decided to investigate in detail the formation of supramolecular aggregates in solution, after taking also into consideration (a) the facile and scalable synthesis of this family of compounds, (b) the intrinsic potential as highly substituted scaffolds and (c) the isosteric relationship with ureas, which represent ubiquitous synthons in supramolecular chemistry72 including the formation of softgels.73 | ||
| Scheme 1 One-pot synthesis of squaramide 1 in MeOH. | ||
In this context, unsymmetrical substituted squaramide 1 was easily accessible following our own developed procedure in a one-pot synthesis using equimolar amounts of commercially available 3,4-dimethoxycyclobut-3-ene-1,2-dione (11), 4-tert-butylaniline (12a) and N-((1R,2R)-2-aminocyclohexyl)-4-methylbenzenesulfonamide (13a) in methanol at room temperature (Scheme 1). After 4 hours of reaction, squaramide 1 was obtained in very good yield (80%) after simple filtration as a powdered material with mesoscale ordering as indicated by PXRD analysis (Fig. S16, ESI†).
To correlate the structural functionalities of 1 with its gelation properties, we designed and synthesized the library of squaramides outlined in Fig. 1. Specifically, different analogous squaramides 7–10 were designed by incorporating substituents with different steric and electronic properties, following the same synthetic procedure.60,71 For this purpose, squaramides 7 and 9, with electron-withdrawing groups, or squaramide 8 with an electron-donating substituent in the aromatic ring, or 10 in the absence of the aromatic ring, were synthesized with the aim of modifying the basicity and/or conferring distinctive solvation properties to these organic compounds, which is believed to play a key role on the gelation phenomena.16–27 In all these cases (1, 7–10), the stereochemical configuration and the structural core were maintained. Moreover, squaramides 2 and 3 were included for comparison with 1, since we also observed that the viscosity of MeOH increased during their preparation. Additionally, and encouraged by our last work with urea-based compounds as efficient gelators,74 we also carried out the preparation of squaramide 4, which is the equivalent squaramide to the best urea compound used in our previous work.74 Related with compound 4, squaramide 5, lacking the substituent in the aromatic ring, or 6, bearing a methoxy group, were also explored.
 | ||
| Fig. 1 Library of additional squaramides 2–10 synthesized for this work. | ||
All synthesized squaramides 1–10 were appropriately characterized after purification by a simple and straight filtration (see Experimental section).
Solubility and gelation properties
The gelation ability of compound 1 was initially screened for 34 different solvents using the heating–cooling protocol within a broad concentration range (Table 1 and Table S1, ESI†). For instance, squaramide 1 was found to be insoluble in dichloromethane, chloroform, ethyl acetate, diethyl ether, dibutyl ether, toluene, glycerol, 1,3-dichlorobenzene and water upon heating and/or sonication. Unexpectedly, 1 showed an unusual selectivity to form stable gels in a number of alcoholic solvents (alcogels), including primary, secondary, tertiary, lineal and branched alcohols. The only exception to this rule was the gel obtained in acetone. However, it should be considered that although acetone is present at >99% keto form at equilibrium, the formation of hydrogen bonding gel networks could stabilize to some extend the enol form of the acetone (2-propenol). In sharp contrast, no gelation tendency was detected for squaramides 2–10. These results suggested the existence of unique molecular interactions and a hydrophilic/hydrophobic balance that favor the self-assembly of 1 in alcohols. This was later confirmed by computational studies (vide infra).The gel nature of the samples was initially confirmed by the absence of flow upon turning the vial upside-down and further confirmed by oscillatory rheological measurements in model solvents (vide infra). The critical gelation concentration (CGC) values estimated for the gels obtained with 1 ranged between 3 and 21 g L−1 (see the Experimental section for the procedure). These values indicate that hundreds or thousands of solvent molecules are immobilized per gelator molecule (e.g., the highest solvent/gelator ratio ∼ 1750:1 was obtained in methanol). As far as we are aware, the only precedent in the literature describing the formation of an organogel with squaramides corresponds to a recent report from Ohsedo and co-workers15 describing a series of squarylium monoalkylamides and dialkylamides. Therein, gelation was only observed in DMF (forming opaque gels) and in some cases only after cooling down the mixture to −15 °C.
Stable gels were classified in 3 different groups depending on the experimental protocol that was used for their preparation. The first group could only be formed by the classical heating–cooling protocol (entries 1–3), the second group could only be formed by applying heating and subsequent ultrasound treatment (entries 4–10) and the third group could be formed by any of these two methods (entries 11–24). Thus, ca. 47% of the obtained gels could be formed by heating–cooling, whereas ca. 82% could be prepared by a heating–ultrasound protocol (bold text). Although there is not an apparent correlation between the type of alcohol and the gel preparation method, heating followed by ultrasound was found to be a very convenient technique for the gelation of branched alcohols within minutes regardless the degree of substitution at the α-carbon. Based on these groups, we selected one model solvent of each group (i.e., methanol, phenyl methanol and hexan-1-ol) for further characterization and comparisons. So obtained alcogels displayed different degree of translucency (Fig. 2). Such optical differences indicate the formation of aggregates of different sizes.
 | ||
| Fig. 2 Representative photographs of upside-down vials containing organogels made of gelator 1 in different alcohols prepared at the corresponding CGC (Table 1) via heating–cooling (H-C) or heating–ultrasound (H-U). | ||
Ultrasound-induced gelation (sonogelation), mainly by solvent cavitation, has attracted much attention over the last few years.75–78 During our work we found that the application of ultrasound after heating not only allowed the fabrication of stable gels that could otherwise (i.e., via heating–cooling) not be formed, but also decreased the gelation time drastically (e.g., entries 11, 15, 19, 21, 23 vs. entries 12, 16, 20, 22, 24, respectively). The most impressive case was pentan-2-ol that needed more than 10 days to be gelled by heating–cooling and only 1 min by heating–ultrasound even at lower concentration (entry 21 vs. entry 22). This is very appealing since the application of ultrasound to an already isotropic solution seems counterintuitive. However, sonication of the hot clear solution prior gelation may hinder the self-assembled system (non-entangled nanofibers) to fall into its lowest energy level making possible the formation of new gel networks. In addition, both the CGC was reduced in practically all cases in which the heating–ultrasound protocol was used instead of heating–cooling. This was also accompanied by certain reduction of the Tgel values. The case of hexan-1-ol should not be considered prematurely as an exception because the heating–cooling protocol gave a combination of gel and crystals (entry 17 vs. entry 18). Remarkably, the dynamic nature of the gels was evidenced by a gradual increase over time of the Tgel for the gel prepared by heating–ultrasound reaching eventually the same value obtained by heating–cooling (i.e., Tgel (fresh gel made in propan-1-ol) = 56 °C; Tgel (3-month old gel made in propan-1-ol) = 63 °C). As observed with the solid squaramide gelator 1, the xerogels obtained by freeze-drying the corresponding alcogels were found to have mesoscale ordering as demonstrated by PXRD analysis (Fig. S16 and Table S2, ESI†). However, clear differences were observed between both spectra. The peaks observed for the solid sample (i.e., as synthesized 1) are centered at 2θ = 4.41°, 6.71°, 11.51°, 16.35° and 17.90°, corresponding to lattice spacings d of 20 Å, 13.16 Å, 7.68 Å, 5.42 Å and 4.95 Å, respectively (calculated from Bragg's law). In contrast, the peaks centered at 11.51° and 17.90° disappeared completely in the xerogel sample, whereas all other peaks slightly shifted to low angles (i.e., 3.96°, 6.18° and 15.70°, respectively; these peaks correspond to d values of 22.28 Å, 14.3 Å and 5.64 Å, respectively). This suggests that the interactions of the solvent molecules with the gelator (or gelator-based aggregates) during gelation cause disorder and expansion of the potential crystal packing of the gelator, at least to some extent.
In addition, UV-vis analysis during the sol-to-gel transition did not show a shift of the absorption maximum (Fig. S17, ESI†), suggesting that the aggregation mode of 1 in solution (and probably in the solid state, vide infra) resembles that in the gel phase.
:1 v/v water/hexan-1-ol by heating and subsequently submitted to sonication for 1 min. After 30 min of resting, only the organic phase was completely gelled, which was stiff enough to hold the liquid water phase upon inversion of the vial. Due to the partial solubility of hexan-1-ol in water (i.e., 5.9 g L−1 at 20 °C), the aqueous phase was not completely clear after gelation of the alcohol. However, after 24 h the water phase got clearer and the organic phase more turbid (Fig. 3). This was presumably due to the disruption of the microemulsion and diffusion of the tiny fraction of gelator molecules that could be trapped into the aqueous phase. Phase-selective gelation using water-soluble alcohols such as methanol was not successful. The addition of 10–20% water either before or after gel formation resulted in the collapse of the gel after several days caused by a thick precipitation (Fig. S14, ESI†).
 | ||
| Fig. 3 Phase-selective gelation and change in opacity of hexan-1-ol/water mixture after 24 h (gelator 1, c = 7 g L−1). Total volume = 2 mL. Yellow arrow indicates the boundary between phases. | ||
Thermal and temporal stability of alcogels
In general, the supramolecular gels were thermoreversible and stable to multiple heating–cooling cycles. The gel-to-sol transition temperatures (Tgel) were estimated by the inverse flow method (IFM). Due to the dependence of this method with the cooling rate and thermal history of the sample, we confirmed the good agreement of the Tgel with the first endothermic transition observed by differential scanning calorimetry (DSC) of a model gel (Fig. S5, ESI†).As usually observed in physical gels, Tgel values increased as the gelator concentration increased until reaching a plateau before visible crystal nucleation. For example, Tgel of the gel made in methanol increased ca. 23 °C upon increasing ca. 3-fold the concentration of 1 with respect to the CGC (7 g L−1). Crystal formation was observed over 22 g L−1 within 10 min (Fig. 4). Although these results might be in agreement with the formation of more entangled networks at higher gelator concentrations, the change in Tgel is likely due to solution thermodynamics where the phase boundary, or liquidus line, increases at low concentration and levels off at higher concentration.
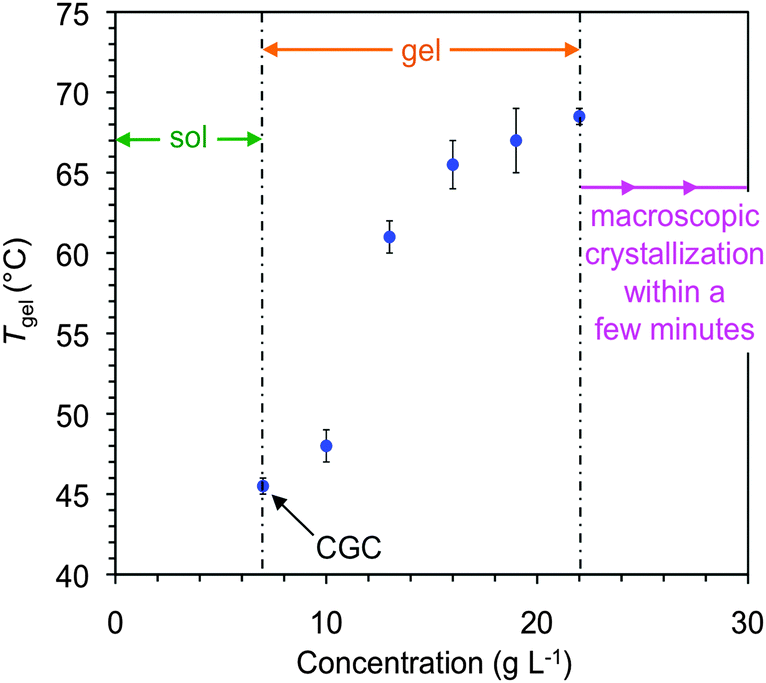 | ||
| Fig. 4 Phase diagram and variation of Tgel as a function of the concentration of gelator 1 in methanol. | ||
Very interestingly, in several cases we have observed an enhancement of both the gelation kinetics and the gel stability when the gels were reformed after being thermally destroyed for the first time. We believe that the gel-to-sol thermal transition could be controlled to avoid the complete dissociation of the supramolecular network (even when behaving as a fluid from the rheological standpoint). This could preserve the thermodynamically more stable aggregates through a self-sorting mechanism, thus providing a more robust starting platform for rebuilding the gel structure. We believe that such “preformation hypothesis” could be more general and become an important contribution to the gelation theory. We are currently studying this in detail and we will publish our results at due course.
On the other hand, gel-to-crystal transitions have been previously related to Ostwald rule of stages.80,81 The subtle equilibrium between the metastable gel phase and the thermodynamically stable crystalline phase82–85 could also be monitored in several of our alcogels. For instance, macroscopic crystallization in the form of long needles was observed first at the interface between air and the developing gel phase made of 1 in 2-methylbutan-1-ol, hexan-1-ol or propan-1-ol via heating–cooling (Fig. 5). Nevertheless, the robustness of the gel phase allowed in most of the cases the coexistence of crystalline and gel phases for at least one month supporting the inversion of the test tube. In general, the alcogels prepared via heating–ultrasound remained stable for several months without visible crystallization when stored in dark at room temperature.
 | ||
| Fig. 5 (A) Digital photograph showing crystal growth in the gel made of 1 in 2-methylbutan-1-ol (c = 4.7 g L−1) by heating–cooling. (B) Optical microscope picture of the crystals observed in (A). (C) Gradual crystal growth observed during the gelation of hexan-1-ol using 1 (c = 9.1 g L−1) and the heating–cooling protocol. The gelator concentration corresponds to the CGC (Table 1). | ||
Although gel-to-gel transitions have already been reported,86–89 it is still a rare phenomenon. The gels made in phenylmethanol showed an optical transition from transparent to opaque within 30 min at the CGC (Fig. 6A). This behavior is typically observed during the growth of fibrilar aggregates over time. Intriguingly, the uncommon opposite phenomenon was observed with the alcogel prepared in propan-1-ol by heating–cooling. In this case, initial macroscopic crystallization was observed during the gelation process leading to a transient opaque gel, which turned into a thermodynamically stable transparent gel within 12 days (Fig. 6B). However, additional detailed experimentation to determine the exact packing arrangement of the gelator molecules, and prove its gradual change, is still necessary in order to confirm the apparent gel-to-gel transition in this case.
 | ||
| Fig. 6 (A) Transparent-to-opaque transition recorded for the gel made of 1 in phenylmethanol (c = 21 g L−1) within 30 min after gel formation via heating–ultrasound. (B) Opaque-to-transparent transition observed in the gel made of 1 in propan-1-ol (c = 5.2 g L−1) within 12 days after gel formation via heating–cooling. The concentrations of gelator correspond to the CGC (Table 1). | ||
Influence of gelator enantiopurity on gel formation
Chirality plays a fundamental role in the formation of supramolecular gel networks.90 Therefore, we also examined the influence of the enantiomeric purity of 1 on its gelation ability. Samples with different enantiomeric excesses (ee) were prepared by combining (R,R)-1 with the appropriate amount of the racemic compound, which was prepared following the general synthetic procedure using the corresponding racemic diamine (see Experimental section). Stable-to-inversion gels could only be formed with the enantiomerically pure (R,R)-1. The squaramide with 80% ee yielded a weak partial gel after 88 h. Macroscopic crystallization of the gelator was observed after 9 days. The use of 1 with ee < 80% led to snow flake-like precipitation of 1 without any gel formation (Fig. 7). | ||
| Fig. 7 Influence of enantiomeric purity of gelator 1 on its gelation ability in methanol (c = 7 g L−1) monitored over time. Enantiomeric excesses are given in percentages. | ||
Driving forces for molecular aggregation
The comparison of FTIR spectrum of solid squaramide 1 with those of the xerogel (prepared by freeze-drying the corresponding alcogel) and 1 in solution revealed only slight alteration with small frequency shifts to lower wavelengths (Δλ < 4 cm−1) for characteristic peaks (e.g., stretching absorptions bands for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ∼1658–1794 cm−1; secondary amines ∼3210–3345 cm−1; aromatic C–H ∼2910–3005 cm−1; aromatic C–N ∼1265–1370 cm−1) (Fig. S4, ESI†). This suggests that 1 may also be aggregated in the solid state involving intuitive hydrogen bonding and π-stacking interactions similarly to its analogue urea-based organogelator.74 This is also in agreement with the results obtained by UV-vis measurements during the sol-to-gel transition (vide supra).
O ∼1658–1794 cm−1; secondary amines ∼3210–3345 cm−1; aromatic C–H ∼2910–3005 cm−1; aromatic C–N ∼1265–1370 cm−1) (Fig. S4, ESI†). This suggests that 1 may also be aggregated in the solid state involving intuitive hydrogen bonding and π-stacking interactions similarly to its analogue urea-based organogelator.74 This is also in agreement with the results obtained by UV-vis measurements during the sol-to-gel transition (vide supra).
On the other hand, temperature-dependent 1H-NMR experiments could provide additional information regarding phase interconversions. However, it should be noted that NMR signals of gelator molecules are unlikely to be observed in the gel phase due to long correlation times. Therefore, the observed signals are typically ascribed to small amounts of gelator molecules dissolved in the immobilized solvent.91,92 Preliminary NMR measurements of the alcogel made of 1 in d4-methanol with increasing temperature from 25 °C to 60 °C showed gradual chemical shifts (δ) of almost all protons of 1 suggesting at least conformational changes and/or expected modification in the aggregation pattern (e.g., through hydrogen bonding, π-stacking and/or electrostatic interactions) (Fig. S6, ESI†).
In contrast to the PXRD pattern of compound 1, non-gelators 2–10 showed additional peaks after 17° (Fig. S16 and Table S2, ESI†). Some of these compounds showed even higher crystallinity than the squaramide gelator 1 (e.g., compounds 5, 7). On the other hand, the two major peaks at low angle (around 4° and 6°) found in the xerogel of 1 do not appear in the other solid compounds, except compound 7 that also showed two peaks in the same region as well as the new additional peaks at higher angle. In other compounds (e.g., compounds 6, 8) only one of the two mentioned peaks was present together with the additional peaks at higher angle. Thus, although we can not provide a unique reason why squaramide 1 is the only one that forms gels, it seems that the complex gelation phenomenon, frequently defined as a “frustrated crystallization”, finds here a unique example, where both the structure of the gelator and the solvent must play a crucial and synergistic role in turning the phase balance towards gelation in the case of compound 1vs.2–10.
In order to gain a clearer insight into the potential aggregation mode of 1 and its unique gelation ability, comparative PCM-M06L/6-31+G(d,p) calculations in methanol were carried out on model dimers of 1, 3, 7, 8 and 10. It should be remarked that this methodology, which is not appropriated to evaluate why only alcohols and acetone give gels, is suitable to describe precisely specific interactions and evaluate differences among the interaction patterns in the different dimers. Moreover, 1, 3, 7, 8 and 10 are similar enough to represent the solvent using a polarized continuum dielectric medium, like the PCM one. Accordingly, these calculations do not intend to ascertain the role of the solvent in the gelation mechanism but differences among molecules in a given solvent. For simplicity, analyses of the results were restricted to the minima of lower relative energy, which correspond to those with more attractive interaction energies (ΔEi). Thus, 13 of the 22 minima obtained for 1 exhibit ΔEi values lower than −6 kcal mol−1. More specifically, ΔEi ranges from −26.1 to −6.1 kcal mol−1 (Fig. 8).
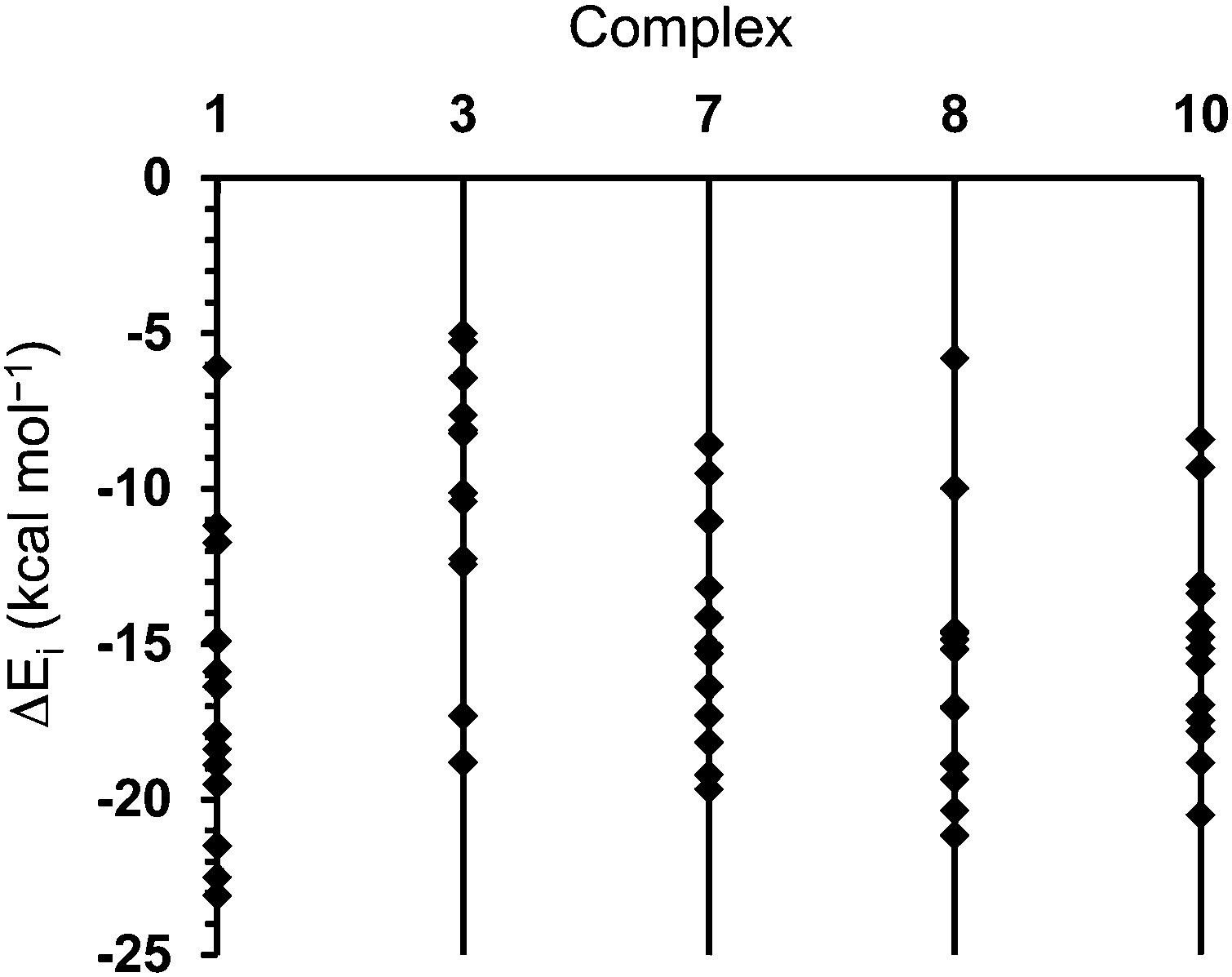 | ||
| Fig. 8 For the most stable complexes of each family, representation of the interaction energy (ΔEi) of complexes calculated for 1, 3, 7, 8 and 10 with ΔEi ≤ 5.0 kcal mol−1. | ||
Inspection of the lowest energy dimer (Fig. 9A) revealed that the squaramide moiety of each molecule is involved in different intermolecular interactions. In addition to the conventional N–H⋯O hydrogen bond, which involves the squaramides of the two molecules, this dimer also exhibits stabilizing N–H⋯π and C–H⋯π intermolecular interactions. In addition to such interactions, the two molecules involved in this complex show intramolecular π⋯π stacking interactions, as is illustrated for one of the two monomers (Fig. 9A, right). These attractive interactions involve three aromatic rings forming a multiple phenyl⋯phenyl and squaramide⋯phenyl stacking, which provide extra stability to the complex. Moreover, careful inspection of the complexes ΔEi < −15 kcal mol−1 (9 of the 13 minima represented in Fig. 8) revealed that similar inter- and intramolecular interactions coexist in all cases. However, although in all cases intramolecular interactions involve the stacking of the three aromatic rings, intermolecular interactions typically consists of two CO⋯H–N hydrogen bonds involving the squaramides or a single SO⋯H–N hydrogen bond. This is illustrated in Fig. 9B and C, which display the second (ΔEi = −22.5 kcal mol−1) and third minima (ΔEi = −21.5 kcal mol−1) in the energy ranking, respectively.
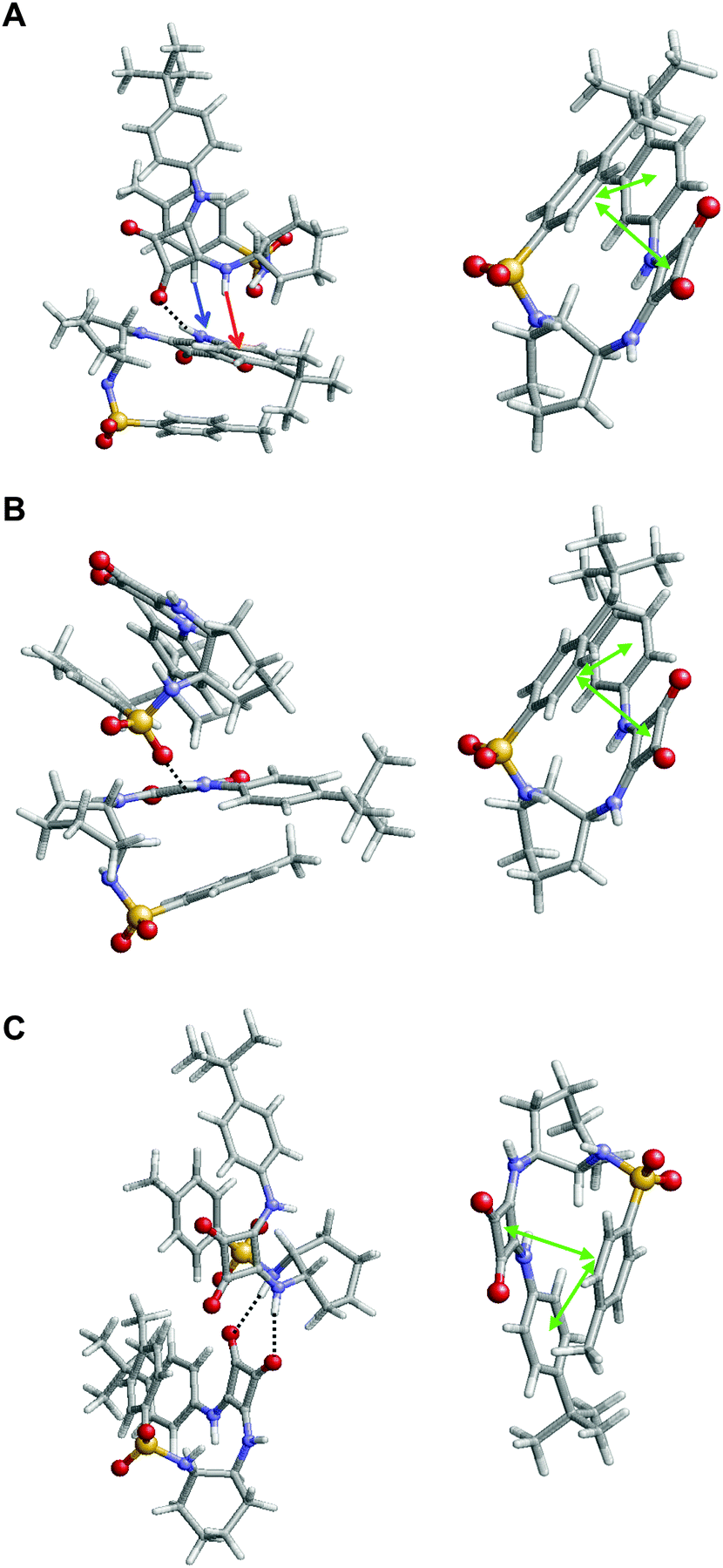 | ||
| Fig. 9 Representation of the three most stable complexes obtained for 1: (A) ΔEi = −26.1 kcal mol−1; (B) ΔEi = −22.5 kcal mol−1 and (C) ΔEi = −21.5 kcal mol−1. Intermolecular (left) and intramolecular (right) interactions are displayed for the complex and a single molecule of the complex, respectively. Hydrogen bonds are represented by black dashed lines, N–H⋯π and C–H⋯π interactions by read and blue arrows, respectively, and intramolecular π⋯π stacking interactions by green double arrows. | ||
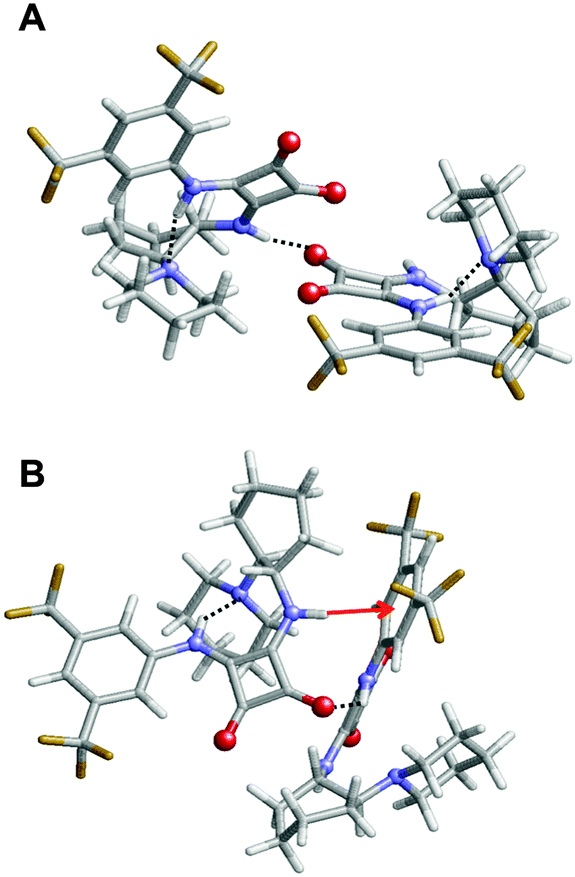 | ||
| Fig. 10 Representation of the two most stable complexes obtained for 3: (A) ΔEi = −18.8 kcal mol−1 and (B) ΔEi = −17.3 kcal mol−1. Intermolecular and intramolecular interactions are displayed for each complex. Inter- and intramolecular hydrogen bonds are represented by black dashed lines, and N–H⋯π interactions by read arrows. | ||
Complexes of 3 tend to be stabilized by intermolecular N–H⋯O hydrogen bonds and N–H⋯π interactions, while intramolecular interactions consists of N–H⋯N hydrogen bonds (Fig. 10). It should be noted that, in comparison with 1, the lack of the third aromatic ring absence precludes the formation of intramolecular π⋯π stacking interactions. Comparison of complexes obtained for 1 and 3 suggests that both the lower number of intermolecular hydrogen bonds and the lack of intramolecular π⋯π stacking interactions are responsible for the relatively high ΔEi values and poor stability of the latter with respect to the former.
In comparison with 3, the interaction patterns of complexes derived from 7, 8 and 10 are more similar to those observed for 1, even though there are also some differences among the formers and the latter. Thus, the lower energy complexes calculated for 7 and 10 exhibited two N–H⋯O intermolecular hydrogen bonds (Fig. S19 and S20, ESI,† respectively), while those derived from 8 show only one of such interactions (Fig. S21, ESI†). Furthermore, no N–H⋯π and C–H⋯π intermolecular interactions were detected in 7, 8 and 10. Accordingly, intermolecular interactions seem to be weaker for 8 than for 7, 10 and, specially, 1.
Regarding to intramolecular interactions 7, 8 and 10 display π⋯π stacking, even though they also present some relevant differences. Specifically, such π⋯π interactions are apparently weaker for 7 and 10 than those obtained for 1 and 8. Thus, three aromatic rings participate in each π⋯π interaction identified in complexes of the two latter compounds (Fig. 9 and Fig. S21, ESI†), while only two aromatic rings are involved in those found in the complexes of the formers (Fig. S19 and S20, ESI†). The reduction in the intensity of π⋯π interactions for 7 and 10 has been attributed to the repulsive effects provoked by the fluoride substituents and to replacement of an aromatic ring by an aliphatic chain, respectively. These results clearly indicate that dimers calculated for 1 show a large and complex network of inter- and intramolecular interactions that provides an additional stability to the complex. This network of interactions becomes weaker and smaller for the complexes that involve the rest of compounds and, therefore, such extra stability disappears.
Additional PCM-M06L/6-31+G(d,p) calculations were performed for dimers of 1 in water and chloroform (ε = 4.7 and 78.4, respectively). Results were similar to those obtained in methanol suggesting that the influence of the solvent on the gelation ability of this compound is not due to simple electrostatic effects induced by the polarization of the solvent but to the role of the first solvation shell in the network of inter- and intramolecular interactions discussed above. Unfortunately, continuum models, like PCM, are unable to describe the effects associated to solute⋯solvent interactions in the first solvation shell. Therefore, considering the remarkable selectivity of the gelator for alcoholic solvents, the differences in PXRD patterns (e.g., as synthesized gelator vs. xerogel) and the standard gelation mechanism associated to supramolecular gels,1,43–50 we should not dismiss the potential role of at least some solvent molecules acting, for instance, as bridges between gelator molecules. This has been proved by additional PCM-M06L/6-31+G(d,p) calculations on complexes formed by two interacting molecules of 1 and two methanol molecules positioned between them (Fig. S22, ESI†). It is worth noting that each molecule of 1 interacts simultaneously with methanol and with the second molecule of 1. Unfortunately, the theoretical methodology used in this work is suitable to examine the ability of the different compounds to form intermolecular interactions but it is not appropriated for a systematic exploration of this bridge-like gelation mechanism. Thus, due to the huge number of possibilities, other theoretical approaches based on statistical sampling procedures would be more appropriated for such purpose.
Morphological properties of alcogels
Field emission scanning electron microscopy (FESEM) of the corresponding xerogels showed a remarkable influence of the solvent on the morphologies. For instance, straight laths of ca. 0.2–1.4 μm in width were observed for gel made in phenylmethanol (Fig. 11B), whereas dense fibrilar networks with entangled fibers of ca. 10–20 nm in diameter were characteristic of the gels made in hexan-1-ol (Fig. 11C and D) and methanol (Fig. 11A). The exact molecular mechanism associated with each morphology remains unclear. No major differences were apparently observed for the gel phase obtained either by heating–cooling or heating–ultrasound for the gel made in hexan-1-ol (Fig. 11C, D and Fig. S8, ESI†). However, transmission electron microscopy (TEM) seems to reveal a less dense fibrilar network for the gel prepared by heating–ultrasound (Fig. 12 and Fig. S9, ESI†).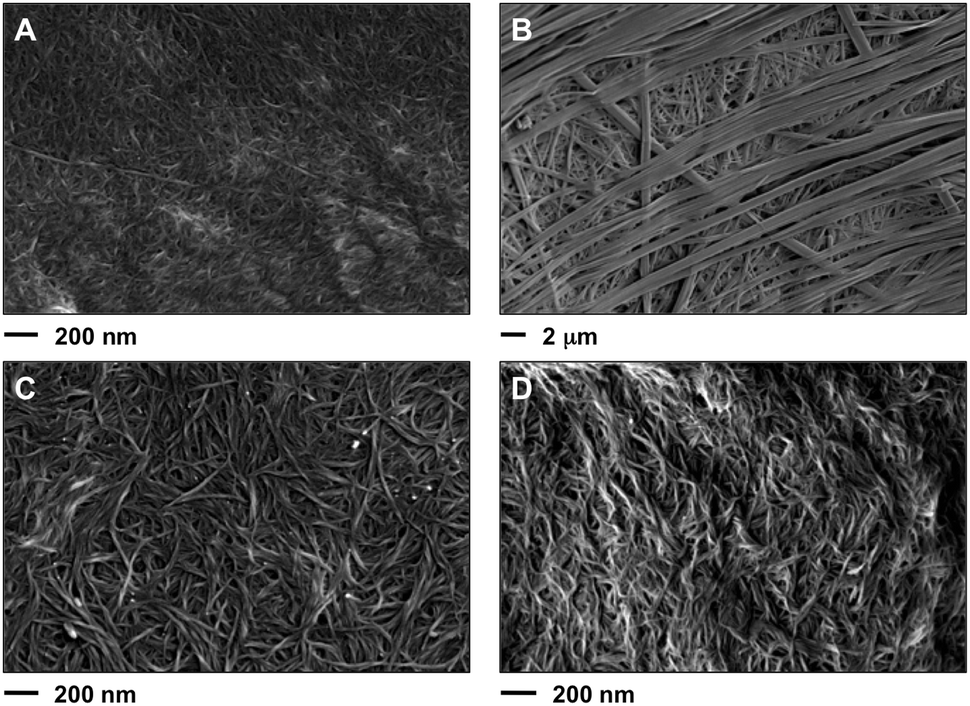 | ||
| Fig. 11 Representative FESEM images of the xerogels prepared by freeze-drying of the corresponding organogels made of 1 in different solvents at their CGC (Table 1). (A) Methanol (heating–cooling, c = 7 g L−1), (B) phenylmethanol (heating–ultrasound, c = 21 g L−1), (C) hexan-1-ol (heating–ultrasound, c = 7 g L−1) and (D) hexan-1-ol (heating–cooling, c = 9.1 g L−1). Note: the selected images are representative of the bulk material. For additional images see ESI.† | ||
 | ||
| Fig. 12 Representative TEM images of xerogels of the corresponding organogels made of 1 in different solvents at the CGC (Table 1): (A) methanol (heating–cooling, c = 7 g L−1), (B) hexan-1-ol (heating–ultrasound, c = 7 g L−1). Note: the selected images are representative of the bulk material. For additional images see ESI.† | ||
As expected, complementary fibril structures with average heights between ca. 4 nm and 10 nm were also observed by atomic force microscopy (AFM) (Fig. 13). Certain fiber helicity was detected by FESEM and AFM imaging (Fig. S10, ESI†). Although we have recorded images of the nanostructures by different techniques in order to identify any possible artifact, it should be emphasized that important changes in the structures could occur during preparation of the samples and, therefore, the interpretation of the images should always be done carefully without overselling.
 | ||
| Fig. 13 Representative AFM images of the gel made of 1 in methanol (heating–cooling) at the CGC (c = 7 g L−1). Note: the selected images are representative of the bulk material. For additional images see ESI.† | ||
In addition, the anisotropic and thermoreversible features of the alcogels allowed turning on/off their birefringence under polarized light (Fig. S11, ESI†). The ability to control the refractive index of soft materials constitutes an important property for applications in optical devices.93
Rheological properties of alcogels
The viscoelastic properties of the model gels were confirmed by oscillatory rheological measurements (Fig. S7, ESI†). The storage (G′) and loss modulus (G′′) were measured at room temperature as a function of the shear strain (dynamic strain sweep, DSS) and the frequency (dynamic frequency sweep, DFS) to define the linear viscoelastic regime of the material (Fig. 14 and Fig. S7, ESI†). Although all gels should be defined as weak gels according to the moduli values, reasonably constant values of the tanδ (G′′/G′) during the measurements indicated a relative good tolerance of the materials towards low external forces. Moreover, the value of the storage modulus was always ca. one order of magnitude higher than the loss modulus. The aging stability of the gels at room temperature was confirmed by dynamic time sweep (DTS) measurements within the predetermined linear limits (i.e., 0.1% strain, 1 Hz frequency).
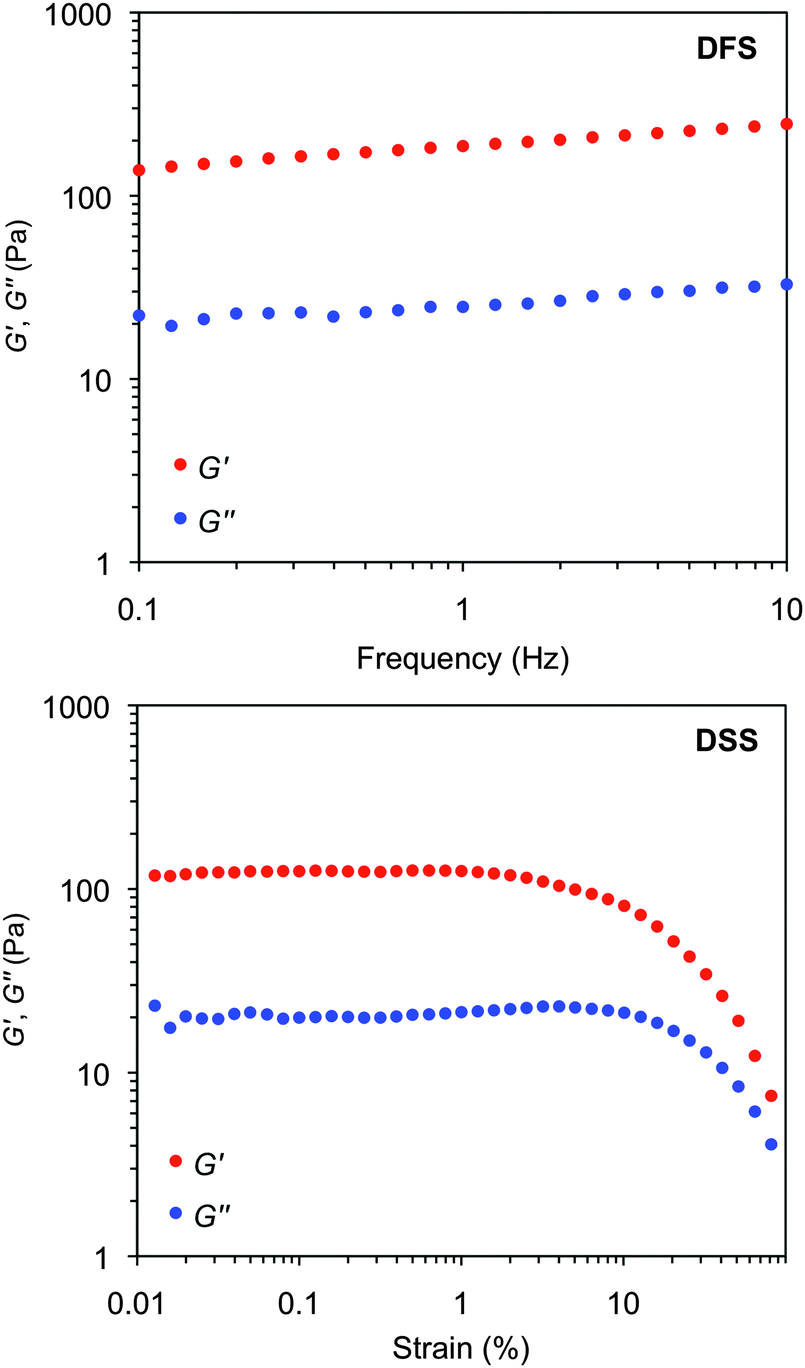 | ||
| Fig. 14 Representative oscillatory rheological plots of the model gel made of 1 in methanol at CGC (c = 7 g L−1). Top: DFS experiment. Bottom: DSS experiment. | ||
Furthermore, these gels were also found to be thixotropic,94 a key property for real-life applications of many gel-based materials.95 This behavior was confirmed by a three-steep rheological loop test consisting of (1) application of shear strain as defined by DTS experiments (G′ > G′′), (2) subsequent increase of the strain until the gel collapses (G′ < G′′) and (3) return to the starting strain value (G′ > G′′). Full recovery of the gel strength was observed within seconds after each cycle (Fig. 15). This self-healing behavior was also macroscopically observed upon vigorous shaking of the vial containing the bulk gel followed by a resting period (Fig. S18F, ESI†).
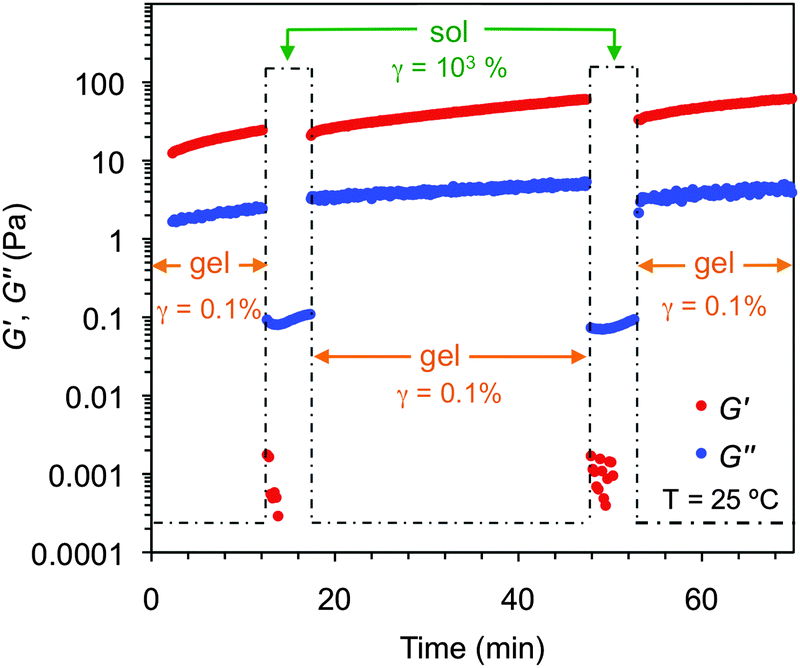 | ||
| Fig. 15 Rheological loop test of the gel made of 1 in butan-1-ol at CGC (c = 8.4 g L−1). Steps: (1) 1 Hz, 0.1% strain, 10 min; (2) 0.1 Hz, 1000% strain, 5 min; (3) 1 Hz, 0.1% strain, 30 min. Steps 2 and 3 were repeated for a second cycle. | ||
Responsiveness to external ions
Since squaramide derivatives are known to coordinate to anions at the N–H groups and to cations at the carbonyl moiety, we expected a response of the alcogels to the presence of external ions. In order to confirm this, a small amount of different metal salt solutions in methanol (i.e., FeCl3, Ni(OAc)2·3H2O, NiCl2·6H2O, Cu(NO3)2 and CuCl2) were layered over 1 mL of the model gel made of 1 in methanol (c = 10 g L−1). Potential complexation of the ions with the gelator molecule may disrupt the nanofibers causing the gradual collapse of the gel network. As a matter of fact, this was observed in preliminary experiments upon addition of either 30 mM FeCl3 or 60 mM NiCl2·6H2O stock solutions within 3.5 h and 6 h, respectively (Fig. 16C and D). Interestingly, the addition of 60 mM CuCl2 or Ni(OAc)2·3H2O did not cause visible decomposition of the gel network within 72 h (Fig. 16A and B), suggesting different effects of metal ions and counterions on the gel stability. The response of the gel was independent on the procedure used for the preparation of the hybrid material (Fig. 16E). Future research is still necessary to elucidate the exact nature of the metal complexes and the dominant mechanism for the gel destruction. | ||
| Fig. 16 Effect of external ions on the stability of the gel made of 1 in methanol (c = 10 g L−1). (A) 60 mM CuCl2: stable gel. (B) 60 mM Ni(OAc)2·3H2O: stable gel. (C) 30 mM FeCl3: gel decomposition in 3.5 h. (D) 60 mM NiCl2·6H2O: gel decomposition within 6 h. (E) Left: Addition of 1 to metal solution (30 mM FeCl3) followed by heating–cooling. Right: Addition of metal solution (30 mM FeCl3) to preformed gel via heating–cooling. Gel colours correspond to the colours of the metal salt solutions. | ||
Conclusions
The foregoing results demonstrate that chiral N,N′-disubstituted squaramide 1 self-assembles selectively in a large variety of alcohols at concentrations ranging from 3 to 21 g L−1. This phenomenon leads to the formation of nanostructured supramolecular alcogels with the ability to response to thermal, mechanical, optical and chemical stimuli. Experimental gelation tests and computer modeling of a series of structurally related squaramides (2–10) demonstrated the existence of a unique combination of non-covalent molecular interactions and favorable hydrophobic/hydrophilic balance in 1 that drive the growth of stable gel networks. The application of a heating ultrasound protocol instead of the classical heating–cooling cycle allows not only the formation of alcogels that could otherwise not be formed, but it also decreases both CGC and gelation times in comparison to the heating–cooling treatment. Moreover, the nature of the solvent has a remarkable effect on the morphology of the aggregates (e.g., straight laths, entangled fibers) and some gels also exemplify the strong competition between gelation and macroscopic crystallization typically observed in weak physical gels. Thixotropic behavior and phase selective gelation of non-miscible alcohol/water mixtures are other interesting features of these alcogels.This work opens new possibilities for the use of squaramides in materials synthesis beyond their classical applications in catalysis, sensors and medicine. However, additional examples of other squaramide-based gelators with broader gelation abilities and higher mechanical stabilities are still necessary in order to establish the real potential of this building block for the preparation of supramolecular gels. Investigations related to the described “preformation hypothesis” within the overall gelation theory for similar systems, as well as the study of the apparent gel-to-gel transitions are currently underway in our laboratories and the results will be published at due course.
Acknowledgements
Universität Regensburg, Deutsche Forschungsgemeinschaft (DFG, 9209720), Ministerio de Economía y Competitividad (MINECO) and European FEDER funds (MAT2012-34498), Universidad de Zaragoza (JIUZ-2014-CIE-07), Consejo Superior de Investigaciones Científicas (CSIC, PIE-201580I010) and Diputación General de Aragón (DGA) (Research Group E-104) are acknowledged for financial support. We thank the Servicio General de Apoyo a la Investigación-SAI (Universidad de Zaragoza) for electron microscopy, the Laboratorio de Microscopías Avanzadas at Instituto de Nanociencia de Aragón (LMA-INA) for AFM imaging and Centre de Supercomputació de Catalunya (CESCA) for the computational resources provided. J. V. A.-R. thanks DGA for a predoctoral fellowship. D. D. D. thanks DFG for the Heisenberg Professorship Award.Notes and references
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed.
- J. R. Merritt, L. L. Rokosz, H. N. Kingsley, B. Kaiser, W. Wang, T. M. Stauffer, L. E. Ozgur, A. Schilling, G. Li, J. J. Baldwin, A. G. Taveras, M. P. Dwyer and J. P. Chao, Bioorg. Med. Chem. Lett., 2006, 16, 4107 CrossRef CAS PubMed.
- J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890 CrossRef PubMed.
- J. V. Alegre-Requena, Synlett, 2014, 298 CAS.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS.
- F. R. Wurm and H.-A. Klok, Chem. Soc. Rev., 2013, 42, 8220 RSC.
- R. Prohens, A. Portell, C. Puigjaner, S. Tomàs, K. Fujii, K. D. M. Harris, X. Alcobé, M. Font-Bardia and R. Barbas, Cryst. Growth Des., 2011, 11, 3725 CAS.
- R. Prohens, A. Portell and X. Alcobé, CrystEngComm, 2012, 12, 4548 CAS.
- R. Prohens, A. Portell, C. Puigjaner, R. Barbas, X. Alcobé, M. Font-Bardia and S. Tomàs, CrystEngComm, 2012, 14, 5745 RSC.
- A. Portell, M. Font-Bardia and R. Prohens, Cryst. Growth Des., 2013, 13, 4200 CAS.
- A. Portell and R. Prohens, Cryst. Growth Des., 2014, 14, 397 CAS.
- B. Soberats, L. Martinez, E. Sanna, A. Sampedro, C. Rotger and A. Costa, Chem. – Eur. J., 2012, 18, 7533 CrossRef CAS PubMed.
- C. López, E. Sanna, L. Carreras, M. Vega, C. Rotger and A. Costa, Chem. – Asian J., 2013, 8, 84 CrossRef PubMed.
- Y. Ohsedo, M. Miyamoto, A. Tanaka and H. Watanabe, New J. Chem., 2013, 37, 2874 RSC.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237 CrossRef CAS.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263 CrossRef CAS.
- O. Gronwald and S. Shinkai, Chem. – Eur. J., 2001, 7, 4328 CrossRef CAS.
- K. Hanabusa, Springer Ser. Mater. Sci., 2004, 78, 118 CAS.
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006 Search PubMed.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS PubMed.
- X. Y. Liu, Top. Curr. Chem., 2005, 256, 1 CrossRef CAS PubMed.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS.
- G. C. Maity, J. Phys. Sci., 2007, 11, 156 Search PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649 RSC.
- A. Vintiloui and J.-C. Leroux, J. Controlled Release, 2008, 125, 179 CrossRef PubMed.
- J. H. Jung and S. Shinkai, Top. Curr. Chem., 2004, 248, 223 CrossRef.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed.
- S. Li, V. T. John, G. C. Irvin, S. H. Bachakonda, G. L. McPherson and C. J. O'Connor, J. Appl. Phys., 1999, 85, 5965 CrossRef CAS.
- T. Kato, Science, 2002, 295, 2414 CrossRef CAS PubMed.
- J. Puigmarti-Luis, V. Laukhin, A. P. del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238 CrossRef CAS PubMed.
- W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374 CrossRef CAS.
- C. Sanchez and M. Llusar, Chem. Mater., 2008, 20, 782 CrossRef.
- A. Friggeri, K. J. C. van Bommel and S. Shinkai, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006, pp. 857–893 Search PubMed.
- D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427 RSC.
- T. Tanaka, Sci. Am., 1981, 244, 110 CrossRef.
- Polymer Gels: Fundamentals and Biomedical Applications, ed. D. DeRossi, Y. Kajiwara, Y. Osada and A. Yamauchi, Plenum Press, New York, 1991 Search PubMed.
- S.-K. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151 RSC.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS PubMed.
- P. Xie and R. Zhang, J. Mater. Chem., 2005, 15, 2529 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC.
- M. George, R. Mathew and R. G. Weiss, Mol. Gels, 2006, 449 CAS.
- D. K. Smith, Chem. Commun., 2006, 34 RSC.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160 CrossRef CAS PubMed.
- A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem. – Asian J., 2011, 6, 266 CrossRef CAS PubMed.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38 RSC.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400 CrossRef CAS.
- Polymer Gels and Networks, ed. Y. Osada and A. R. Khokhlov, Marcel Dekker, New York, 2002 Search PubMed.
- Synthesis, Characterization, and Theory of Polymeric Networks and Gels, ed. S. M. Aharoni, Plenum Press, New York, 1992 Search PubMed.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, Adv. Mater., 2002, 14, 198 CrossRef CAS.
- X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336 CrossRef CAS PubMed.
- J. H. Shin, M. L. Gardel, L. Mahadeva, P. Matsudaira and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9636 CrossRef CAS PubMed.
- P. Terech, I. Furman and R. G. Weiss, J. Phys. Chem., 1995, 99, 9558 CrossRef CAS.
- H. Ihara, M. Takafuji and T. Sakurai, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific, California, CA, 2004, vol. 9, pp. 473–495 Search PubMed.
- Low Molecular Mass Gelators, ed. F. Fages, Top. Curr. Chem., Springer-Verlag, Berlin, Heidelberg, vol. 256, 2005 Search PubMed.
- J. V. Alegre-Requena, E. Marqués-López and R. P. Herrera, RSC Adv., 2015, 5, 33450 RSC.
- H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775 CrossRef CAS PubMed.
- H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- K. Remya and C. H. Suresh, J. Comput. Chem., 2013, 34, 1341 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- E. Marqués-López, J. V. Alegre-Requena and R. P. Herrera, EP14382260.9, 2014 Search PubMed.
- Encyclopedia of Supramolecular Chemistry, ed. J. W. Steed and J. L. Atwood, Marcel Dekker, New York, vol. 2, 2004 Search PubMed.
- M. Yamanaka, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 33 CrossRef CAS , and references therein.
- E.-M. Schön, E. Marqués-López, R. P. Herrera, C. Alemán and D. D. Díaz, Chem. – Eur. J., 2014, 20, 10720 CrossRef PubMed.
- D. Bardelang, Soft Matter, 2009, 5, 1969 RSC.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346 RSC.
- S. Pan, S. Luo, S. Li, Y. Lai, Y. Geng, B. He and Z. Gu, Chem. Commun., 2013, 49, 8045 RSC.
- J. M. Malicka, A. Sandeep, F. Monti, E. Bandini, M. Gazzano, C. Ranjith, V. K. Praveen, A. Ajayaghosh and N. Armaroli, Chem. – Eur. J., 2013, 19, 12991 CrossRef CAS PubMed.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185 RSC.
- V. J. Anderson and H. N. W. Lekkerkerker, Nature, 2002, 416, 811 CrossRef CAS PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem. – Eur. J., 2010, 16, 3176 CrossRef CAS PubMed.
- E.-M. Schön, S. Roelens and D. D. Díaz, CrystEngComm, 2015, 17, 8021 RSC , and references therein.
- B. Roy, P. Bairi and A. K. Nandi, Soft Matter, 2012, 8, 2366 RSC.
- N. M. Dixit and C. F. Zukoski, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 061501 CrossRef PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080 RSC.
- A. Baral, S. Basak, K. Basu, A. Dehsorkhi, I. W. Hamley and A. Banerjee, Soft Matter, 2015, 11, 4944 RSC.
- A. Kotlewski, B. Norder, W. F. Jager, S. J. Picken and E. Mendes, Soft Matter, 2009, 5, 4905 RSC.
- V. A. Mallia, P. D. Butler, B. Sarkar, K. T. Holman and R. G. Weiss, J. Am. Chem. Soc., 2011, 133, 15045 CrossRef CAS PubMed.
- X. Yu, Q. Liu, J. Wu, M. Zhang, X. Cao, S. Zhang, Q. Wang, L. Chen and T. Yi, Chem. – Eur. J., 2010, 16, 9099 CrossRef CAS PubMed.
- H. Sato, E. Nogami, T. Yajima and A. Yamagishi, RSC Adv., 2014, 4, 1659 RSC.
- L. Frkanec and M. Zinic, Chem. Commun., 2010, 522 RSC.
- V. Caplar, M. Zinic, J.-L. Pozzo, F. Fages, G. Mieden-Gundert and F. Vögtle, Eur. J. Org. Chem., 2004, 4048 CrossRef CAS.
- R. E. Fischer, B. Tadic-Galeb and P. R. Yoder, Optical System Design, McGraw Hill, 2nd edn, 2008 Search PubMed.
- K. Lalitha, Y. S. Prasad, V. Sridharan, C. U. Maheswari, G. John and S. Nagarajan, RSC Adv., 2015, 5, 77589 RSC.
- P. Kirilov, F. Gauffre, S. Franceschi-Messant, E. Perez and I. Rico-Lattes, J. Phys. Chem. B, 2009, 113, 11101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopy, rheology, additional experimental and calculation details, pictures and tables. See DOI: 10.1039/c5sm02997j |
| This journal is © The Royal Society of Chemistry 2016 |