
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of partially denatured insulin with a DSPC floating lipid bilayer†
A. J. C.
Dennison
*ab,
R. A. L.
Jones
c,
R. A.
Staniforth
d and
A. J.
Parnell
c
aUniversity Grenoble Alpes, IBS, F-38044 Grenoble, France. E-mail: andrewdennison@live.com
bInstitut Laue-Langevin, 73 Rue des Martyrs, 38000, Grenoble, France
cDepartment of Physics and Astronomy, The University of Sheffield, Sheffield S3 7RH, UK
dDepartment of Molecular Biology and Biotechnology, The University of Sheffield, Sheffield S3 7RH, UK
First published on 5th November 2015
Abstract
The carefully controlled permeability of cellular membranes to biological molecules is key to life. In degenerative diseases associated with protein misfolding and aggregation, protein molecules or their aggregates are believed to permeate these barriers and threaten membrane integrity. We used neutron reflectivity to study the interaction of insulin, a model amyloidogenic protein, with a DSPC floating lipid bilayer. Structural changes consistent with protein partitioning to the membrane interior and adsorption to a gel phase model lipid bilayer were observed under conditions where the native fold of the protein is significantly destabilised. We propose that the perturbation of the membrane by misfolded proteins involves long term occupation of the membrane by these proteins, rather than transient perforation events.
I. Introduction
A wide range of incurable diseases (including Alzheimer's, Parkinson's and type II diabetes) are associated with the misfolding of proteins. Common to the pathology of each of these diseases are amyloid aggregates. The current consensus is that the toxicity mechanisms causing the onset and progression in these diseases arise from cell damage and death caused by the presence of oligomeric forms of amyloidogenic proteins.1–3 These toxic species carry out their activity at the cell surface and pathways as to how this leads to cell death are currently the focus of much investigation. Although a number of protein receptor systems have been postulated,4 studies using simple vesicular systems5 have shown that only the phospholipid component is necessary for the proteins' ability to traverse the membrane bilayer.This membrane damage must therefore be an adsorption-driven process, with structural rearrangement either preceding or following the adsorption and incorporation of oligomers or monomers respectively. Either way, surface properties6,7 including those of lipids8 have been shown to play an active role in the unfolding and misfolding process, accelerating fibrillisation rates in comparison to those in the bulk. At interfaces there are significant physical and chemical differences in the local conditions to those in bulk solutions. FTIR9 and neutron reflectivity10 studies have previously shown how interfacial processes can alter conformation and structure in adsorbed protein films.11,12
To study the nature of the interaction of proteins at a membrane surface, we carried out an experiment in which we directly probed the interaction of a fibril-forming protein with a model lipid membrane using neutron reflectometry. The protein we used was insulin, which has frequently been used as a model system for protein fibrillisation due to its wide availability and textbook amyloid behaviour: well characterised13 insulin fibrils are readily formed by heating under acidic conditions.14 Insulin amyloids have also been isolated in vivo, although the extent of their involvement in pathology is probably limited to injection sites in diabetic patients,15 albeit that improper storage of insulin hormone can compound this problem and lead to reduced efficacy and shelf-life.
Neutron reflectometry (NR) is a structural probe which is extremely well suited to the study of model lipid membranes and biological processes occurring at these interfaces.16,17 This technique is capable of the high resolution determination of the layer thicknesses and composition, by exploiting the large differences in scattering between H2O and D2O, multiple measurements of the same sample can provide a unique solution to the interfacial structure for adsorbed films. In order to investigate the initial phase of penetration, conditions which accelerate the formation of intermediate states in the misfolding of insulin18 while still allowing the sample to remain unchanged during the long measurement times required for NR. A floating lipid membrane was chosen as a model membrane because a planar geometry is needed for NR and, with this type of membrane mimic, the bilayer is decoupled from the interactions with the underlying silicon substrate by a thin layer of water.
II. Experimental
Sample preparation
Insulin from bovine pancreas, 1-(trimethoxysilyl)-propylacrylate, guanidine hydrochloride, glycine, AAPH, triethylamine and all solvents used were obtained from Sigma-Aldrich. Lipids and modified deuterated lipids as shown in Fig. 1 were supplied by Avanti Polar Lipids, Alabaster, TN, USA. All materials were used without further purification except for toluene, which was dried using CaH2, and distilled at reduced pressure to remove water, this was stored in glassware with a young's tap to minimise moisture uptake. D2O was obtained from Flurochem, Glossop, UK.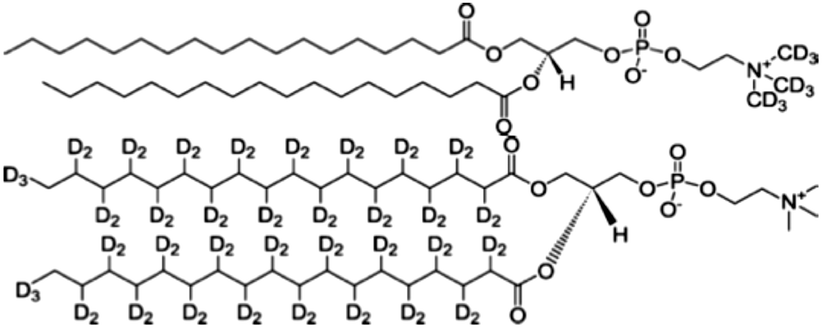 | ||
| Fig. 1 Molecular structure of the two deuterated lipids: d9-DSPC the headgroup deuterated lipid (top) and d70-DSPC with deuterated tails (bottom). | ||
Grafted lipopolymer films on silicon were produced following the method described by Hughes et al.19 with some variation; firstly silicon blocks (Crystran, Poole, UK) were cleaned using the RCA-1 process (70 °C for 10 minutes in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 H2O:H2O2 30%:NH3 30%) in order to remove any surface organic contaminants, then the blocks were dried using nitrogen gas and heated to 50 °C in the reaction vessel under vacuum, in order to remove any adsorbed water, an essential step before the deposition of the silane.
:1:1 H2O:H2O2 30%:NH3 30%) in order to remove any surface organic contaminants, then the blocks were dried using nitrogen gas and heated to 50 °C in the reaction vessel under vacuum, in order to remove any adsorbed water, an essential step before the deposition of the silane.
The blocks were then sealed in a PTFE reaction dish with a 25 mM solution of 1-(trimethoxysilyl)-propylacrylate (Sigma-Aldrich, UK) in dry toluene (Sigma-Aldrich, UK; HPLC grade, further dried using calcium hydride and distilled) with 21.5 mM of triethylamine (Sigma-Aldrich, UK). A period of 18 hours incubation in the solution was used for the formation of the self-assembled monolayer (SAM) after which the blocks were cleaned using toluene, isopropanol and water before being blown dry with N2 gas. The process of making these layers is relatively delicate due the necessity for the Langmuir–Schaeffer dip to be performed with the sample absolutely parallel to the subphase. This is achieved by either manual alignment or by the use of a Langmuir trough, which has the facility to laser align the sample and the subphase. Good results are possible with both techniques, however more reliably reproducible dips are possible using the Laser alignment method.
Floating lipid bilayers of 1,2-distearoyl-sn-glycero-3-phosphocholine-N,N,N-trimethyl-d9 (d9-DSPC) were deposited at 45 mN m−1 using a combination of Langmuir–Blodgett and Langmuir–Scheaffer deposition using a Langmuir-Trough (Nima, UK) (Fig. 2).
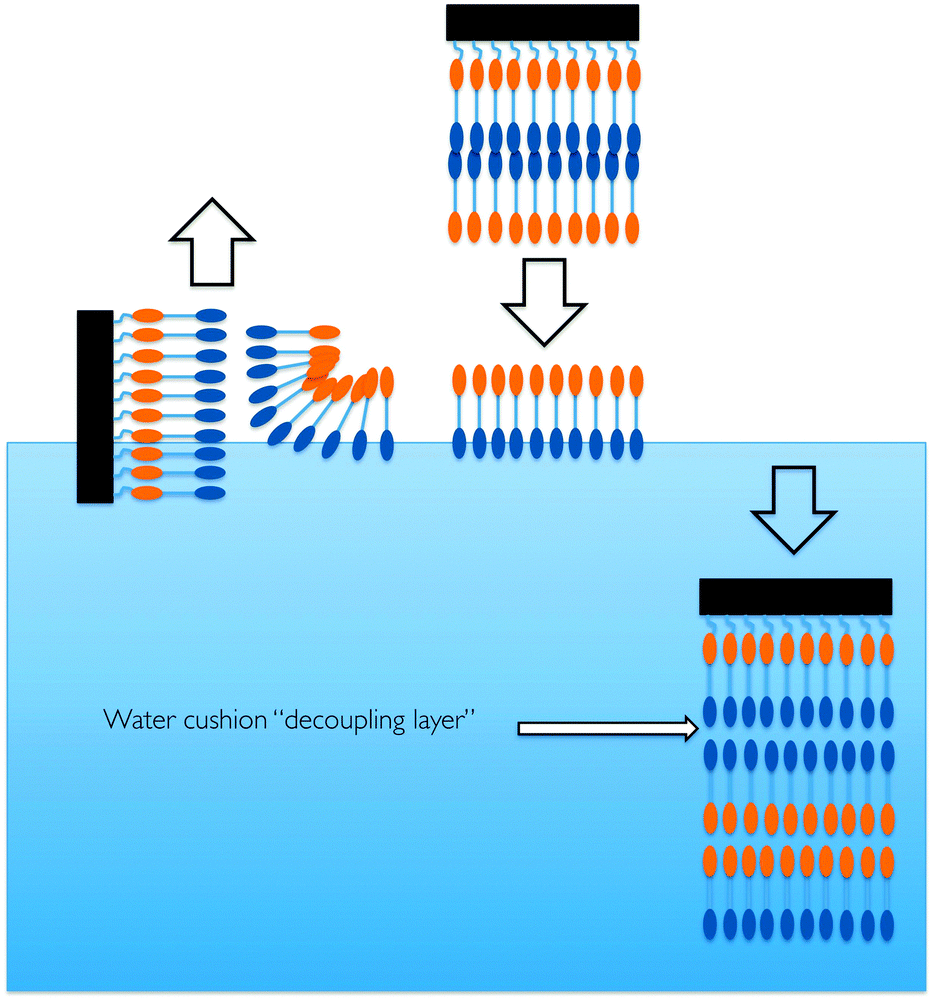 | ||
| Fig. 2 A cartoon representation of the process of generating a floating lipid bilayer (after Hughes et al.).19 | ||
For the final Langmuir–Schaefer dip the pressure-jump measured by the area change for the trough was less than 2 cm2 indicating transfer ratios of 95% or better for all samples. The surface quality at all stages for production of the grafted lipopolymer films was monitored using spectroscopic ellipsometry and qualitative assessment of the contact angle.
The spectroscopic ellipsometry was performed using a Wollam M2000V spectroscopic ellipsometer at a fixed incident angle of 70°. The data was fitted using the Woolam Complete EASE software package using a constrained model where the native oxide layer thickness was fixed after the initial measurement and the surface film was constrained to a Cauchy model. Additional assessment of the surface morphology by atomic force microscopy was also carried out.
Small angle X-ray scattering (SAXS)
SAXS data was collected on beamline B21 at the Diamond light source, the solution scattering was measured using the automated sampling system available at B21, this system fills a cuvette, collects the data and then automatically cleans the cuvette prior to the next solution. The solutions were made in the same way as the for the NR experiments insulin at 1 mg ml−1 with 50 mM glycine buffer acidified to pH 2.3 using concentrated hydrochloric acid. The data for the samples is presented in Fig. S6 (ESI†) for the 0 M, 1.5 M and 3 M Gdn·HCL. Using the software Sasview a Guinier fit was used to extract the radius of gyration of Insulin.Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed on a Brookhaven Instruments 200SM laser light scattering goniometer using a HeNe (125 mW, 633 nm) laser. Single five minute exposure scans over a period of 13 hours were performed and particle sizes were estimated using the CONTIN multiple pass method of data analysis at an angle of 90°.Neutron reflectometry and data fitting
Neutron reflectivity measurements were performed on the D17 instrument at the Institut Laue-Langevin (ILL) and it was used in its standard time-of-fight mode using a 4% resolution in Q. Two angles (0.7° and 2.5°) were used to cover the Q-range to 0.2 Å−1. Normalisation of the detector was performed by running a cuvette of light water (H2O) and the data sets from the two angles were joined using the Cosmos software. Light water contrasts were run for significantly longer than those in D2O buffers, but due to the lower scattering in water it was not possible to obtain the same quality of statistics due to time constraints. Bilayers were prepared 3 days before the experiment and stored in sealed sample cells, which had been cleaned in Decon-90 (Decon Laboratories Ltd) surfactant and washed thoroughly with ultra pure water (18 MΩ cm) and alcohol in water at room temperature, until the beginning of the experiment this ensured that the possibility of bacterial contamination was greatly reduced.All buffers and protein solutions were prepared freshly using Millipore ultra pure water and freshly opened D2O (Flurochem). All buffers were acidified using concentrated HCl (Sigma Aldrich) to pH 2.3. Bovine insulin (Sigma Aldrich, I5500) solutions were made at a concentration of 1 mg ml−1 and were mixed gently using a pipette so as not to cause bubbles (as bubbling can cause interfacial aggregation).
Different contrasts for each measurement were prepared using D2O and H2O solutions of insulin at 1 mg ml−1 with 50 mM glycine buffer acidified to pH 2.3 using conc. HCl and the specified Gdn·HCl concentration. The sample was allowed an incubation period of 5 hours to equilibrate with protein or change of contrast with the protein present before the reflectivity measurement.
The data were model fitted and where possible simultaneously co-refined with both contrasts using the Motofit reflectivity fitting package for Igor Pro. The final fitting of the sample at 3.5 M concentration used models where the substrate and the covalently attached monolayer were totally constrained from the initial structure. Remaining layer parameters then employed the simultaneous co-refinement used for solving the initial structure. Mutation and recombination values for the fit search were optimized after.
In addition to the fitting of the structure of the bilayer in 3.5 M Gdn·HCl, null hypothesis fitting was also performed. For this, reflectivity curves for the membrane both before and after exposure to the protein were simultaneously fit. This fitting procedure resulted in a significantly increased value of χ2.
Reflectivity curves were fit by two different methods: simultaneously fitting in both contrasts using fairly unconstrained models initially and a genetic algorithm to find solutions. This was performed alongside fitting of individual contrasts using much tighter physical constraints. Both methods gave fairly similar results with some interesting differences due to which regions each contrast was most sensitive to. The a priori knowledge of the molecular detail of the bilayer allows some of these differences to be explained. All fits were derived from using the values from Hughes et al. as an initial condition but with a modification to headgroup SLD to account for isotopic substitution in this region.
The fitting process requires physical constraints both to make sense and to limit available parameter space. It should be noted that in the case of lipids the splitting of the molecule into head and tail regions requires further assumptions on where the neutrons ‘see’ the head to finish and tail to begin. This however is an unavoidable simplification due to the slab fitting of molecules, which are not actually slabs of homogeneous scattering material.
Values for the SLD limits of buffers containing Gdn·HCl were calculated by weighing a 100 ml volumetric flask, adding the necessary mass of the salt, weighing again and then filling to 100 ml before a further weighing. Using the known density of water it was possible to obtain measured values for the density of the salt and from this calculate accurately the SLD for both the dry salt and the buffer.
III. Results and discussion
We investigated whether the adsorption of a model amyloidogenic protein to a lipid membrane is altered by destabilisation of the native state20 as the sampling of many molecular conformations by non-native monomers means there is considerably more flexibility for conformational rearrangement. During these measurements we have focused on the unfolding event immediately prior to formation of oligomers by using the denaturant guanidine hydrochloride (Gdn·HCl). We used three solution conditions in the measurement – insulin in pH 2.3 glycine buffer without guanidine, 1 M and 3.5 M Gdn·HCl. The choice of pH was made so that our results are comparable with the acidic conditions used by others to study insulin fibrillisation. The choice of the other two conditions were 1 M as a high-salt, natively folded condition and the 3.5 M point because Gdn·HCl titration of insulin shows significant changes between around 2.75 M and 3.25 M as revealed by circular dichroism.18 This transition to an expanded monomeric state18 is an on-pathway step in the formation of the amyloid state. Another effect of Gdn·HCl at lower concentrations is to significantly reduce the Debye length in solution, as it is a salt. This is important to highlight as at low concentrations the enhanced screening of electrostatic forces can actually stabilize proteins rather than destabilize them.21DLS measurements confirmed that under these solution/temperature conditions and timescales no detectable bulk protein aggregation occurs. This provides evidence that there are no aggregates larger than the instrument resolution limit of 30 nm (see ESI†). While this does not rule out the presence of oligomeric species it is clear that formation of fibrils does not occur over the experimentally relevant timescale. The radius of gyration of the protein as measured by SAXS at 0 M Gdn·HCl was shown to be 11.7 Å ± 0.5 Å, sadly this was only possible for the 0 M dataset, as the change in electron density upon the addition of Gdn·HCl reduces the overall effective contrast in the system. This result is in good agreement to the size of the insulin monomer measured in 20% acetic acid solution.18 This previous study found that the effect of guanidine hydrochloride to the solution caused expansion of the Rg under solution conditions of 20% acetic acid and disassociation of hexameric insulin as a function of Gdn·HCl concentration therefore we expect that insulin was monomeric under all solution conditions used. The lack of detectable signal in the SAXS results for the samples containing Gdn·HCl can be seen as a secondary validation of the result seen by DLS – that initially there are no aggregates present as with rising molecular weight the signal from SAXS is increased. It should be noted that the previous study by Ahmad was conducted at higher concentration and this facilitated their measurement.
It has been suggested that line defects in bilayers have the potential to act as sites which allow partitioning of the protein into the membrane structure from solution through the interaction of exposed hydrophobic residues22 facilitating irreversible adsorption events. This should be accelerated when defects are long-lived and the protein is in a state that allows significant freedom to explore conformational space. For this reason lipid bilayers in the gel phase were used to provide a model system for this.
Initial reflectivity measurements for the measured bilayers in 1 M and 3.5 M Gdn·HCl at pH 2.3 showed profiles similar to those shown in ref. 19, however the initial structure for the sample without the salt present was significantly different to the rest. A satisfactory fit for the sample without salt was not possible using simultaneous co-refinement, however separate fits of the individual contrasts converged on structural solutions with features consistent with a lipid bilayer with a significantly larger reservoir next to the grafted substrate (not shown). In this case the usefulness of the data as a control is therefore of limited use to the study.
The reason for bilayer instability in the 0 M Gdn·HCl case is most likely due to electrostatic effects caused by the low pH. The pKa of the phosphatidylcholine headgroup is quoted as being between 1 and 2 in ref. 23 and as this pH is approached a fraction of the lipids will become charged. This charging will not only cause like-charge repulsive effects but also possibly affect headgroup hydrogen bonding. The combination of repulsion between headgroups, possible perturbation of H-bonding and also layer–layer repulsion due to the grafted layer becoming similarly charged. The importance of the headgroup–headgroup hydrogen bonding network is illustrated by phase-separation observed in PC membranes under conditions of moderate pH perturbation.24 The salt effect on the membrane is consistent with the effect of monovalent salts on membrane properties being to increase membrane rigidity25 with rising salt concentration. This increased membrane rigidity, combined with enhanced screening of electrostatic effects is what we would attribute this stability to, although the exact contribution of each is impossible to determine based on these measurements. The estimated Debye screening length in the salt-free conditions was 13.6 Å in the 50 mM buffer, compared to 1.6 Å and 3 Å for the samples with Gdn·HCl present. This increase in the Debye screening length in solution means that while the samples in guanidine have screening lengths smaller than the headgroup size, the sample without would therefore not screen nearest neighbour electrostatic effects.
After incubation of the floating lipid bilayer with insulin in 1 M and 3.5 M Gdn·HCl buffers for 5 hours no change was observable in the 1 M sample with identical reflectivities for both contrasts, the 3.5 M sample showed significant change in reflectivity together with a shift of the fringe minimum to lower Q. The comparison of the lack of change in conditions where the main effect of the added guanidine is that of charge screening (1 M) to those where the protein is partially denatured (3.5 M) shows that the protein stability in solution has a strong effect on its propensity to adsorb to a lipid bilayer.
Fig. 3 highlights the lack of any structural changes for the sample in 1 M Gdn·HCl, where the main action of the low denaturant concentration is that of Debye screening. This is juxtaposed against what we observe for the sample in 3.5 M guanidine (Fig. 4); clear changes in both contrasts are visible.
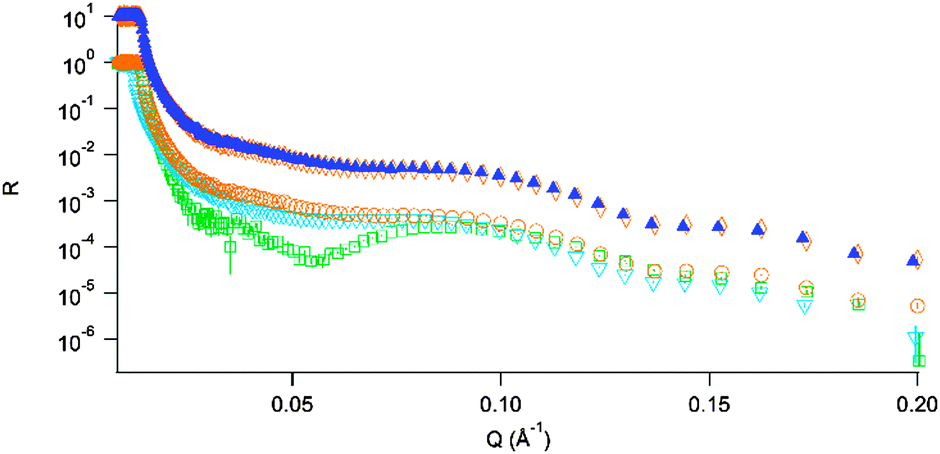 | ||
| Fig. 3 Upper, offset curves: DSPC bilayer before ▲ and after ◇ exposure to insulin in 1 M Gdn·HCl. Lower curves: initial reflectivity profiles for the 3 bilayers; 0 M Gdn·HCl; □, 1 M Gdn·HCl; ▼, 3.5 M Gdn·HCl; ○. | ||
 | ||
| Fig. 4 Top: Reflectivity profiles and fits for the bilayer in 3.5 M Gdn·HCl for the different conditions: initial, D2O; ▲, final, D2O; ○, initial H2O; □, final H2O; ▼. Bottom: Best fit structures for the initial bilayer in 3.5 M Gdn·HCl and after exposure to insulin in the same conditions. | ||
The shift in the position of critical reflection for the sample incubated in the D2O solution was due to an incomplete exchange of the sample cell. The adsorption occurring at the outer solution–lipid interface has an increase in roughness consistent with the radius of gyration of monomeric insulin plus the original roughness of the bilayer. A significant change in the SLD of the bilayer is also visible in these conditions. This could be explained by incorporation of protein into the bilayer core, probably in a non-native conformation. Similar observations were recently made by van Maarschalkerweerd26 where α synuclein was found to partition to a lipidic phase resulting in lipid–protein co-aggregates in the bulk. Interestingly, this study also found that the lipids in these aggregates also had a “gel like character”.
An upper limit for protein concentration in the lipid bilayer for the sample in 3.5 M Gdn·HCl was estimated to be 35% at the outer leaflet and 11.6% in the membrane interior using the approach described in ref. 27 and the value from the change in scattering length density. This method properly accounts for the changes in scattering length density for the protein due to the effects of H–D exchange in both side-groups and backbone due to the solution conditions. Comparing the two conditions where Gdn·HCl is present, it is clear that in these conditions, where the Debye length is comparable, partitioning to the membrane is favoured at higher Gdn·HCl concentration where the protein has greater conformational flexibility. This is favourable because the protein has increased disorder and flexibility to explore the different conformational arrangements, allowing exposure of hydrophobic residues, which stabilise the membrane-bound state. As the negative result from the DLS rules out bulk aggregation The presence of an adsorbed protein film is consistent with the results from ref. 26 and 28 and has additional significance in the context of phosphatidylcholine derived stabilisers for insulin29 where binding in the headgroup region has also been observed.
IV. Conclusions
A comparison between the behaviour of insulin in the 1 M and 3.5 M Gdn·HCl shows that, in conditions which partially unfold the protein, there appears to be evidence that the protein inserts itself in the bilayer and adsorbs at the outer leaflet of the membrane. This change occurs throughout the membrane in both the tail and head regions there is also an increased interfacial roughness at the top sample surface. This accumulation of material could explain in part the acceleration of fibrillisation at membrane surfaces observed by others. There is no evidence as to whether this is caused by individual proteins or aggregates, although complementary measurements found no evidence for aggregates in the bulk solution phase.The alternative hypothesis of adsorption to the membrane surface and a destabilisation of the membrane leading to an increased amount of membrane edges would also lead to a similar rise in scattering length density in the aliphatic region. However, both of these results would be consistent with significant alterations of membrane properties due to incubation with the partially folded protein.
The lack of adsorption in 1 M Gdn·HCl is consistent with what would be expected based on previous results such as the X-ray reflectivity study by Perriman30 and QCM-D measurements by Glasmastar.31
Acknowledgements
We would like to thank Robert Cubitt and Robert Barker at the Institut Laue-Langevin for their help and support during experiment 8-02-471 and to the partnership for soft condensed matter for access to sample preparation equipment in Grenoble. We also thank James Doutch for help with the SAXS measurements on beamline B21 at the Diamond Light Source (UK). We are grateful to the EPSRC for a doctoral training award that made this work possible.Notes and references
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- C. G. Glabe, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS PubMed.
- L. Haataja, T. Gurlo, C. J. Huang and P. C. Butler, Endocr. Rev., 2008, 29, 303–316 CrossRef CAS PubMed.
- J. Lauren, D. A. Gimbel, H. B. Nygaard, J. W. Gilbert and S. M. Strittmatter, Nature, 2009, 457, 1128–1132 CrossRef CAS PubMed.
- T. L. Williams, I. J. Day and L. C. Serpell, Langmuir, 2010, 26, 17260–17268 CrossRef CAS PubMed.
- S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696, DOI:10.1073/pnas.0701250104.
- M. I. Smith, J. S. Sharp and C. J. Roberts, Biophys. J., 2007, 93, 2143–2151 CrossRef CAS PubMed.
- J. S. Sharp, J. A. Forrest and R. A. L. Jones, Biochemistry, 2002, 41, 15810–15819 CrossRef CAS PubMed.
- S. Adams, A. M. Higgins and R. A. L. Jones, Langmuir, 2002, 18, 4854–4861 CrossRef CAS.
- S. A. Holt, D. J. McGillivray, S. Poon and J. W. White, J. Phys. Chem. B, 2000, 104, 7431–7438 CrossRef CAS.
- A. Ball and R. A. L. Jones, Langmuir, 1995, 11, 3542–3548 CrossRef CAS.
- W. Norde and A. C. I. Anusiem, Colloids Surf., 1992, 66, 73–80 CrossRef CAS.
- J. L. Jimenez, E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9196–9201 CrossRef CAS PubMed.
- J. L. Whittingham, D. J. Scott, K. Chance, A. Wilson, J. Finch, J. Brange and G. Guy Dodson, J. Mol. Biol., 2002, 318, 490 CrossRef.
- S. Yumlu, R. Barany, M. Eriksson and C. Rocken, Hum. Pathol., 2009, 40, 1655–1660 CrossRef CAS PubMed.
- J. H. Lakey, J. R. Soc., Interface, 2009, 6, S567–S573 CrossRef CAS PubMed.
- J. Penfold, Curr. Opin. Colloid Interface Sci., 2002, 7, 139–147 CrossRef CAS.
- A. Ahmad, I. S. Millett, S. Doniach, V. N. Uversky and A. L. Fink, Biochemistry, 2003, 42, 11404–11416 CrossRef CAS PubMed.
- A. V. Hughes, J. R. Howse, A. Dabkowska, R. A. L. Jones, M. J. Lawrence and S. J. Roser, Langmuir, 2008, 24, 1989–1999 CrossRef CAS PubMed.
- B. Vestergaard, M. Groenning, M. Roessle, J. S. Kastrup, M. v. de Weert, J. M. Flink, S. Frokjaer, M. Gajhede and D. I. Svergun, PLoS Biol., 2007, 5, e134 Search PubMed.
- A. K. Bhuyan, Biochemistry, 2002, 41, 13386–13394 CrossRef CAS PubMed.
- S. Grudzielanek, V. Smirnovas and R. Winter, Chem. Phys. Lipids, 2007, 149, 28–39 CrossRef CAS PubMed.
- R. Koynova and M. Caffrey, Biochim. Biophys. Acta, Rev. Biomembr., 1998, 1376, 91–145 CrossRef CAS.
- S. Suresh and J. M. Edwardson, Biochem. Biophys. Res. Commun., 2010, 399, 571–574 CrossRef CAS PubMed.
- G. Pabst, A. Hodzic, J. Strancar, S. Danner, M. Rappolt and P. Laggner, Biophys. J., 2007, 93, 2688–2696 CrossRef CAS PubMed.
- A. van Maarschalkerweerd, V. Vetri, A. E. Langkilde, V. Fodera and B. Vestergaard, Biomacromolecules, 2014, 15, 3643–3654 CrossRef CAS PubMed.
- Y. M. Efimova, A. A. van Well, U. Hanefeld, B. Wierczinski and W. G. Bouwman, J. Radioanal. Nucl. Chem., 2005, 264, 271–275 CrossRef.
- C. C. Lee, Y. Sun and H. W. Huang, Biophys. J., 2012, 102, 1059–1068 CrossRef CAS PubMed.
- I. Amar-Yuli, D. Azulay, T. Mishraki, A. Aserin and N. Garti, J. Colloid Interface Sci., 2011, 364, 379–387 CrossRef CAS PubMed.
- A. W. Perriman, M. J. Henderson, C. R. Evenhuis, D. J. McGillivray and J. W. White, J. Phys. Chem. B, 2008, 112, 9532–9539 CrossRef CAS PubMed.
- K. Glasmastar, C. Larsson, F. Hook and B. Kasemo, J. Colloid Interface Sci., 2002, 246, 40–47 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic ellipsometry of grafted lipid film. See DOI: 10.1039/c5sm02502h |
| This journal is © The Royal Society of Chemistry 2016 |