
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Buffering agents modify the hydration landscape at charged interfaces†
William
Trewby
,
Duncan
Livesey
and
Kislon
Voïtchovsky
*
Durham University, Stockton Road, DH1 3LE, UK. E-mail: kislon.voitchovsky@durham.ac.uk
First published on 28th January 2016
Abstract
Buffering agents are widely used to stabilise the pH of solutions in soft matter and biological sciences. They are typically composed of weak acids and bases mixed in an aqueous solution, and can interact electrostatically with charged surfaces such as biomembranes. Buffers can induce protein aggregation and structural modification of soft interfaces, but a molecular-level picture is still lacking. Here we use high-resolution atomic force microscopy to investigate the effect of five commonly used buffers, namely 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 2-(N-morpholino)ethanesulfonic acid (MES), monosodium phosphate, saline sodium citrate (SSC) and tris(hydroxymethyl)aminomethane (Tris) on the hydration landscape of Muscovite mica in solution. Mica is an ideal model substrate due to its negative surface charge and identical lattice parameter when compared with gel-phase lipid bilayers. We show that buffer molecules can produce cohesive aggregates spanning over tens of nanometres of the interface. SSC, Tris and monosodium phosphate tend to create an amorphous mesh layer several molecules thick and with no preferential ordering. In contrast, MES and HEPES adopt epitaxial arrangements commensurate with the underlying mica lattice, suggesting that they offer the most suitable solution for high-resolution studies. To confirm that this effect persisted in biologically-relevant interfaces, the experiments were repeated on a silica-supported lipid bilayer. Similar trends were observed for this system using atomic force microscopy as well as ellipsometry. The effect of the buffering agents can be mitigated by the inclusion of salt which helps displace them from the interface.
1 Introduction
Electrostatic forces play a fundamental role in soft matter,1 and in particular biological systems where they mediate the folding and stability of proteins2 and DNA,3 control interactions between molecules,4 ensure efficient charge and energy transfer across and along biomembranes,5,6 and help shape large molecular assemblies while controlling their mechanical and dynamic properties.7–9 These processes usually take place in solution and hence depend critically on the solution's ionic strength and pH-the concentration of protons or hydroxide ions in solution. In biological assays as well as certain industrial processes,10,11 pH is controlled by buffering agents; molecular macro-ions with dimensions of the order of nanometres (see Fig. 1) that partially dissociate in aqueous solutions. They are intended to ensure that the in vitro environment allows for optimal function of the molecules in solution, and mimics that of the natural world. However, evidence suggests that buffers may actively alter the dynamics and aggregation of proteins in solution and affect the surface of biomembranes.12 Biological membranes are largely composed of zwitterionic or negatively charged lipids13 whose organisation in the membrane depends on specific electrostatic and hydration interactions. These interactions are controlled by the type and quantity of counter-charges in solution.14–16 Buffer molecules can therefore exert a great deal of influence over biomembranes; Tris (structure given in Fig. 1), for example, can induce micrometre-scale ripple phases in neutral lipid bilayers.12 The underlying mechanism behind this is still unclear. Aside from the structural rearrangement of soft interfaces, buffer molecules are also likely to create a layer that prevents normal interaction of solute molecules with the interface, and add constraints to the membrane's local mechanical properties, in a comparable manner to interfacial ionic networks.8,17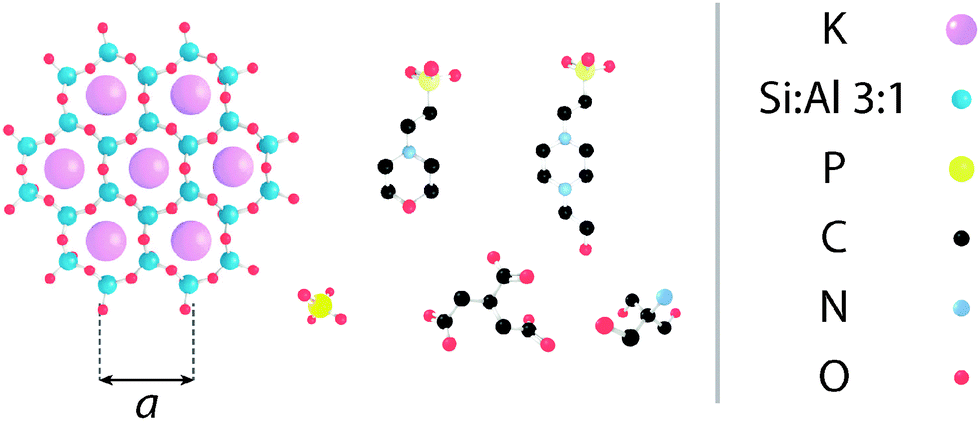 | ||
| Fig. 1 Molecular structure of the substrate and buffering molecules used in this study, shown approximately to scale. (left) The hexagonal lattice of muscovite mica's [100] surface, as presented to the AFM tip (a = 5.199 Å45); (L-R, top) HEPES; MES; (L-R, bottom) monosodium phosphate; SSC; Tris. | ||
To the best of our knowledge, none of these effects have been systematically studied. This is due, in part, to the difficulty in deriving local information about the interface at the nanoscale. Here we use high-resolution amplitude-modulation atomic force microscopy (AM-AFM)18 to tackle the question of buffer aggregation and layering at charged interfaces.
AFM can operate with sub-nanometre lateral resolution, making it an ideal tool to investigate interfacial effects; it has previously been used to investigate the nature of water adsorption to Muscovite mica surfaces in ambient conditions,19–21 which has helped elucidate the behaviour of water at solid interfaces. It is the technique's ability to image while fully immersed in liquid that allows biological interfaces to be characterised in their native environment. Recent advances in AFM have improved the resolution, making possible the imaging of individual lipids,22,23 proteins8,24,25 and the secondary structure of DNA nucleotides26,27 in solution. The technique was also successfully used to map the strongly adsorbed interfacial water making up the hydration shell of various solids28–30 and biological membranes.31,32 Singe adsorbed metal ions could also be identified both on solids33–37 and lipid bilayers23 in aqueous solution.
Here, we compare the aggregation and layering of five commonly used buffers, namely 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 2-(N-morpholino)ethanesulfonic acid (MES), monosodium phosphate, saline sodium citrate (SSC) and tris(hydroxymethyl)aminomethane (Tris) at the surface of mica in solution. The choice of mica is motivated by its identical lattice parameters and symmetry when compared to gel-phase lipid bilayers, its negative surface charge, and its overall robustness. Its negative surface charge occurs due to dissociation of potassium ions from its [100] surface (see Fig. 1) and results in a streaming zeta potential of approximately −80 mV38 at pH 7.0. The molecular organisation of supported lipid bilayers can vary tremendously, depending on the method of preparation, the specific composition and the system's history and environment,39,40 making direct, molecular-level comparison between buffers difficult. In the present case, the use of mica allows for a direct comparison between the different buffers while retaining some structural characteristics of bilayers.
The biological relevance of the study was then confirmed by investigating the effect of buffering agents on a supported lipid bilayer (SLB) formed on a silicon/silicon dioxide wafer with AM-AFM as well as ellipsometry.41 By analysing the change in amplitude of in- and out-of-plane polarised light upon reflection, ellipsometry measurements gave a non-invasive, large-scale method of studying the buffers' behaviour at the solid–liquid interface. The lipid 1,2-dipalmitoyl-sn-glycero-3-phosphate (DPPA), which is anionic and in the gel phase (Tm ∼ 67 °C) at physiological pH and temperature was chosen due to its importance for dynamic biological processes such as signal transduction42 and fusion,43 especially in plants.44
In this paper hydration forces are to be understood in their literal sense: the forces that arise from the local organization of water molecules at interfaces and around ions and molecules. These nanoscale forces often dominate the adsorption or organization of charged molecules at interfaces34,46 and play a fundamental role in regulating biological function.47,48
2 Materials and methods
Buffering agents
The buffers that were investigated were all purchased from Sigma-Aldrich (St. Louis, MO, USA) and are listed below (see Fig. 1 for structures):• HEPES: 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (powder form), 99.95% purity, pKa = 7.56
• MES: 2-(N-morpholino)ethanesulfonic acid hydrate (powder form), 99.95% purity, pKa = 6.27
• SSC: saline-sodium citrate (powder blend), pKa = 3.13
• Tris: trizma base (2-amino-2-(hydroxymethyl)-1,2-propanediol), 99.99% purity, pKa = 8.07
• Monosodium phosphate: Na2HPO4, 99.90% purity, pKa = 7.20
• PBS: phosphate-buffered saline (powder form), pKa = 7.20
All quoted pKas were listed in ref. 49 and are valid at 25 °C. Buffers were made up to a concentration of 10 mM and titrated to a pH of 7.0 at 25 °C with 0.17 M KOH and 0.5 M HCl. The solutions were then sonicated for 10 minutes in order to ensure complete solution of the buffering molecules and removal of any dissolved gases. Approximately 50 μL of the buffer solution was deposited on the mica and a similar quantity on the cantilever tip using a pipette. This ensured no air bubbles could become trapped as the cantilever was submerged. A capillary bridge was then formed between the mica and cantilever by bringing the two into close proximity.
Atomic force microscopy
A commercial Cypher ES AFM (Asylum Research, Santa Barbara, USA) equipped with direct laser excitation (blueDrive™) and thermal control was used for all experiments. The blue drive and temperature control result in highly stable imaging parameters, making direct comparison between buffers more meaningful. We also improved comparability by using the same cantilever for the duration of the experiment. The cantilever (Arrow UHFAuD, NanoWorld, Switzerland) had a stiffness k ∼ 1.8 Nm−1 (calculated from the cantilever's thermal spectrum50) and was driven at its fundamental resonance frequency of 391.07 kHz in liquid. The measurements on mica were conducted sequentially over one day. The experiments on the supported lipid bilayer were carried out at a later date, so maintaining identical tip-sample conditions was non-trivial. Instead, a new Arrow cantilever was used for this imaging, to ensure there were no contaminants or artefacts due to tip degradation. Prior to imaging, the cantilever was immersed in a bath of ultrapure water (18.2 MΩ, <5 ppm organics, Merck Millipore, Watford, UK), followed by isopropyl alcohol (Sigma-Aldrich, 99.7% purity), followed by ultrapure water again for a total of 30 minutes to remove as much organic matter from the tip as possible, while minimising physical tip alterations. Grade I Muscovite mica (SPI supplies, USA) was used and freshly cleaved with adhesive tape before all measurements.The AFM was operated in amplitude-modulation mode with the cantilever and tip fully immersed in the liquid. In amplitude-modulation, the probe's cantilever is oscillated near resonance and the amplitude is kept constant while scanning. The phase difference between the driving oscillation and that of the probe is allowed to vary freely. The working amplitude was adjusted to gain the best image quality possible between 1.0 nm and 1.5 nm while the free oscillation amplitude was kept constant at 1.6 nm. This ensured that the tip mainly probed the solvation layers of the surface.51 In these conditions, the phase contrast can be related to the local solvation free energy of the liquid for the surface.52,53 In Fig. 2, 3, 5 and 6, original scan sizes were (100 × 100) nm2, (20 × 20) nm2, (20 × 20) nm2 and (100 × 100) nm2, respectively. All images were taken at a constant scan rate (lines-per-second) of 4.88 Hz.
Since the goal was to offer a direct comparison between the buffering agents, our emphasis was placed on keeping imaging conditions as similar as possible for each buffer rather than achieving the highest resolution. This is necessary because AFM measurements are by definition perturbative and the AFM tip influences the observations.33,52,54 Using the same cantilever and identical imaging conditions does not negate tip effects, but improves comparability.
Image analysis
The AFM images were produced by Asylum Research software package (ver. 13.17.101) (Asylum Research, Santa Barbara, USA) for Igor (ver. 6.3.7.2, Wavemetrics, Lake Oswego, USA), before being flattened line-by-line with a first-order polynomial. The root-mean squared roughness, Rq, values were calculated according to the formulawhere z(xk,yl) is the measured height at point (xk,yl) and k and l sum over the points and lines of each image. In our case, the raw images produced had M = N = 256. Each data point in Fig. 4 was taken as the average Rq of 5 images, with the error bars representing the standard deviation of that set. The presented figures were slightly low-pass filtered using FFT-based analysis to remove unwanted high-frequency noise.
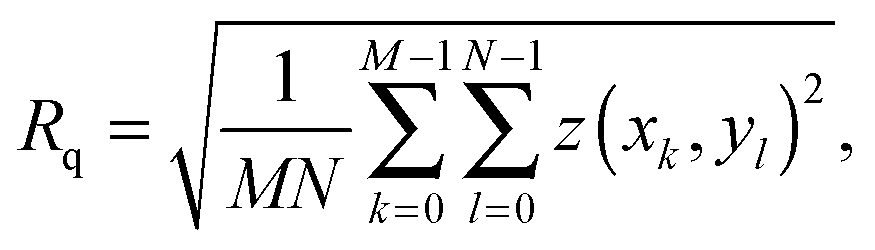
Silica-supported lipid bilayer formation
Supported lipid bilayers (SLBs) of the anionic lipid 1,2-dipalmitoyl-sn-glycero-3-phosphate (DPPA) (Avanti Polar Lipids Inc., AL, USA) were formed via the vesicle fusion method.55,56 Briefly, a chloroform solution containing DPPA at 1 mg mL−1 was pipetted into a 10 mL vial and dried under nitrogen until there was no visible fluid remaining. It was then placed under vacuum for >4 hours to ensure complete evaporation. The lipid film was subsequently re-hydrated with Milli-Q ultrapure water to a concentration of 3 mg mL−1 and gently sonicated until the solution became uniformly milky and opaque due to the formation of multi-lamellar vesicles. The solution was then extruded at least 19 (but always an odd number) times using a Mini-Extruder kit (Avanti Polar Lipids Inc., AL, USA) with a 100 nm filter (Whatman, GE Healthcare Life Sciences, Buckinghamshire, UK) to form small, unilamellar vesicles (SUVs). The solution was then diluted with 150 mM NaCl to a concentration of 0.1 mg mL−1. This resulted in a salt concentration of ∼145 mM which would encourage the SUVs to fuse and spread onto the silica substrate. All glassware, and components had been cleaned thoroughly by sonication with ultrapure water, then isopropyl alcohol, and water again for ten minutes each before coming into contact with the lipids.A silicon wafer with a native silicon dioxide layer was cleaned thoroughly by sonication in diluted Deacon-90 (Deacon Laboratories, Sussex, UK) detergent for ten minutes, followed by a similar treatment with ultrapure water, isopropyl alcohol and then ultrapure water once more to ensure the surface had no contaminants on it that could affect the formation of the lipid bilayer. The silica was then made hydrophilic via exposure to an argon plasma at 1 mbar for 30 s. Immediately afterwards, ∼80 μL of the SUV/NaCl solution was pipetted onto the wafer before being sealed in a petri-dish, heated to 77 °C for 1 hour and cooling to room temperature at a rate of 10 °C per hour. The purpose of this heat-ramp was to ensure that the lipids were allowed to relax in their fluid state before being examined, minimising the possibility of kinetic traps existing, such as in the case of the “ripple” phase.57 When exchanging the buffering fluid, the bilayer was copiously rinsed, with at least ten times the initial volume (∼100 μL) covering the silicon wafer.
Ellipsometry
Ellipsometry provides a label-free, non-perturbative method of investigating thin films forming interfaces.41 It works on the principle that the parallel and perpendicular coefficients of reflection of monochromatic light (Rp and Rs respectively) are very sensitive to the presence of chemical layers formed at the reflective surface. Ellipsometric measurements typically probe the ratio between these complex reflectivities, ρ, such thatwhere the ellipsometric angles ψ and Δ give an indication of the extent to which a surface is modified. Measurements were carried out on a picometer phase-modulated ellipsometer (Beaglehole Instruments) using a helium-neon laser (λ = 589 nm), with an incident angle of 70°.
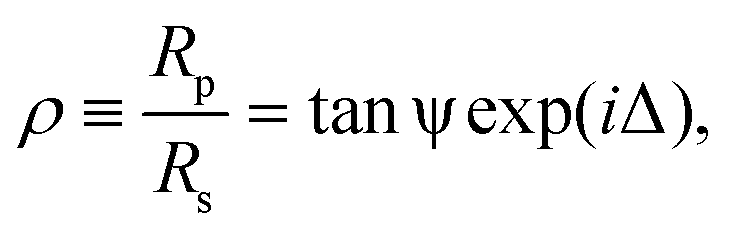
After the SLB was formed on the silica surface (see above), the wafer was gently rinsed with 150 mM NaCl solution to remove any unfused vesicles. It was then entirely submerged in the NaCl solution in a Petri dish so as to remove aberration that would be produced by a curved droplet surface. Each data-point presented is the average of five sets (each at different points on the silica) of ten ellipsometric measurements. When altering buffering agents, care was taken not to expose the sample to air (and potentially destroy the SLB) by exchanging fluids using a pipette tip. At least twice the volume of the Petri dish was used when rinsing the bilayer, to ensure no residual salt or buffer remained at its surface. The sample was then left to equilibrate for 30 minutes before continuing the measurement.
3 Results and discussion
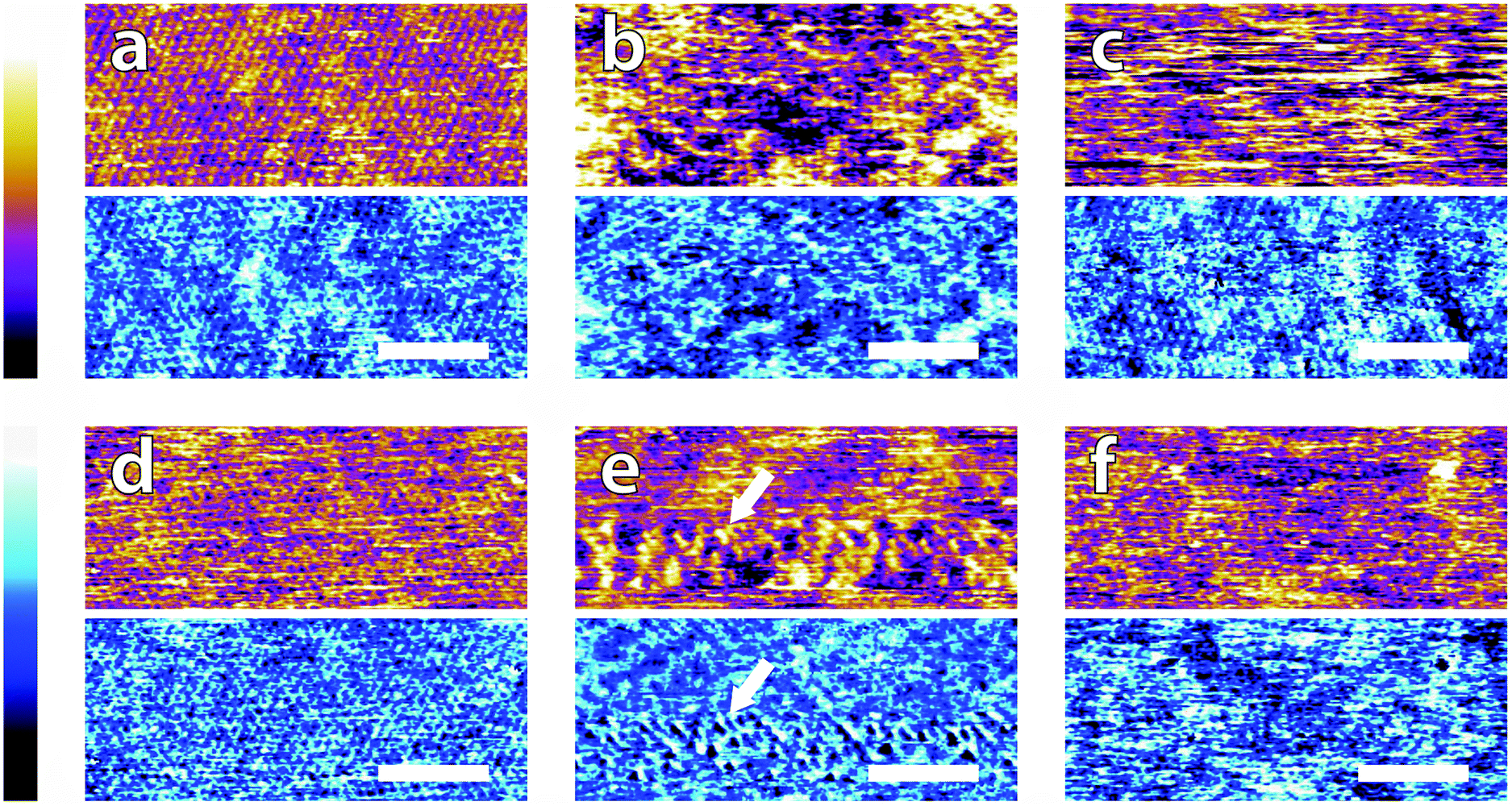
In order to systematically evaluate the impact of buffers at the interface, selected AFM images of the height and phase contrast obtained at different magnifications are shown. Those taken at lower magnification (100 nm) are shown in Fig. 2. The height scans (orange/purple) all span a comparable z-range, with the displayed colour scale ranging from 150 pm (white) to 0 pm (black). Images taken in buffer (Fig. 2a–e), show distinct differences, despite sharing an identical substrate of freshly-cleaved mica. The buffering molecules, which are comparable in size to the mica lattice parameter (see Fig. 1), directly interact with the substrate and work to screen the surface potential, producing epitaxial hydration structures that can be resolved by the AFM tip.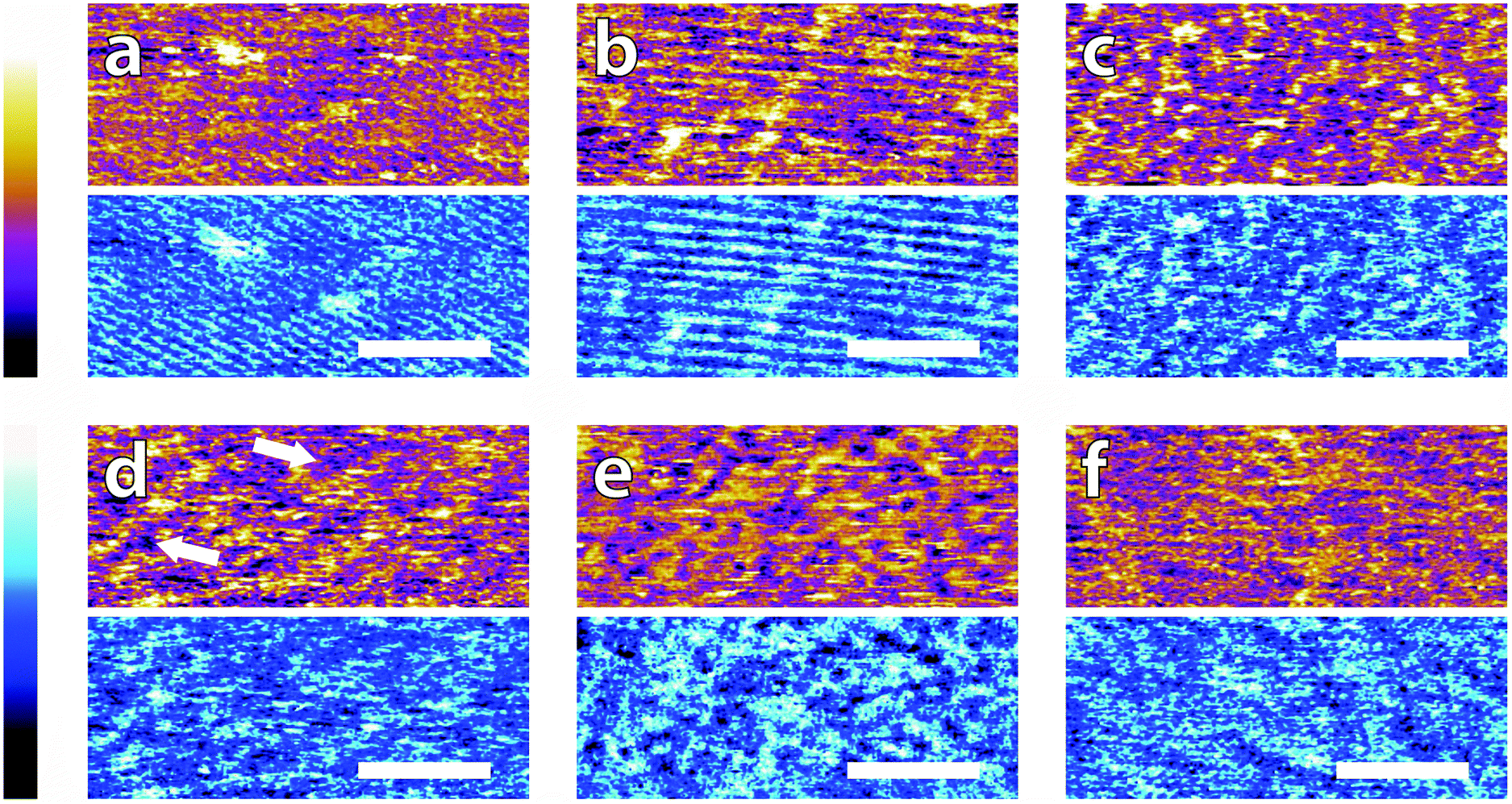 | ||
| Fig. 2 Low resolution AFM images of mica topography (orange-purple, upper) and phase contrast (blue, lower) in buffer solutions. In MES (a) and HEPES (b) rows are clearly visible in topography and in phase. No regular structure is visible in monosodium phosphate (c), SSC (d) and Tris (e). Some structures are visible forming a mesh on the surface (arrows in d). De-ionised water (f) was imaged as a control. Rows related to the mica lattice are hardly visible at that scale. In all images, the scale bar represents 30 nm. The topographic colour scale represents relative height variations ranging over 150 pm from black to white. The phase colour scale corresponds to variations of 10° (black to white). | ||
These structures vary from case to case. In both MES and HEPES, (Fig. 2a and b) diagonal, periodic rows are clearly visible both in topography and in phase. Their periodicity was taken perpendicular to the rows – i.e. running from bottom-left to top-right in Fig. 2a and close to the vertical in Fig. 2b. Fourier analysis revealed that they had a similar periodicity of 3 ± 1 nm. This value, about six times large than the periodicity of mica's lattice,45 suggests an organised epitaxial layer composed of buffer molecules adsorbed on the underlying mica surface. In contrast, images acquired in monosodium phosphate, SSC and Tris (Fig. 2c–e) exhibit a homogeneous texture with regular features exhibiting a characteristic size of approximately 10 nm, but with occasional superstructures such as the elongated formations of Fig. 2d (arrows). The lack of any regularly repeating features in these images implies that the buffer molecules are not able to organise themselves in a way commensurate with the underlying mica lattice. However, the existence of this typical lengthscale indicates that a stable buffer layer has formed at the interface and that the layer is cohesive enough to withstand the imaging process. Buffer molecules occasionally become attached to, or influence the motion of the cantilever during the imaging process, resulting in temporary loss of resolution (typically over one line). This leads to horizontal discontinuities in the image that can be observed in parts of Fig. 2e (see also; Fig. S1, ESI†). Fig. 2f shows the mica surface imaged in ultrapure water as a control. Features related to the mica lattice can be resolved, consistent with the picture of a well-defined hydration structure at the surface of hydrophilic lattices.58
Overall, the low magnification images show that the buffer molecules adsorb on mica and affect the hydration landscape of the interface. They can form cohesive layers strong enough to withstand the AFM imaging process. These layers are not necessarily commensurate with the underlying mica lattice, but still able to modify the surface in a manner characteristic of each buffer. Atomic-level details are however not visible at this scale, so higher magnification is necessary.
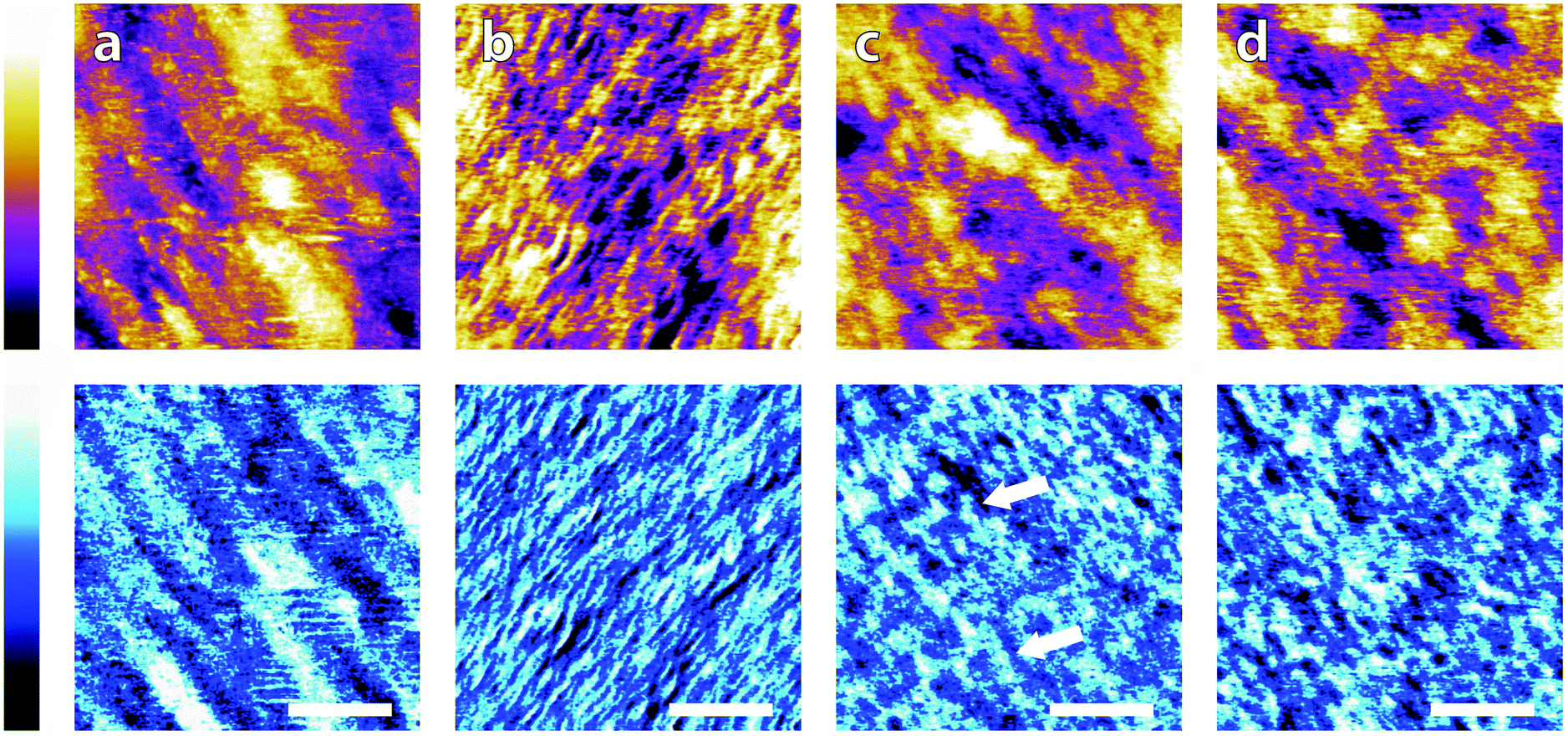
Fig. 3 illustrates representative high-resolution images (20 nm) of the interface in the different buffer solutions. Interestingly, details of the mica lattice are often visible, but large features in phase and/or topography suggest that the adsorbed buffer layer identified in Fig. 2 is still present in most cases.
 | ||
| Fig. 3 High magnification AFM images of mica topography (upper) and phase (lower) in buffer solutions. In MES (a) the hexagonal symmetry of the lattice can be clearly resolved despite infrequent horizontal defects. Regular epitaxial modulations are visible in the phase. In HEPES (b) the symmetry is again clear, but modulated by large height variations in both topography and phase. In Monosodium phosphate (c) the topography image displays no atomic features but they can be occasionally resolved in the corresponding phase image. In SSC (d) the mica lattice is perceptible with larger features similarly to in (a). In Tris (e), large features consistent with those in Fig. 2e are visible in the upper part of the image. Occasionally, the tip jumps (arrow) revealing a structured mesh. In ultrapure water (f) atomic-level features are visible alongside point-like deformities. The scale bar is 5 nm. Colour scales: the height is 150 pm and phase is 10°. | ||
Again, clear variations between the different buffers are observed. In MES and SSC (Fig. 3a and d respectively), clear atomic-level resolution images were obtained but their quality was often inconsistent, with many horizontal discontinuities and instabilities (see Fig. S2 and S3, ESI†). HEPES (Fig. 3b) produced very stable and reproducible images, showing a mesh-like network developing epitaxially on the surface of mica. Of the five buffers used, HEPES demonstrates the greatest height variations whilst retaining atomic-level detail. This is evident from comparison of the roughnesses of the scans presented in Fig. 4; HEPES has a roughness of approximately 50 pm at the (20 × 20) nm2 scale, second only to monosodium phosphate (which did not allow the same atomic-level detail). Conversely, the HEPES image at the 100 nm scale demonstrated a similar roughness to the other buffering agents. This is related to the fact that in the latter case, the cantilever was scanning at a constant rate over a larger surface – that is, the faster motion of the tip reduced its ability to pick out the sub-nanometre details. Monosodium phosphate (Fig. 3c), provided the worst image quality when compared to the other buffers and ultrapure water (Fig. 3f). Occasionally, the phase signal could resolve details of the mica lattice, but nothing was visible in the topography. Images tended to be very unstable, as reflected in the extremely high roughness values of the images (Fig. 4). Finally, the Tris-buffered solution (Fig. 3e) tended to induce some bi-stability while imaging in both topography and phase, with regions revealing atomic-level details while other showed some aggregates adsorbed on the surface (arrows, lower half of Fig. 3e). These larger surface features are in good agreement with those observed at lower magnification (Fig. 2f).
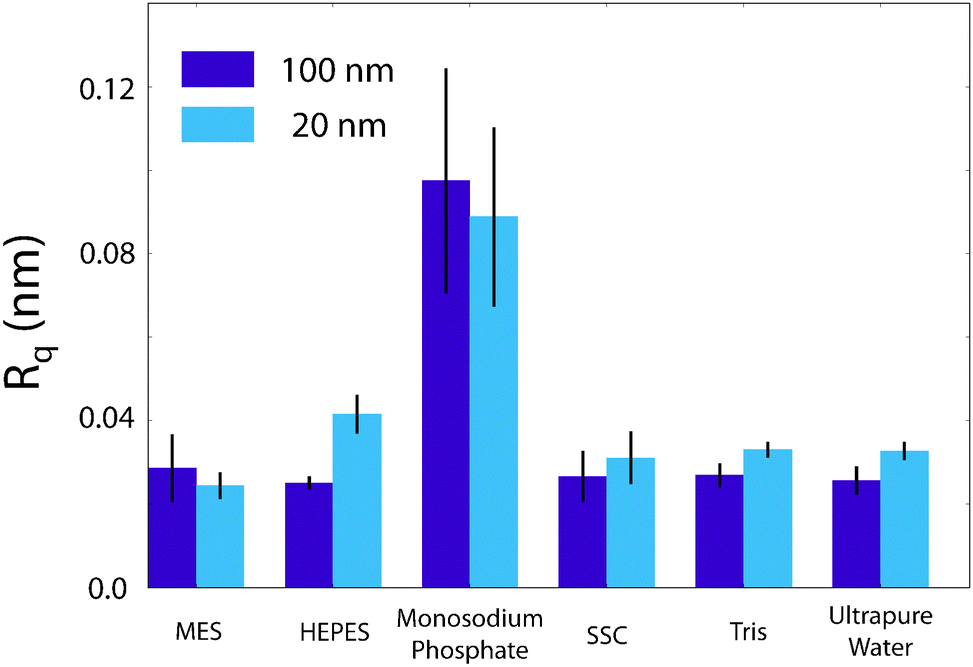 | ||
| Fig. 4 Comparison of the root-mean squared roughness, Rq, of the buffer/mica interface as imaged by the AFM. The effect of monosodium phosphate upon the AFM imaging process is clearly elucidated by its large roughness. HEPES demonstrates an anomalous increase in roughness from 25.1 pm to 41.6 pm at (100 × 100) nm2 and (20 × 20) nm2 scans respectively. This is contrasted with the other buffers, where the 100 nm roughness is equal to that at 20 nm. It is likely related to the increase in resolution at this lengthscale that leads to the mesh being observed in Fig. 3b. Error bars represent standard deviation of 5 consecutive scans in each buffer. | ||
Generally, higher magnification images tend to reveal more details about the mica lattice compared to the images shown in Fig. 2. AFM measurements being in essence perturbative,33 the tip is more likely to displace or remove adsorbed buffer molecules in the high-resolution scans where more time is spent over each area of the surface imaged. Nonetheless, in most cases, the mica lattice is superimposed upon larger scale features that reflect the features imaged in Fig. 2. This confirms that the buffer molecules tend to form a structured layer on the surface. This layer is templated by the mica surface, but lateral interactions between buffer molecules must also play an important role, as visible in the formation of mesh-like structures in HEPES and Tris.
Imaging in ultrapure water proved difficult for achieving atomic-level resolution images of the muscovite lattice and it was necessary to reduce the cantilever's free and working amplitudes by about 20%. This can be explained by a lack of stable hydration sites at the surface of the tip in solution,51 compared to the strong hydration landscape of mica. Although the cantilever's silicon nitride surface was cleaned (see Materials and methods), no surface modification was conducted hence limiting the stability of tip's hydration structure.54 This limitation affects the cantilever's vertical resolution59 and is detrimental for high resolution imaging. The existence of multiple, well-defined hydration layers at the surface of mica28,60,61 exacerbated the problem, and several large (33 nm) domains superimposed to the lattice structure are visible in Fig. 3f. These domains are likely induced by the tip probing different hydration states of the surface.
It is common knowledge in the AFM community that the addition of salt to aqueous solutions aids resolution, presumably through the formation of better-defined, more symmetrical hydration structures on both surfaces. The concentration and type of charges in solution can strongly influence the electrical double-layer forces of submerged solids39 and therefore play a significant role for AFM resolution.62 However, the use of small oscillation amplitudes while imaging here leads to the resolution being dominated by short-range hydration effects, including those of adsorbed buffer ions.
Hydration forces cannot adequately be described by the continuum theories of double layers,51 and are sensitive to the nature of the ionic species in solution. In particular the different hydration structure of adsorbed ions can dramatically influence the resolution achieved.8,33 In the case of buffers, stock solutions typically contain a wide range and density of salts aside from the buffering agent. This salt can compete with the charged buffer molecules for the charged interface and partially mitigate the effects observed in Fig. 2 and 3. Here we quantify the impact of increased salt concentrations by comparing three solutions; ultrapure water, a monosodium phosphate buffer and a phosphate-buffered saline (PBS) solution. The PBS solution is routinely used in biology to mimic physiological conditions and its buffering effect comes from the same monosodium phosphate molecule as that presented in Fig. 2f and 3f, but with the addition of 140 mM NaCl. The results are presented in Fig. 5.
 | ||
| Fig. 5 Comparison of the effect of ionic content on image quality in buffered solutions. Topographic images are presented in the upper portion and phase in the lower. (a) Ultrapure water, (b) monosodium phosphate buffer; again no atomic-scale features are observable in the topography. (c) Phosphate-buffered saline solution; the addition of 140 mM NaCl to the buffer results in a dramatic increase in image quality. Arrows indicate variability in imaging conditions; from individual lattice points (white) to row-like formations (red). Each image's scale bar is 5 nm and the colour scales are 150 pm for the height and 10° for the topography. | ||
The image in ultrapure water, Fig. 5a, is consistent with Fig. 2 and 3; the underlying lattice of mica is visible but the imaging is at times unstable, with several horizontal imperfections. In monosodium phosphate (Fig. 5b), the image quality is noticeably reduced and the mica lattice can only be resolved in part of the image, as in Fig. 3d. The low resolution is in part due to buffer molecules loosely adsorbed on the surface that interfere with the tip during the imaging process but also may be a result of non-specific adsorption to the cantilever itself. It is not possible to diagnose this while imaging, as the effects would be indistinguishable from one another. The phase in Fig. 5b is nonetheless consistent, as found previously. This could indicate a strong hydration layer formed on the mica, beneath the loosely adsorbed buffer molecules, but a definitive conclusion requires further, independent confirmation. In contrast, the image obtained in PBS buffer (Fig. 5c) is by far the clearest, demonstrating atomic-level resolution over the entire image. The overall imaging stability was confirmed with larger-scale images (see Fig. S4, ESI†) as well as the roughness measurement of 0.018 ± 0.002 nm, smaller than every previous buffer measured (Fig. 4). However, despite the regularity of Fig. 5c, the surface is not entirely homogeneous, with the lower portion of the phase image exhibiting individuated lattice sites (white arrow), while the upper half only rows can be discerned (red arrow). This is another indication that, although the resolution is improved, the buffer molecules are still interfering with the solid–liquid boundary, but in competition with Na+ ions. Significantly, in PBS, many surface features are visible both in phase and in topography, reflecting the different hydration states of the adsorbed buffer and Na+ ions.63
Fig. 5 illustrates the negative impact that buffer molecules have on high-resolution AFM scans, and also the effect of ionic content on imaging. Metal ions are considerably smaller than buffer molecules and as such can displace the latter at the interface and help create more stable hydration structures which are reflected by the improved imaging conditions. Adsorption of ions at charged interfaces is however a competitive process33,60 and buffer molecules will always be present, although in smaller quantities. The principal mechanism for buffer interference with biological systems is therefore likely to involve specific interactions and steric effects not solely based on electrostatic interactions. The extended mesh-like network visible in HEPES and Tris involve also inter-molecular interactions which are likely to survive in the presence of salt. This is visible in monosodium phosphate where the large features visible in the phase in Fig. 5b remain after the addition of salt (Fig. 5c), albeit less well-defined. In the context of AFM imaging, the adsorption of sodium ions at mica's surface allows for a well-defined and stable hydration in PBS and outweighs the negative impact of having extra molecules in solution, but the results are based on measurements with a relatively stiff cantilever (see Materials and methods), and loosely bound buffer molecules may still be visible when ultrasoft imaging conditions are used, for example over delicate biological samples.
To confirm whether the data obtained up to this point was indeed relevant for biological interfaces, the effect of the buffering solutions upon a silica-supported lipid bilayer (SLB) of DPPA was investigated by AFM. The bilayer was formed on a silicon/silicon dioxide substrate to enable ellipsometry measurements to be carried out that would otherwise have been hindered by the interference between the upper and lower plane of mica64 and required either advanced averaging techniques or assumptions about the refractive index to be made.65 The lower surface charge density of silicon dioxide66 also ensured the anionic lipid bilayer, which otherwise may have been repelled from the mica surface,67 remained stable. A selection of representative lower-magnification (100 nm) images of the SLB in different buffer molecules is presented in Fig. 6. There are much larger height variations when compared with the images in Fig. 2; the z-scale ranges from 0–500 pm rather than 0–150 pm. This is due to the intrinsic roughness of the silica substrate (see Fig. S5, ESI†), rather than any adhered layer. This roughness inhibited high-resolution comparisons between the different buffer solutions similar to those in Fig. 3 and 5, and for this reason, only three of the previously-investigated buffers were compared; HEPES (Fig. 6b), Tris (Fig. 6c) and monosodium phosphate (Fig. 6d) as these produced the most characteristic changes in topography previously. They were compared to the SLB in 150 mM NaCl– that is, the solution it was formed in. Distinctive changes in topography upon the exchanging of buffers are still evident.
 | ||
| Fig. 6 Comparison of buffering agents on a silica-SLB with that of a monovalent salt. (a) 150 mM NaCl; the surface demonstrates similar features to that of the bare silica surface (SI), indicating that the bilayer follows the support straightforwardly (b) 10 mM HEPES; clear wrinkling is induced by the exchange of buffers, with a coherent direction (bottom-left to top-right) and approximate width of 7 nm. (c) 10 mM Tris; the phase image displays non-uniform mesh-like structures across the surface (arrows) of ∼15 nm – midway between that of NaCl and HEPES – in a similar manner to its behaviour on mica (Fig. 2e) while the height is dominated by the large-scale topography. (d) 10 mM monosodium phosphate; SLB demonstrates similar features to those when immersed in Tris; a homogeneous network-like structure is found in the phase trace, but the topography more closely resembles that of the SLB in NaCl. Topography is shown in orange/purple (colour scale range = 500 pm in all), with the corresponding phase shown below in blue (colour scale range = 10° in all) and scale bars represent 30 nm. | ||
The image in 150 mM NaCl, Fig. 6a, demonstrates a smoothly-varying topography over a lengthscale similar to that of bare silica (Fig. S5, ESI†) and a root-mean squared roughness of 107 pm. This is much smaller than the roughness observed on the bare silicon/silicon dioxide surface (see Fig. S5, ESI†) of 137 pm, implying that the formation of an SLB on the surface smooths out some of the rougher features. The phase image agrees with the topography inasmuch as the features picked out have the same orientation and are of similar size. These observations imply the bilayer follows the topography of the support well, without any extra anomalous surface features. This is to be expected from the high concentration of the salt, which allows the negative lipids plentiful access to cations in solution, minimising any strong electrostatic forces present. After rinsing the bilayer and cantilever with the HEPES solution and leaving 30 minutes for equilibration (see Materials and methods) the bilayer was imaged again. In this case, Fig. 6b, strong modifications to the surface topography are observed; the SLB is wrinkled and accordingly has a much higher surface roughness of 157 pm. These wrinkles have a width of between 5 and 10 nm, which is of the same order of magnitude as the bilayer thickness. It is likely that the wrinkling is induced by electrostatic interactions between the charged lipid headgroups. Specifically, HEPES is much less able to screen the repulsive headgroups when compared to sodium ions, resulting in a much greater area per phosphate group. This is not wholly balanced by the area of the lipids' acyl chains, resulting in a non-zero spontaneous curvature. The evidence that the wrinkling is a physical response of the bilayer, rather than a templating effect as on mica comes from observations of a similar phenomenon by Li et al.68 Their experiments used neutral lipid bilayers supported on a gold electrode. They found that ripples could be reversibly induced on a DMPC membrane by altering the potential applied to the electrode and concluded that the wrinkling was due to the lipids' asymmetric environment and mismatched headgroup/chain area. Since in our case, the change was induced by a reduction in salt in solution, it seems likely that similar mechanisms are at play here.
The effects of Tris buffer and monosodium phosphate (Fig. 6c and d) acting on the DPPA bilayer are qualitatively similar to each other, yet distinct from Fig. 6b; both topographies demonstrate equal roughnesses (Rq = 107 pm) and similar morphologies to those observed in the sodium chloride buffer. The reason for the discrepancy between HEPES and the other buffers is not clear, but could well be related to HEPES' zwitterionic form as zwitterions have been shown to have little effect on the ionic strength of a solution.69 There are clear features observed in the phase scans of Tris and monosodium phosphate; each buffer induces small (∼15 nm) features on the bilayer surface (highlighted by arrows) which mimic those observed when the buffers were present on mica (Fig. 2c and e). These similarities, at least in phase, indicate an equivalent templating process of the buffering ions occurring at the membrane as occurred on mica. The fact that there does not seem to be any major effect on the topography could be linked to the already relatively large roughness of silica, or possibly to the specific way the tip-sample interactions are modified by the buffer and SLB.
One way to confirm whether the observed changes in topography and phase represent physical modifications to the bilayer-buffer interface or to the AFM interactions while imaging is to make use of a large-scale technique that does not perturb the system. To this end, ellipsometry was performed on a DPPA/silica system, prepared identically as before (see Materials and methods) and the characteristic angles, ψ and Δ, presented in Fig. 7. Typically, these would be analysed over a range of wavelengths and fitted to a model to gain information about the adsorbed film thickness and optical properties of the layer but given the uncertainty about the true nature of the adlayer and complexity of the lipid layer itself, this is somewhat beyond the scope of this article. Instead, the extent to which the angles depended on the buffering agent was measured at a single wavelength (589 nm). The data show a clear difference in both ψ and Δ between the solutions with buffer in and that with solely NaCl. The buffered solutions are of comparable magnitude, which is somewhat intriguing given that the topography scans of Fig. 6a, c and d are qualitatively very similar. Although the ψ values with buffer molecules are not equal within errors they are much better grouped than that of NaCl. The Δ data points are not so distinct, but the values for Tris and monosodium phosphate are much closer together than they are to HEPES or NaCl, which could explain the similar templating effect seen in the phases of Fig. 6c and d. Ellipsometric measurements on similar systems such as a lipid monolayer-coated silicon dioxide wafer70 or a phosphoryl choline-modified polymer on silica71 do not agree with our values of ψ or Δ – it was assumed that this was due in part to the different nature of their samples – but their results indicate that changes in the ellipsometric angles of ∼0.5° are significant in relation to the formation of thin films. This indicates that the difference in phase scans observed in the same images did indeed represent a layering of buffer molecules on the bilayer surface. However, without further models or analysis, no stronger conclusions can be drawn.
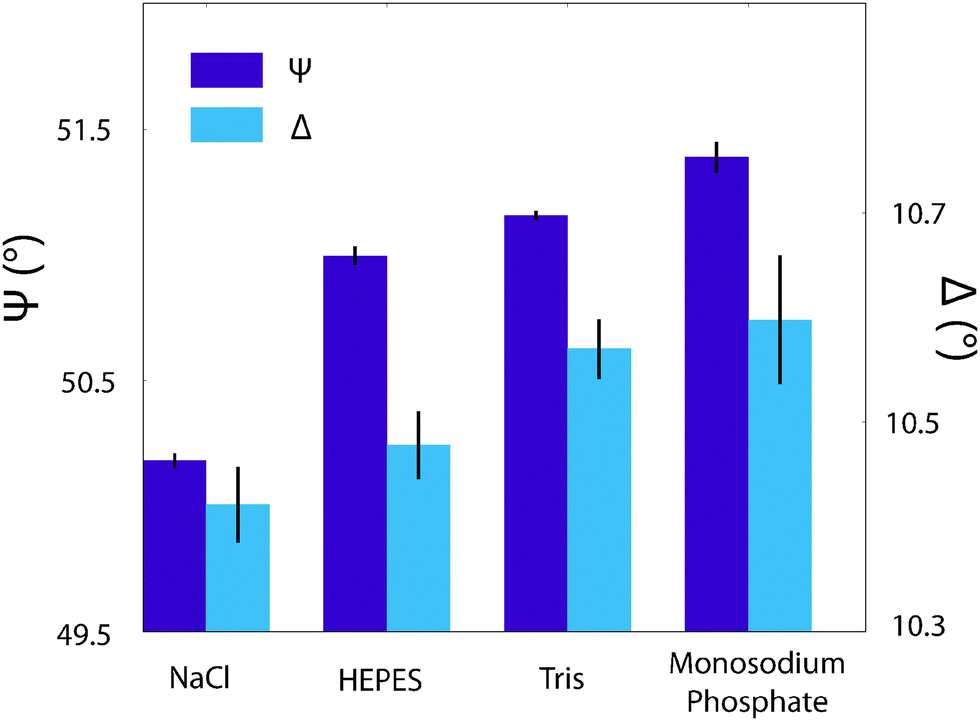 | ||
| Fig. 7 The effect of buffering agents on the interfacial properties of a silica-supported lipid bilayer as probed by ellipsometry. The angles ψ and Δ are as defined in the Materials and methods section. There is a clear discrepancy in the ψ values between the sample immersed in a NaCl solution and those immersed in buffers, which have much smaller separations. This suggests a distinct interfacial modification in agreement with Fig. 6. The Δ values' trend is less clear, but Tris and monosodium phosphate are equal within errors, which correlates with the AFM images produced in Fig. 6. | ||
4 Conclusion
We have used AM-AFM to investigate the interface between mica and five different buffering agents with molecular-level resolution. The buffer molecules produce cohesive aggregates on the charged substrate. MES and HEPES can form epitaxial lattice-like arrangements that are commensurate with the underlying mica structure, suggesting that they offer the most suitable solution for high-resolution studies. SSC, Tris and monosodium phosphate conversely formed an amorphous mesh layer with no preferential ordering. In particular monosodium phosphate interferes with the imaging process to such an extent that no atomic-level details is visible in the topography. The effects of the buffering agents are however mitigated by the adjunction of salt which can displace them from the interface. We have also shown that buffers directly interact with biological membranes, demonstrating with both AM-AFM and ellipsometry the altered interface that is produced by inclusion of a buffer. Buffering molecules actively assemble at charged surfaces, sometimes creating cohesive layers that involve interactions between buffer molecules. Further studies will examine how these effects influence the behaviour and dynamics of the biomimetic systems they are commonly used in.Acknowledgements
The authors acknowledge funding from the Engineering and Physical Sciences Council (UK) (grant 1452230), The European Council (MC-CIG grant 631186) and Durham University (UK). The authors would also like to thank Prof. Colin Bain and Marie-Capucine Pope for their advice regarding ellipsometry on mica and silica surfaces.References
- R. A. L. Jones, Soft Condensed Matter, OUP, Oxford, 1st edn, 2002 Search PubMed.
- E. Lacroix, A. R. Viguera and L. Serrano, J. Mol. Biol., 1998, 284, 173–191 CrossRef CAS PubMed.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS PubMed.
- Z. S. Hendsch and B. Tidor, Protein Sci., 1994, 3, 211–226 CrossRef CAS PubMed.
- S. McLaughlin and D. Murray, Nature, 2005, 438, 605–611 CrossRef CAS PubMed.
- W. N. Green and O. S. Andersen, Annu. Rev. Physiol., 1991, 53, 341–359 CrossRef CAS PubMed.
- K. Voïtchovsky, S. Antoranz Contera, M. Kamihira, A. Watts and J. F. Ryan, Biophys. J., 2006, 90, 2075–2085 CrossRef PubMed.
- S. Antoranz Contera, K. Votchovsky and J. F. Ryan, Nanoscale, 2010, 2, 222–229 RSC.
- D. Lacoste, G. I. Menon, M. Z. Bazant and J. F. Joanny, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 28, 243–264 CrossRef CAS PubMed.
- Q. Zhang, C. S. Dandeneau, X. Zhou and G. Cao, Adv. Mater., 2009, 21, 4087–4108 CrossRef CAS.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- J. Mou, J. Yang and Z. Shao, Biochemistry, 1994, 33, 4439–4443 CrossRef CAS PubMed.
- D. Marsh, Handbook of Lipid Bilayers, CRC Press, 2nd edn, 2013 Search PubMed.
- A. a. Gurtovenko and I. Vattulainen, J. Phys. Chem. B, 2008, 112, 1953–1962 CrossRef CAS PubMed.
- J. Song, J. Franck, P. Pincus, M. W. Kim and S. Han, J. Am. Chem. Soc., 2014, 136, 2642–2649 CrossRef CAS PubMed.
- H. I. Petrache, T. Zemb, L. Belloni and V. A. Parsegian, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7982–7987 CrossRef CAS PubMed.
- S. Garcia-Manyes, G. Oncins and F. Sanz, Biophys. J., 2005, 89, 1812–1826 CrossRef CAS PubMed.
- R. García and R. Pérez, Surf. Sci. Rep., 2002, 47, 197–301 CrossRef.
- C. Spagnoli, K. Loos, A. Ulman and M. K. Cowman, J. Am. Chem. Soc., 2003, 125, 7124–7128 CrossRef CAS PubMed.
- K. Xu, P. Cao and J. R. Heath, Science, 2010, 329, 1188–1191 CrossRef CAS PubMed.
- J. Hu, X.-d. Xiao, D. Ogletree and M. Salmeron, Surf. Sci., 1995, 344, 221–236 CrossRef CAS.
- U. M. Ferber, G. Kaggwa and S. P. Jarvis, Eur. Biophys. J., 2011, 40, 329–338 CrossRef CAS PubMed.
- T. Fukuma, M. Higgins and S. Jarvis, Phys. Rev. Lett., 2007, 98, 106101 CrossRef PubMed.
- C. a. Bippes and D. J. Muller, Rep. Prog. Phys., 2011, 74, 086601 CrossRef.
- D. Fotiadis, S. Scheuring, S. A. Müller, A. Engel and D. J. Müller, Micron, 2002, 33, 385–397 CrossRef CAS PubMed.
- A. Pyne, R. Thompson, C. Leung, D. Roy and B. W. Hoogenboom, Small, 2014, 1–5 Search PubMed.
- C. Leung, A. Bestembayeva, R. Thorogate, J. Stinson, A. Pyne, C. Marcovich, J. Yang, U. Drechsler, M. Despont, T. Jankowski, M. Tschöpe and B. W. Hoogenboom, Nano Lett., 2012, 12, 3846–3850 CrossRef CAS PubMed.
- T. Fukuma, Y. Ueda, S. Yoshioka and H. Asakawa, Phys. Rev. Lett., 2010, 104, 016101 CrossRef PubMed.
- K. Kobayashi, N. Oyabu, K. Kimura, S. Ido, K. Suzuki, T. Imai, K. Tagami, M. Tsukada and H. Yamada, J. Chem. Phys., 2013, 138, 184704 CrossRef PubMed.
- H. Imada, K. Kimura and H. Onishi, Langmuir, 2013, 29, 10744–10751 CrossRef CAS PubMed.
- E. T. Herruzo, H. Asakawa, T. Fukuma and R. García, Nanoscale, 2013, 5, 2678–2685 RSC.
- H. Asakawa, S. Yoshioka, K. I. Nishimura and T. Fukuma, ACS Nano, 2012, 6, 9013–9020 CrossRef CAS PubMed.
- M. Ricci, P. Spijker and K. Voïtchovsky, Nat. Commun., 2014, 5, 4400 CAS.
- M. Ricci, P. Spijker, F. Stellacci, J. F. Molinari and K. Voïtchovsky, Langmuir, 2013, 29, 2207–2216 CrossRef CAS PubMed.
- I. Siretanu, D. Ebeling, M. P. Andersson, S. L. S. Stipp, A. Philipse, M. C. Stuart, D. van den Ende and F. Mugele, Sci. Rep., 2014, 4, 4956 CAS.
- K. Kimura, S. Ido, N. Oyabu, K. Kobayashi, Y. Hirata, T. Imai and H. Yamada, J. Chem. Phys., 2010, 132, 194705 CrossRef PubMed.
- S.-H. Loh and S. P. Jarvis, Langmuir, 2010, 26, 9176–9178 CrossRef CAS PubMed.
- P. J. Scales, F. Grieser and T. W. Healy, Langmuir, 1990, 6, 582–589 CrossRef CAS.
- R. P. Richter, R. Bérat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- T. C. Anglin and J. C. Conboy, Biophys. J., 2008, 95, 186–193 CrossRef CAS PubMed.
- K. Vedam, Thin Solid Films, 1998, 313–314, 1–9 CrossRef CAS.
- J. J. Shin and C. J. Loewen, BMC Biol., 2011, 9, 85 CrossRef CAS PubMed.
- J. Faraudo and A. Travesset, Biophys. J., 2007, 92, 2806–2818 CrossRef CAS PubMed.
- X. Wang, S. P. Devaiah, W. Zhang and R. Welti, Prog. Lipid Res., 2006, 45, 250–278 CrossRef CAS PubMed.
- J. Fraxedas, Water at Interfaces: a Molecular Approach, CRC Press, 2014 Search PubMed.
- T. Fukuma, B. Reischl, N. Kobayashi, P. Spijker, F. F. Canova, K. Miyazawa and A. S. Foster, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 155412 CrossRef.
- M. Grossman, B. Born, M. Heyden, D. Tworowski, G. B. Fields, I. Sagi and M. Havenith, Nat. Struct. Mol. Biol., 2011, 18, 1102–1108 CAS.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047–16051 CrossRef CAS PubMed.
- D. R. Lide, CRC handbook of chemistry and physics, CRC press, 2004 Search PubMed.
- H. J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1–7 CrossRef.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 3rd edn, 2011 Search PubMed.
- K. Voïtchovsky, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 022407 CrossRef PubMed.
- K. Voïtchovsky, J. J. Kuna, S. A. Contera, E. Tosatti and F. Stellacci, Nat. Nanotechnol., 2010, 5, 401–405 CrossRef PubMed.
- S. M. R. Akrami, H. Nakayachi, T. Watanabe-Nakayama, H. Asakawa and T. Fukuma, Nanotechnology, 2014, 25, 455701 CrossRef CAS PubMed.
- M.-P. Mingeot-Leclercq, M. Deleu, R. Brasseur and Y. F. Dufrêne, Nat. Protoc., 2008, 3, 1654–1659 CrossRef PubMed.
- I. Reviakine and A. Brisson, Langmuir, 2000, 16, 1806–1815 CrossRef CAS.
- A. Alessandrini and P. Facci, Soft Matter, 2014, 10, 7145–7164 RSC.
- P. Fenter and N. C. Sturchio, Prog. Surf. Sci., 2005, 77, 171–258 CrossRef.
- R. García, Amplitude Modulation Atomic Force Microscopy, Wiley-VCH, 2010 Search PubMed.
- C. Park, P. Fenter, K. Nagy and N. Sturchio, Phys. Rev. Lett., 2006, 97, 016101 CrossRef PubMed.
- J. I. Kilpatrick, S.-H. Loh and S. P. Jarvis, J. Am. Chem. Soc., 2013, 135, 2628–2634 CrossRef CAS PubMed.
- D. J. Müller, D. Fotiadis, S. Scheuring, S. a. Müller and A. Engel, Biophys. J., 1999, 76, 1101–1111 CrossRef.
- S. S. Lee, P. Fenter, K. L. Nagy and N. C. Sturchio, Langmuir, 2012, 28, 8637–8650 CrossRef CAS PubMed.
- A. Naderi, J. Iruthayaraj, A. Vareikis, R. Makuška and P. M. Claesson, Langmuir, 2007, 23, 12222–12232 CrossRef CAS PubMed.
- D. Beaglehole and H. K. Christenson, J. Phys. Chem., 1992, 96, 3395–3403 CrossRef CAS.
- Y. Xie, The Nanobiotechnology Handbook, CRC Press, 2013, p. 692 Search PubMed.
- K. El Kirat, V. Duprès and Y. F. Dufrêne, Biochim. Biophys. Acta, 2008, 1778, 276–282 CrossRef CAS PubMed.
- M. Li, M. Chen, E. Sheepwash, C. L. Brosseau, H. Li, B. Pettinger, H. Gruler and J. Lipkowski, Langmuir, 2008, 24, 10313–10323 CrossRef CAS PubMed.
- E. Stellwagen, J. D. Prantner and N. C. Stellwagen, Anal. Biochem., 2008, 373, 407–409 CrossRef CAS PubMed.
- T.-L. Phang and E. I. Franses, Langmuir, 2006, 22, 1609–1618 CrossRef CAS PubMed.
- E. F. Murphy, J. R. Lu, J. Brewer, J. Russell and J. Penfold, Langmuir, 1999, 15, 1313–1322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm02445e |
| This journal is © The Royal Society of Chemistry 2016 |