
A terminally protected dipeptide: from crystal structure and self-assembly, through co-assembly with carbon-based materials, to a ternary catalyst for reduction chemistry in water†
Daniela
Mazzier
a,
Francesco
Carraro
a,
Marco
Crisma
b,
Marzio
Rancan
c,
Claudio
Toniolo
ab and
Alessandro
Moretto
*ab
aDepartment of Chemical Sciences, University of Padova, 35131 Padova, Italy. E-mail: alessandro.moretto.1@unipd.it
bInstitute of Biomolecular Chemistry, Padova Unit, CNR, 35131 Padova, Italy
cInstitute for Energetics and Interphases, CNR, 35131 Padova, Italy
First published on 7th October 2015
Abstract
A terminally protected, hydrophobic dipeptide Boc-L-Cys(Me)-L-Leu-OMe (1) was synthesized and its 3D-structure was determined by single crystal X-ray diffraction analysis. This peptide is able to hierarchically self-assemble in a variety of superstructures, including hollow rods, ranging from the nano- to the macroscale, and organogels. In addition, 1 is able to drive fullerene (C60) or multiwalled carbon nanotubes (MWCNTs) in an organogel by co-assembling with them. A hybrid 1-C60–MWCNT organogel was prepared and converted (through a high vacuum-drying process) into a robust, high-volume, water insoluble, solid material where C60 is well dispersed over the entire superstructure. This ternary material was successfully tested as a catalyst for: (i) the reduction reaction of water-soluble azo compounds mediated by NaBH4 and UV-light with an overall performance remarkably better than that provided by C60 alone, and (ii) the NaBH4-mediated reduction of benzoic acid to benzyl alcohol. Our results suggest that the self-assembly properties of 1 might be related to the occurrence in its single crystal structure of a sixfold screw axis, a feature shared by most of the linear peptides known so far to give rise to nanotubes.
1. Introduction
Since J. M. Lehn's pioneering contributions, the bottom-up self-assembly strategy has emerged as one of the most applicable approaches to drive small molecule arrangements.1–4 Following a hierarchical process, under appropriate conditions the combination of a variety of non-covalent interactions may allow properly designed molecules to form stable, ordered aggregates that can evolve into nano-, micro- and macroscale materials characterized by discrete 3D-architectures with specific shapes, dimensions and functions, exploitable in areas ranging from chemistry to materials science and medicine.5,6 In this connection, peptides represent powerful building blocks on the nanoscale because of their modularity, molecular diversity, which can be expanded beyond the repertoire of genetically coded amino acids, and secondary structure variety.7–17 More specifically, nanotubes and nanofibers predominate as molecular architectures among the self-assemblies formed by short, unprotected peptides18–21 of which the phenylalanine homo-dipeptide H-Phe-Phe-OH is the most extensively investigated example (for review, see ref. 7). Notably, such architectures may adsorb or entrap other molecules, thus generating ordered hybrid materials.22 Conversely, only a few examples of terminally-protected peptides, the crystal structures of which are characterized by a nanotube architecture, were reported, but their self-assembly properties have not been further explored.23–26Here, we present our data for the hydrophobic, terminally protected dipeptide Boc-L-Cys(Me)-L-Leu-OMe (1) [where Boc is tert-butoxycarbonyl, Cys(Me) is S-methyl cysteine, and OMe is methoxy] (Fig. 1A) that was crystallographically characterized and was subsequently found to be able to hierarchically self-assemble producing nano-, micro- and macroscale tubes and organogels that we eventually exploited to incorporate guest molecules such as gold nanoparticles, C60 fullerene and multiwalled carbon nanotubes (MWCNTs). Moreover, from an organogel composed of 1, C60 and MWCNTs, by a simple vacuum-drying process, we generated an ordered, robust, carbon-based non-covalently assembled material, which displayed high performances when used as a catalyst in reduction chemistry.
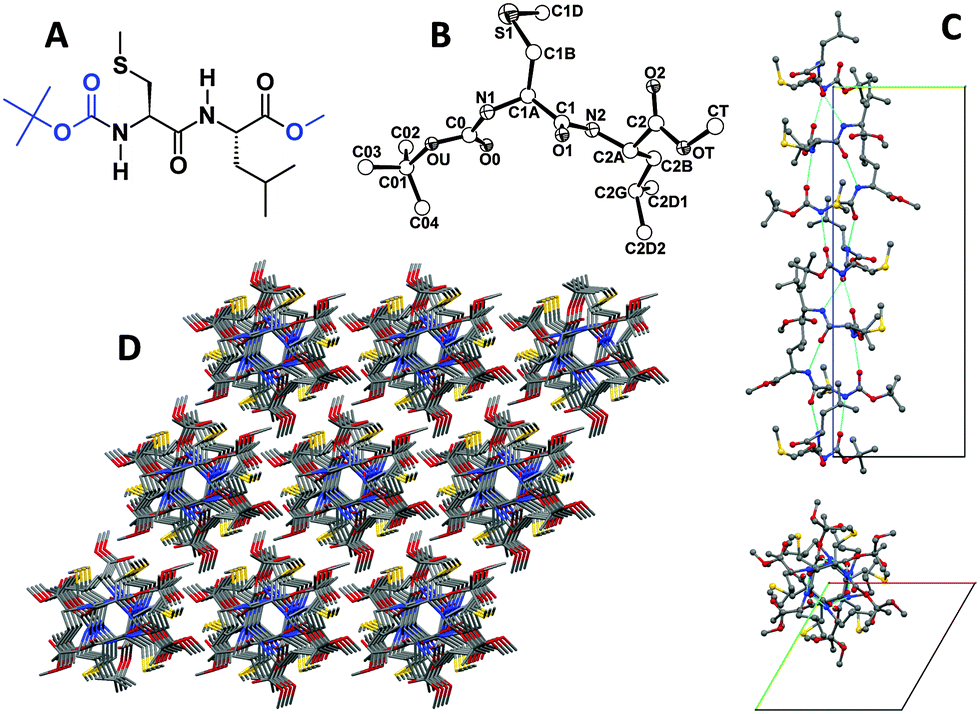 | ||
| Fig. 1 (A) Chemical formula of dipeptide 1. (B) X-Ray diffraction structure of 1 with atom numbering. (C) Packing mode of 1 in the crystal. Intermolecular H-bonding generates helical rows of molecules (see the text for details). A single row is shown as viewed perpendicular (upper part) and parallel (lower part) to the sixfold screw axis. Intermolecular H-bonds are indicated by dashed lines. (D) Overall packing of 1 molecules as viewed nearly along the c axis. Carbon, grey; nitrogen, blue; oxygen, red; sulphur, yellow. | ||
2. Materials and methods
2.1 X-Ray diffraction
(i) Single crystal X-ray diffraction data were collected using an Agilent Technologies Gemini E four-circle kappa diffractometer equipped with a 92 mm EOS CCD detector, using graphite monochromated Cu Kα radiation (λ = 1.54178 Å). Data collection and reduction were performed with the CrysAlisPro software (Agilent Technologies). The structures were solved by ab initio procedures of the SIR 2002 program,27 and refined by full-matrix least-squares on F2, using all data, by applying the SHELXL-97 program.28 Additional details may be found in the ESI.† (ii) Powder X-ray diffraction measurements were carried out by means of a Bruker D8 Advance diffractometer equipped with a Göbel mirror and a Cu Kα source (40 kV, 40 mA). Powder pattern indexing was performed by using the N-TREOR29 software in the framework of the EXPO201330 package.2.2 Self-assembly investigations
(i) Self-assembly from solvent evaporation. Typically, from 500 mg to 1 g of 1 were dissolved in 10 to 20 mL of the appropriate solvent (MeOH, CH3CN or EtOAc). The resulting clear solution was allowed to stand in a large crystallization vessel at controlled temperature (25 °C). After evaporation of the solvent macroscopic rods were obtained. (ii) Self-assembly from water/EtOAc interfacial crystallization. Typically, from 100 to 200 mg of 1 were dissolved in 10 to 20 mL of EtOAc. The resulting clear solution was gently layered over mQ water in a crystallization vessel at controlled temperature (25 °C). After evaporation of EtOAc, micrometric rods were collected from the water solution. (iii) Self-assembly from MeOH/water. Typically from 0.1 to 1 mL of a MeOH solution of 1 (2 mg mL−1) were added to 1 mL of H2O and allowed to stand in a sealed vessel for 1 to 3 days.2.3 Organogel preparations
(i) Pristine 1-organogel was prepared as follows. Dipeptide 1 was added in small amounts to toluene to a concentration of 85 mg mL−1. Then, the resulting suspension was heated at 60 °C under sonication, for 5 min. After standing at rt, the clear solution turned into an organogel. (ii) The 1-C60 organogel was prepared as follows. Typically, from 5 to 8 mg of C60 were added to 1 mL of toluene and the suspension was heated at 70 °C up to complete solubilization. Then, 85 mg of 1 were added to the C60 solution in small amounts to a concentration of 85 mg mL−1. The resulting suspension was heated at 60 °C under sonication for 10 min. After standing at rt, the clear, dark-red solution turned into an organogel. (iii) The 1–MWCNT organogel was prepared as follows. MWCNTs, 20 mg, were suspended in 1 mL of toluene and sonicated for 5 min. Then, 1 was added to the suspension in small amounts to a concentration of 85 mg mL−1. The resulting suspension was heated at 65 °C under sonication for 20 min. The black mixture gradually turned into an organogel.2.4 1-C60–MWCNT composite/catalyst preparation
Typically, 5 mL of the 1-C60 organogel and 5 mL of the 1–MWCNT organogel (prepared as described above) were separately melted in their sol state. The two solutions were combined and heated at 75 °C under sonication for 5 min. The dark mixture gradually turned into a stable organogel. The organogel was directly dried in vacuo. The nanocomposite contains from 2.5 to 5% w/w of fullerene.2.5 Reduction of azo compounds under UV irradiation
Reduction of methyl orange (MO) was carried out in a 50 mL beaker. NaOH solution (1 M, 0.5 mL), distilled water (40 mL), NaBH4 (100 mg), and MO (200 mg) were added. Then, the 1-C60–MWCNT catalyst (100 mg, C60 as 2.5% w/w) was added to form a suspension. The reaction proceeded by stirring under UV irradiation (UV lamp, 365 nm, 50 Hz) at rt for 45 min. After the catalyst was removed by filtration, the reaction mixture was characterized using HPLC-MS analysis. The experiment was repeated 3 times. The reduction procedure of methyl red (MR) and 4-aminoazobenzene (AZ) in water was similar to that described above.2.6 Reduction of benzoic acid under UV irradiation
Reduction of Benzoic Acid was carried out under similar conditions to those described for Azo compounds. The reaction proceeded by stirring under UV irradiation (UV lamp, 365 nm, 50 Hz) at rt for 3 hours. After the catalyst was removed by filtration, the reaction mixture was characterized using HPLC-MS analysis.3. Results and discussion
3.1 X-Ray diffraction
The X-ray diffraction structure of 1 [determined on a single crystal grown from MeOH (methanol)] is illustrated in Fig. 1B.31 The conformation adopted by both α-amino acid residues is semi-extended, and the N–H groups of Cys and Leu point to opposite directions. In the packing mode (Fig. 1C), the (urethane) N1–H group is intermolecularly H-bonded to the (y, −x + y, z + 1/6) symmetry equivalent of the (urethane) O0 atom, and the (amide) N2–H group is H-bonded to the (x − y, x, z − 1/6) symmetry equivalent of the (amide) O1 atom (Table S3, ESI†). As a result, H-bonded molecules wrap around the sixfold screw axis along the c direction, each molecule being connected to the next by two H-bonds. The left-handed, supramolecular sixfold helix thus generated is characterized by a very narrow lumen, about 2.5 Å in diameter (Fig. 1C, bottom). The Cys(Me) and Leu side chains, as well as the N- and C-terminal Boc and OMe groups of each molecule, are located on the external surface. Each helical row of molecules is surrounded by six counterparts (Fig. 1D). Lateral stabilization between rows is provided, in addition to van der Waals interactions, by C–H⋯O interactions taking place between the (Boc) C02-H02B group and the (x − 1, y − 1, z) symmetry equivalent of the (methyl ester) OT atom (Table S3, ESI†).3.2 Self-assembly and gold nanoparticle inclusion
Dipeptide 1 is able to adopt a well-organized, micro-arrangement when allowed to self-assemble under appropriate conditions. Upon slow evaporation of concentrated solutions of 1 in either EtOAc (ethyl acetate), CH3CN or acetone, long, aligned rods (up to 10 cm in length) are formed as shown in Fig. 2A. Interestingly, such rods are characterized by an overall parallelepiped shape and present an empty inner cavity (Fig. 2B). | ||
| Fig. 2 (A) Camera picture of macroscopic rods from 1 obtained after evaporation from a concentrated solution in CH3CN. (B) Camera picture showing details of a macroscopic rod obtained from a CH3CN solution. (C) SEM image of micrometric rods from 1 obtained from a water/EtOAc interfacial crystallization. (D) SEM image of micrometric rods from 1 obtained from a water/MeOH solution. (E and F): SEM images showing details of the rods (obtained from a water/MeOH solution). | ||
Smaller and packed rods of 1 (at the microscale level) were obtained by interfacial crystallization between water and EtOAc, as revealed by SEM images (Fig. 2C). Moreover, by adding a diluted solution of 1 in MeOH to a large excess of water, the formation of micrometric, isolated rods was observed (Fig. 2D). According to a SEM analysis (Fig. 2E and F), even at this microscale level, the rods are characterized by an empty inner cavity while maintaining the overall parallelepiped shape found at the macroscale level.
Peptide 1 was further studied by circular dichroism (CD) in CH3CN solution and as solid-state rods obtained after evaporation from the same solvent (Fig. 3A and B). Notably, 1 shows a strong aggregation propensity in CH3CN solution at concentrations above 1 mg mL−1 (Fig. 3A). This conclusion is justified by the change of the Cotton effect near 195 nm from weakly positive at 0.3 mg mL−1 concentration to strongly negative at concentrations in the range 2.0–10 mg mL−1, accompanied by a marked increase in ellipticity of the positive Cotton effect at about 218 nm. The CD spectra of the most concentrated solutions nicely resemble the spectrum obtained in the solid state (Fig. 3B), i.e. that of the rod structure, although the latter would be entirely red-shifted, albeit slightly.
 | ||
| Fig. 3 (A) CD spectra of 1 at different concentrations in CH3CN solution. (B) Solid-state CD spectra of rods obtained from CH3CN. (C) Comparison of simulated PXRD measurements (from single-crystal X-ray diffraction) with experimental results on rods obtained from the CH3CN and MeOH/water preparation methods. (D) Comparison of Raman spectra of the rods obtained from the EtOAc/water and CH3CN preparation methods. | ||
The powder X-ray diffraction (PXRD) technique was exploited in order to check whether the formation of rods characterized by an empty cavity at the macro- and microscale level (prepared from CH3CN and MeOH/water, respectively) could be ascribed to a supramolecular organization different from that determined at the single crystal level. The PXRD patterns of the rods prepared by the two methods (CH3CN and MeOH/water) are identical to each other all over the 2θ range investigated, and virtually overlap to the simulated pattern of the single crystal X-ray diffraction sample (Fig. 3C). In addition, the values of the unit cell parameters extracted from the two experimental PXRD patterns are in excellent agreement with those of the single crystal (Table 1). These results clearly indicate that, although different preparation methods lead to different morphologies, the 3D-supramolecular organization adopted by 1 in the crystal state is shared by the micro- and macro-hollow rods. Similar conclusions were extracted from a Raman study (Fig. 3D). Indeed, the rods obtained from the EtOAc/water and CH3CN preparation methods exhibit identical Raman signatures.
| a (Å) | b (Å) | c (Å) | α | β | γ | Volume (Å3) | |
|---|---|---|---|---|---|---|---|
| Single crystal | 11.4458(2) | 11.4458(2) | 27.7551(5) | 90 | 90 | 120 | 3148.95(10) |
| MeOH/water | 11.468(4) | 11.468(4) | 27.810(16) | 90 | 90 | 120 | 3167(2) |
| CH3CN | 11.426(4) | 11.426(4) | 27.847(28) | 90 | 90 | 120 | 3148(3) |
To gain in-depth information on the nature of the rod architectures, we investigated the mother liquor remained after the MeOH/water preparation method. The transmission electron microscopy (TEM) analysis of this mixture carried out on stained (uranyl acetate) samples showed the presence of nano- and micrometric rods (Fig. 4A).
 | ||
| Fig. 4 (A) Stained TEM image after filtration of the macroscopic rods from a MeOH/water solution of 1. (B) Stained TEM image showing details of the self-assembled rods. (C) Stained TEM image showing details of the multiwall nature of a nanoscopic rod. | ||
A more detailed investigation of these samples revealed two interesting features. First, we observed the presence of “incompletely folded” rods, which confirmed the tubular nature of such a type of architecture (Fig. 4B). Secondly (Fig. 4C), we noticed a multilayer structure of the external part of the rod. In a few cases, our TEM analysis of the same, but non-stained, sample (Fig. 5A) revealed the typical structure of a multiwalled nanotube characterized by two parallel, dark lines, associated with a higher material density, occurring at the boundaries of the rods. Interestingly, these images are similar to those that can be obtained from the TEM analysis performed on MWCNT samples.32 To prove the existence of such a type of hollow superstructure, we made use of citrate-passivated (water soluble) gold nanoparticles (10 nm diameter)33 which were added to the MeOH/water solution. After the self-assembly process, the results of TEM analysis (non-stained samples, Fig. 5B), energy-dispersive X-ray spectroscopy (EDX) (Fig. 5C), and optical microscopy (polarized light, Fig. 5D and E) clearly showed that the gold nanoparticles are incorporated into the rod structures.
 | ||
| Fig. 5 (A) Non-stained TEM image of a nanorod of 1. (B) Non-stained TEM image showing the encapsulation of water-soluble gold nanoparticles into a rod. (C) SEM image and the EDX pattern of the 1-nanotube/gold nanoparticle hybrid rods. (D) Optical microscopy image of 1-rods prepared from MeOH/water under polarized light. (E) Optical microscopy image of co-assembled 1-rods/gold nanoparticles under polarized light. | ||
Interestingly, we found that 1 is able to interact with citrate-passivated gold nanoparticles, probably via its thioether sulfur atom, resulting in the self-aggregation of the nanoparticles. This hypothesis is supported by comparison of the TEM images (Fig. 6A and B) and the UV-vis absorption spectra (Fig. 6C) of the citrate-passivated gold nanoparticles and the 1-passivated gold nanoparticles. The strong aggregation occurring for 1-passivated gold nanoparticles results in a remarkable red-shift of its plasmonic band as compared with that of the citrate-passivated gold nanoparticles. Moreover, the CD spectra of the 1-passivated gold nanoparticles (Fig. 6D) showed a chiral signature in correspondence to the plasmonic region (400–600 nm), that may arise from the interactions of 1 (which is a chiral molecule) with the gold core. A similar chiral signature of the plasmonic band was previoulsy reported for a 10 nm gold nanoparticle–peptide conjugate.34
 | ||
| Fig. 6 (A) TEM image of citrate-passivated gold nanoparticles. (B) TEM image of 1-passivated gold nanoparticles. (C) UV-vis absorption spectra of citrate-passivated gold nanoparticles (black line) and 1-passivated gold nanoparticles (red line). (D) CD spectra of 1-passivated gold nanoparticles. | ||
In addition, treatment at 400 °C in an oxygen atmosphere of the co-assembled 1-rods/gold nanoparticles described above led to the formation of gold nanorods (about 100–200 nm in length) as detected by TEM (Fig. 7A and B). The UV-vis absorption spectrum of this material (Fig. 6C) is characterized by a relatively sharp band at about 525 nm, accompanied by a much broader absorption centered near 720 nm, features typical of gold nanorods.35
 | ||
| Fig. 7 (A) and (B) TEM images of gold nanorods, obtained by treatment at 400 °C in an oxygen atmosphere of the co-assembled 1-rods/gold nanoparticles, recorded prior and after a sonication process, respectively. (C) UV-vis absorption spectra of gold nanorods in aqueous suspension. | ||
3.3 Organogel formation
To further expand our study of the self-assembly propensity of 1 in organic solvents, we prepared a set of concentrated solutions of 1 in toluene. We observed the formation of an organogel (after a gentle heating followed by a cooling process) when solutions of 1 reach a concentration of 85 mg mL−1. According to a TEM analysis, the organogel is characterized by a dense network composed of long (up to 10 μm) and twisted fibers (Fig. 8A and B).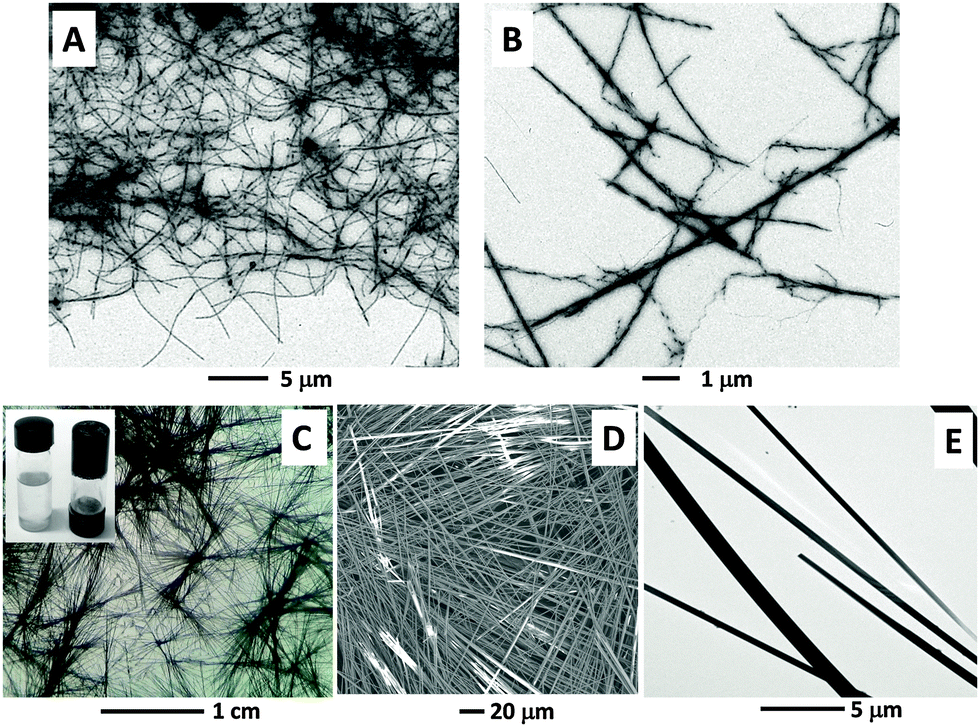 | ||
| Fig. 8 (A and B) Stained TEM images showing the organogel microstructure (fibers) of 1 and details of the twisted nature of the fibers. (C) Optical microscopy image of the 1/MWCNT organogel obtained after evaporation of toluene. Inset: Vials showing a 85 mg mL−1 solution of 1 in toluene at 70 °C (left) and the corresponding organogel formed after sonication in the presence of MWCNTs and cooling to room temperature (right; the vial is upside down). (D and E): SEM and non-stained TEM images, respectively, of the 1/MWCNT composite obtained after evaporation of toluene. | ||
3.4 Multicomponent organogels
Addition of MWCNTs to a solution of 1 (85 mg mL−1) in toluene, followed by sonication, allowed us to obtain 1/MWCNT organogel systems (Fig. 8C, inset). Once deposited on a glass surface, this organogel rapidly crystallized in microscopic black needles, as detected by optical microscopy and SEM (Fig. 8C and D, respectively). Moreover, small and large nanostructures were unraveled also by TEM (Fig. 8E). A comparison of the TEM analyses of the organogel formed by 1 alone (stained sample, Fig. 8A and B) and of the 1/MWCNT organogel (non-stained sample, Fig. 8E), indicates a much higher matter density for the latter. This finding suggests that in the 1/MWCNT organogel the MWCNTs might have been incorporated, and probably ordered, within the rods. We found that addition of a 15% v/v MeOH (a good solvent for 1, but not for MWCNTs) was successful in disassembling the 1/MWCNT organogel system, resulting in a black MWCNT precipitate and a colorless solution (the same effect was also observed after the addition of ethanol, isopropanol, CH3CN and N,N-dimethylformamide). Therefore, we decided to progressively disrupt the rods directly on the TEM-grid by adding increasing amounts of MeOH or CH3CN to the 1/MWCNT organogel drop prior to TEM detection. The resulting TEM images are reported in Fig. 9. In particular, by comparing the TEM images reported in Fig. 8E (needles obtained by evaporation) and Fig. 9A (needles after addition of 0.5% MeOH) it seems to be clear that the addition of MeOH affects the overall morphology. Subsequent additions of larger amounts of MeOH resulted in the collapse of the needle morphology (Fig. 9B and C). This finding was also observed upon addition of CH3CN (Fig. 9D and E). In particular, Fig. 9A–E show the formation of packed (or single) strands (between a 30–100 nm width, and up to a 5 μm length) that originate from the needle structures mentioned earlier. Our most relevant observation is that strands are detected on a non-stained sample, which suggests a high matter density occurring in these superstructures. We hypothesize that these strands are related to the 1–MWCNT co-assembled systems where the MWCNT surfaces are covered by 1. | ||
| Fig. 9 Non-stained TEM images showing the effect of the addition of MeOH (A–C) or CH3CN (D and E) to the 1/MWCNTs organogel microstructure on the TEM-grid. Specifically: additions of 0.5% (A), 2% (B) and 5% (C) MeOH (v/v); additions of 1% (D) and 3% (E) CH3CN (v/v). (F) Non-stained TEM image after sonication of MWCNTs in a solution of 1 at 10 mg mL−1 in toluene. | ||
As a control experiment, we prepared a solution of 1 at a concentration of 10 mg mL−1 in toluene and added MWCNTs. Under these conditions, after a sonication process, no microstructures were formed. However, it was possible to detect (by TEM) partially unbundled MWCNTs (Fig. 9F). The latter observation is important evidence for the co-assembly propensity of 1 with MWCNTs. Thus, 1 seems to be able to partially disperse MWCNTs in toluene solution. Overall, we can likely assume that the formation of 1/MWCNT co-assembled needles goes through a series of steps. Specifically, MWCNTs are first covered by 1, then MWCNTs become disperse in the solution media, and eventually the covered 1–MWCNT complexes align (with the support of the large excess of 1) giving rise to the supramolecular hybrid architectures.
Moreover, the addition of C60 to a solution of 1 (85 mg mL−1) in toluene, followed by sonication, allowed us to obtain 1/C60 organogel systems (Fig. 10A, and the inset). Inspection of a diluted 1/C60 organogel sample deposited on the TEM grid revealed the presence of small particles (from 5- to 15 nm in diameter) spread all over the organogel network (Fig. 10B). These particles are possibly due to the formation of C60 aggregates. The 1/C60 organogel, once deposited on a glass surface, rapidly crystallized in microscopic red/orange needles (Fig. 10C inset) that were characterized by SEM (Fig. 10C).
 | ||
| Fig. 10 (A) Non-stained TEM images showing the 1/C60 organogel microstructure (formed in toluene). Inset: Vials showing a 85 mg mL−1 solution of 1 in toluene at 70 °C (left) and the corresponding organogel formed after sonication in the presence of C60 and cooling to room temperature (right; the vial is upside down). (B) TEM details showing the presence of small particles (in a diluted sample) incorporated into the microstructure network. (C) SEM image of the 1/C60 organogel microscopic needles obtained after rapid crystallization. Inset: Camera picture of microscopic red/orange needles. (D) SEM image of the 1/C60/MWCNTs composite obtained from the corresponding organogel in toluene after the high vacuum-drying process. Inset: Camera picture of the 1/C60/MWCNTs composite. (E) SEM details of the 1/C60/MWCNT composite showing the presence of ordered co-assembled rods. | ||
The organogels formed by 1 alone, 1/C60, and 1/MWCNTs are characterized by a transition temperature for the sol–gel process (Tsol–gel) of 55, 47, and 85 °C, respectively. These data indicate that, in comparison to 1, the gel stability is only marginally affected by the presence of fullerene, but it is strongly increased by MWCNTs.
3.5 Three-component catalyst
With the aim of preparing a robust material with catalytic properties, we decided to combine 1/C60 and 1/MWCNT carbon-based organogels. In particular, the two gels were melted in their sol (liquid) forms and combined together. After a sonication process followed by standing at room temperature, a novel 1/C60/MWCNT organogel was obtained, which displayed a Tsol–gel of 79 °C. Removal of the solvent by a high vacuum-drying process at room temperature afforded a mechanically robust self-assembled solid material, characterized by a high porosity (300 mg of the material prepared by this process occupy as many as 6 cm3) and insoluble in water (Fig. 10D, and the inset). The vacuum-drying process itself helps keeping the gel size, because the fast evaporation procedure used generates a low temperature. The consequent gel hardening at low temperature is beneficial to retain the size. Furthermore, the fast drying process shortens the time allowed to capillary forces to act on the gel, thus moderating the structure collapse caused by strain accumulation.36 The SEM details reported in Fig. 10E showed the presence of co-assembled rods which exhibit ordered and aligned domains on their surfaces. Remarkably, the same processes performed on the 1 or 1/C60 organogels did not provide any organized material. The interest for nanocarbon materials as catalysts is rapidly growing, as highlighted by recent review articles.37,38 In this connection, Khashab and coworkers39 reported the successful reduction of a set of azo compounds to the corresponding aromatic amines by NaBH4 under UV irradiation in basic water using C60 as a catalyst. It is known that NaBH4 is unable to reduce the azo group without the assistance of a metal catalyst, such as Pd or Ag.40,41 The catalytic effect of C60 was ascribed to the electron acceptor properties of its UV-promoted triplet state, which leads to the activation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. The limitations of such a system are related to the insolubility of C60 in water, which results in its poor dispersion and formation of large aggregates. Indeed, 10 mg of C60 were required in order to catalyze the reduction of 10 mg of methyl orange after 5 hours under illumination in the presence of a large excess of NaBH4.39 We envisioned that our 1/C60/MWCNT hybrid material could be a more efficient catalyst than C60 alone in the reduction of azo compounds. Actually, by using 100 mg of the 1/C60/MWCNT self-assembled solid material (which contains approximately 2.5% w/w of C60) and 100 mg of NaBH4, quantitative conversion of 200 mg of either methyl orange, methyl red, or 4-amino-azobenzene to the corresponding amines was achieved within 45 minutes under UV irradiation at 365 nm in water. The water-insoluble catalyst was easily recovered by filtration. Chemical selectivity was observed as indicated in Fig. 11, which was identical to that reported in the previous study.39 A comparison of our results with those reported for the same reaction by Khashab and coworker39 strongly supports the view that in our catalyst C60 is molecularly well dispersed on a large porous surface, thus allowing a more effective interaction with the azo compounds. These findings prompted us to explore additional potential applications for our 1/C60/MWCNT co-assembled solid material as a catalyst in NaBH4-mediated reduction reactions. NaBH4 is commonly used to reduce aldehydes and ketones to give the related alcohols, but is not reactive enough to reduce carboxylic acids or esters. We found that, under experimental conditions closely related to those exploited for the reduction of azo compounds, our catalyst is able to promote the reduction of benzoic acid to benzyl alcohol (4 hours, 96% conversion, see ESI†).
N bond. The limitations of such a system are related to the insolubility of C60 in water, which results in its poor dispersion and formation of large aggregates. Indeed, 10 mg of C60 were required in order to catalyze the reduction of 10 mg of methyl orange after 5 hours under illumination in the presence of a large excess of NaBH4.39 We envisioned that our 1/C60/MWCNT hybrid material could be a more efficient catalyst than C60 alone in the reduction of azo compounds. Actually, by using 100 mg of the 1/C60/MWCNT self-assembled solid material (which contains approximately 2.5% w/w of C60) and 100 mg of NaBH4, quantitative conversion of 200 mg of either methyl orange, methyl red, or 4-amino-azobenzene to the corresponding amines was achieved within 45 minutes under UV irradiation at 365 nm in water. The water-insoluble catalyst was easily recovered by filtration. Chemical selectivity was observed as indicated in Fig. 11, which was identical to that reported in the previous study.39 A comparison of our results with those reported for the same reaction by Khashab and coworker39 strongly supports the view that in our catalyst C60 is molecularly well dispersed on a large porous surface, thus allowing a more effective interaction with the azo compounds. These findings prompted us to explore additional potential applications for our 1/C60/MWCNT co-assembled solid material as a catalyst in NaBH4-mediated reduction reactions. NaBH4 is commonly used to reduce aldehydes and ketones to give the related alcohols, but is not reactive enough to reduce carboxylic acids or esters. We found that, under experimental conditions closely related to those exploited for the reduction of azo compounds, our catalyst is able to promote the reduction of benzoic acid to benzyl alcohol (4 hours, 96% conversion, see ESI†).
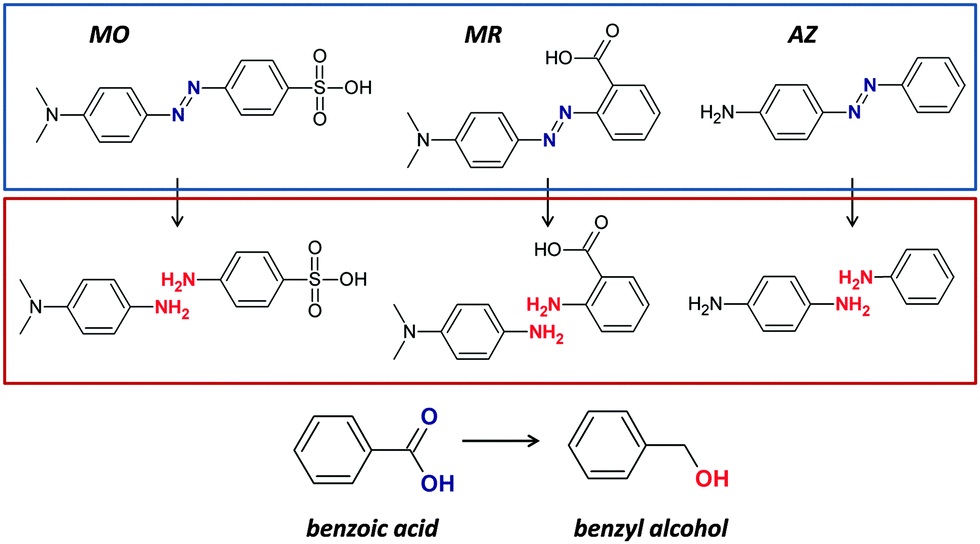 | ||
| Fig. 11 Chemical structures of methyl orange (MO), methyl red (MR), 4-amino-azobenzene (AZ), and benzoic acid, and the corresponding products of reduction reactions. | ||
It has to be noted that although methyl red carries a carboxylic group, its NaBH4-mediated reduction reaction catalyzed by our 1/C60/MWCNT co-assembled solid material did not led to any detectable formation of ortho-aminobenzyl alcohol. In this case the effects of the amino substituent at the ortho position prevent the reduction of the carboxylic function under our experimental conditions.
4. Conclusion
To summarize, we have shown that a terminally protected, hydrophobic dipeptide, Boc-L-Cys(Me)-L-Leu-OMe (1), is able to hierarchically self-assemble in well-defined structures. In particular, evaporation from organic solvents (e.g., EtOAc, CH3CN, acetone) or interfacial (EtOAc/water and MeOH/water) crystallizations led to the formation of hollow rods ranging from the nano- to the macroscale. In addition, an organogel could be obtained using toluene, the microstructure of which is characterized by twisted fibers. The organogel of 1 formed in the presence of C60 or MWCNTs shows a different morphology (needles). Evidence is provided in support of the view that 1, besides helping in dispersing the otherwise insoluble MWCNTs, is able to co-assemble with them. We succeeded in obtaining a novel ternary, co-assembled, carbon-based material by taking advantage of the self-assembly tendency of 1. Specifically, by using a high vacuum-drying process, the 1/C60/MWCNT organogel was converted into a robust, high-volume, water insoluble, solid material where C60 is well dispersed over the entire superstructure. We exploited this new material as a catalyst in the reduction reaction of azo compounds (mediated by NaBH4 and UV-light). Our material proved to perform remarkably better than C60 alone in this reaction. Moreover, we extended its usefulness to a novel application, namely the reduction of benzoic acid to benzyl alcohol.Finally, it is worth recalling that most of the linear peptides reported to form nanotubes are characterized by the occurrence of a sixfold screw axis as a symmetry element in their crystal structures.18,19,23–26 This property is also shared by our terminally protected dipeptide Boc-L-Cys(Me)-L-Leu-OMe (1) that crystallizes in the hexagonal space group P65. However, at variance with 1, its positional isomer Boc-L-Leu-L-Cys(Me)-OMe (2, see ESI†), that crystallizes in the orthorhombic space group P212121, in our hands was unable to generate any superstructure, thus suggesting a strong connection between packing mode and self-assembly behavior. Interestingly, quite recently a strictly related observation was published, namely that sequence exchange in an unprotected dipeptide (Val-Ala vs. Ala-Val) strongly affects its molecular structure and the related self-assembly tendency.42
Acknowledgements
The authors thank Dr F. Bertasi for Raman spectra technical support. Financial support from the University of Padova (PRAT C91 J11003560001) is gratefully acknowledged.References
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- J. M. Lehn, Science, 1993, 260, 1762–1763 CAS.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- A. Rui, A. Mendes and L. Gales, J. Mater. Chem., 2012, 22, 1709–1723 RSC.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651–656 CrossRef CAS.
- X. Y. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037–2050 CrossRef CAS.
- Z. Luo and S. Zhang, Chem. Soc. Rev., 2012, 41, 4736–4754 RSC.
- C. Valéry, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583–9594 RSC.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- M. Mba, A. Moretto, L. Armelao, M. Crisma, C. Toniolo and M. Maggini, Chem. – Eur. J., 2011, 17, 2044–2047 CrossRef CAS PubMed.
- M. Mba, A. I. Jiménez and A. Moretto, Chem. – Eur. J., 2014, 20, 3888–3893 CrossRef CAS PubMed.
- C. H. Görbitz, Chem. Commun., 2006, 2332–2334 RSC.
- C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022–1031 CrossRef PubMed.
- A. K. Das, D. Haldar, R. P. Hedge, N. Shamala and A. Banerjee, Chem. Commun., 2005, 1836–1838 RSC.
- P. P. Bose, A. K. Das, R. P. Hedge, N. Shamala and A. Banerjee, Chem. Mater., 2007, 19, 6150–6157 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- S. Ray, D. Haldar, M. G. B. Drew and A. Banerjee, Org. Lett., 2004, 6, 4463–4465 CrossRef CAS PubMed.
- M. Crisma, C. Toniolo, S. Royo, A. I. Jiménez and C. Cativiela, Org. Lett., 2006, 8, 6091–6094 CrossRef CAS PubMed.
- U. S. Raghavender, Kantharaju, S. Aravinda, N. Shamala and P. Balaram, J. Am. Chem. Soc., 2010, 132, 1075–1086 CrossRef CAS PubMed.
- U. S. Raghavender, B. Chatterjee, I. Saha, A. Rajagopal, N. Shamala and P. Balaram, J. Phys. Chem. B, 2011, 115, 9236–9243 CrossRef CAS PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Altomare, G. Campi, C. Cuocci, L. Eriksson, C. Giacovazzo, A. Moliterni, R. Rizzi and P.-E. Werner, J. Appl. Crystallogr., 2009, 42, 768–775 CAS.
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231–1235 CrossRef CAS.
- Crystal data for Boc-L-Cys(Me)-L-Leu-OMe: C16H30N2O5S; M = 362.48, hexagonal, a = b = 11.4458(2) Å, c = 27.7551(5) Å, V = 3148.95(10) Å3, T = 293(2) K, space group P65, Z = 6, 18290 reflections measured, 2281 independent reflections (Rint = 0.044), R1 = 0.0403 [F ≥ 4σ(F)], wR2 = 0.1083 (F2 all data), goodness-of-fit on F2 = 1.042. Flack parameter −0.01(3). CCDC 1057628.
- A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 571–577 RSC.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- Y. Ying, S. S. Chang, C. L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef.
- Z. Wang, Z. Dai, J. Wu, N. Zhao and J. Xu, Adv. Mater., 2013, 25, 4494–4497 CrossRef CAS PubMed.
- D. R. Dreyer and C. W. Bielawski, Chem. Sci., 2011, 2, 1233–1240 RSC.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- Y. Guo, W. Li, J. Yan, B. Moosa, M. Amad, C. J. Werth and N. M. Khashab, Chem. – Asian J., 2012, 7, 2842–2847 CrossRef CAS PubMed.
- L. Kong, X. Lu, X. Bian, W. Zhang and C. Wang, ACS Appl. Mater. Interfaces, 2011, 3, 35–42 CAS.
- N. Gupta, H. P. Singh and R. K. Sharma, J. Mol. Catal. A: Chem., 2011, 335, 248–252 CrossRef CAS.
- H. Erdogan, E. Babur, M. Yilmaz, E. Candas, M. Gordesel, Y. Dede, E. E. Oren, G. B. Demirel, M. K. Ozturk, M. S. Yavuz and G. Demirel, Langmuir, 2015, 31, 7337–7345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, X-ray crystal structural analysis CCDC 1057628 (1) and 1057629 (2) and catalysis results. See DOI: 10.1039/c5sm02189h |
| This journal is © The Royal Society of Chemistry 2016 |