DOI:
10.1039/C5SM02111A
(Paper)
Soft Matter, 2016,
12, 141-148
A supramolecular gel based on a glycosylated amino acid derivative with the properties of gel to crystal transition†
Received
24th August 2015
, Accepted 25th September 2015
First published on 25th September 2015
Abstract
Here we report the generation of a novel gelator from a glycosylated amino acid derivative, which contained three structural units, an aromatic residue, a carbohydrate moiety and a tert-butyl group in a single molecule. These structural units can promote the supramolecular self-assembly of this gelator in both aprotic and protic solvents via coordinated π–π stacking, multiple hydrogen binding and van der Waals interactions. More importantly, due to their non-equilibrium natures, the organogels formed in DCM, chloroform and ethanol can undergo gel to crystal transition in storage, driven by unbalanced gelator–gelator and solvent–gelator interactions. In this process, the gelators were firstly trapped in a kinetically favorable gel state, and then transferred into a more thermodynamically stable crystal state upon ageing, with the generation of microcrystals in different morphologies.
Introduction
A low-molecular-weight gelator (LMWG) is an interesting new class of small molecules that can form reversible supramolecular gels in solvents via non-covalent hydrogen bonding, van der Waals interactions and π–π stacking.1,2 With the development of advanced soft nanomaterials over the past few years, there has been growing interest in the design and synthesis of LMWGs from a wide spectrum of organic molecules,3–6 such as cholesterols,7 carbamates,8 carbazoles,9,10 amino acids11–13 and carbohydrate derivatives14,15 for the application in chemical sensors, drug delivery, enzyme immobilization and solar cells.16–18
In spite of the large library of LMWGs, there is still a demand for the development of novel gelators from different origins. Among them, amino acids and carbohydrates attracted particular interests for the preparation of LMWGs with various molecular structures and self-assembling properties, owing to their high biocompatibility, versatile molecular structure, and intrinsic chirality and capability for supramolecular interactions. For example, a number of peptide-based LMWGs have been prepared from linear or branched peptide molecules with adjustable self-assembling properties in different organic solvents,19–24 and a series of sugar-based gelators containing various sugars and hydrophobic groups were made with special self-assembling behaviours in both protic and aprotic solvents.25–29
However, the study for the development of novel organo/hydrogelators from glycosylated amino acid is still underexplored. Recent studies have demonstrated that the integration of an amino acid/peptide scaffold containing appropriate hydrophobic groups and saccharides with inherent molecular chirality and multiple hydrogen binding properties holds great promise for the generation of new LMWGs and supramolecular gels with improved structural diversity and functionality.30 Previously, Xu and co-workers developed biocompatible hydrogelators from peptide-monosaccharide conjugates, which showed improved wound healing effects on mice,31 Hamachi et al. displayed the preparation of ambidextrous gelators from glycosylated amino acids, which can self-assemble efficiently in both water and organic solvents,32 and we also demonstrated that the conjugation of an amino-sugar with an amino acid/peptide derivative can generate a novel hydrogelator with multi-functionalities.33
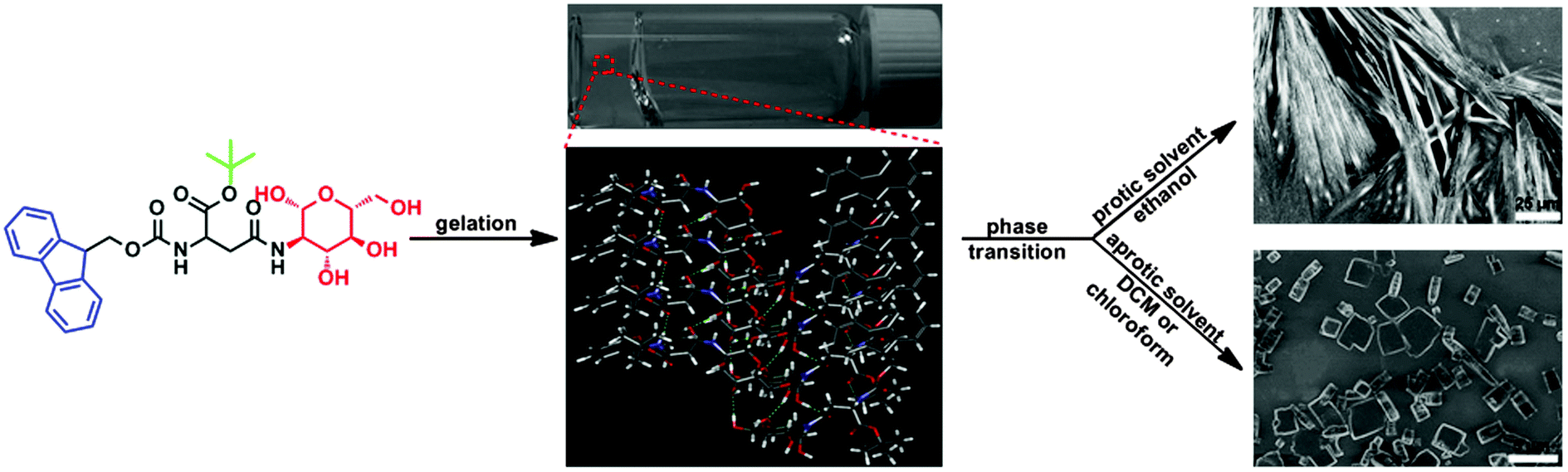
Encouraged by the successful work described above, we sought to design and synthesize a new gelator based on a glycosylated amino acid derivative, and examined its potential for supramolecular self-assembly in different solvents. As shown in Scheme 1, this molecule possessed three basic structural units, including an aromatic moiety (i.e., 9-fluorenylmethoxycarbonyl group), a carbohydrate head-residue (i.e., D-glucosamine) linked to the side chain of Fmoc-Asp-OH and a tert-butyl (Boc) group at the C terminal of this glycosylated amino acid derivative. These structural units can promote supramolecular self-assembly of this molecule in different solvents via π–π stacking, multiple hydrogen binding and van der Waals interactions.
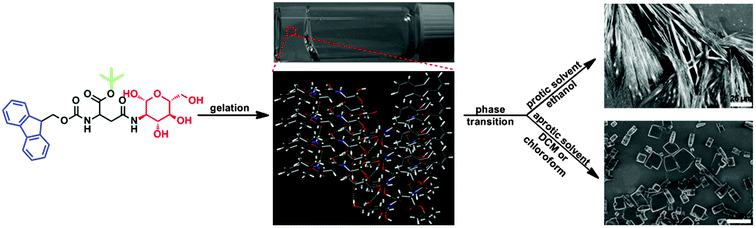 |
| | Scheme 1 Illustration of the self-assembly of Fmoc-Asp(Glc)-OtBu into 1D microfibers and 3D microcubes in both protic and aprotic solvents. | |
During our research, we found that this molecule exhibited high selectivity towards the polarity of solvents for supramolecular self-assembly, and formed stable organogels in dichloromethane (DCM), chloroform, ethanol, 2-propanol and n-butanol, due to balanced gelator–gelator and solvent–gelator interactions.34,35 Moreover, the self-assembled organogels formed in DCM, chloroform and ethanol, respectively, can also undergo gel to crystal transformation in storage, because of the non-equilibrium natures of these self-assembled gels. Recent studies indicated that gelation and crystallization were orthogonal and time-resolved processes under non-equilibrium conditions,36,37 in which crystallization occurred over a period of hours to days, due to the presence of an energetically unfavourable nucleation step, however gelation often took place at a shorter time interval.38,39 Therefore during the supramolecular self-assembly process, the gelators in suitable solvents were firstly trapped in a kinetically favorable gel state, and then transferred into a more thermodynamically stable crystal state upon ageing.40–43 Since the supersaturated condition was the common feature of driving force for supramolecular gelation and crystallization, a study of the gelation system with phase transition properties would be advantageous to gain insight into the relationship between supramolecular gelation and crystallization, and helpful for the design of new gelators with controlled self-assembling properties.44,45
Experimental section
Materials and methods
Fmoc-Asp-OtBu was provided by GL Biochem (Shanghai) Ltd. D-Glucosamine was purchased from Sigma-Aldrich. All the other compounds were obtained from J&K Chemicals and used without further purification. 1H NMR spectra were obtained on a Unity Inova 400 by using DMSO-d6 as a solvent. The CD spectroscopy study was carried out on a JASCO J-810 spectrometer. LC-MS analysis was performed on an Agilent 6120 Quadrupole LC/MS system with an ESI resource. Transmission electron micrograph (TEM) and scanning electron micrograph (SEM) images were recorded on a Tecnai G220 transmission electron microscope and a Hitachi S-4800, respectively. The optical microscopy image was taken using an Olympus IX71 optical microscope. Fourier transform infrared spectroscopy (FTIR) characterization was performed on a Perkin-Elmer spectrophotometer. The rheological study carried out on a Thermo Scientific HAAKE RheoStress 6000 rheometer.
Synthesis of Fmoc-Asp(Glc)-OtBu
Fmoc-Asp-OtBu (4.11 g, 10 mmol), NHS (1.51 g, 12 mmol) and DIC (1.38 g, 12 mmol) were dissolved in 80 mL of DMF, and reacted at room temperature for 8 h. Then a solution of D-glucosamine (2.15 g, 12 mmol) in the mixed solvent of water and DMF was added, and the resulted solution was stirred at room temperature for another 24 h. After evaporation of solvent, the residues were re-dissolved in water and acidified with HCl to pH 2–3. The white precipitates were collected and purified by column chromatography over the silica gel using chloroform/methanol as the eluent to afford the final product (yield: 78%). 1H NMR (400 MHz, DMSO-d6): δ 7.96–7.89 (m, 4H), 7.72–7.71 (d, 3H), 7.43–7.23 (m, 5H), 6.43–6.23 (d, 1H), 4.93–4.90 (t, 2H) 4.59–4.24 (m, 6H), 3.63–3.50 (m, 5H), 3.14–3.07 (d, 1H), 2.60–2.57 (s, 2H), 1.38 (s, 9H); MS: calcd M = 572.50, obsd (M + H)+ = 573.2.
TEM and SEM measurements
The gel was placed on a single-crystal silica plate or a carbon-coated Cu grid and dried under vacuum. The sample for the TEM study was stained with uranyl acetate (2.0% (w/v)). The silica plate loaded with gel was further coated with a thin layer of platinum before SEM measurements. Then TEM and SEM images were recorded using a Tecnai G220 transmission electron microscope and a Hitachi S-4800, respectively.
Rheological measurements
The rheological test was carried out on a Thermo Scientific HAAKE RheoStress 6000 rheometer with a 20 mm parallel plate, and 0.2 mL of each organogel was used for this test. The dynamic strain sweep test was run from 0.1 to 10% with a frequency of 6.282 rad s−1 at 25 °C. The dynamic frequency sweep test was run from 200 rad s−1 to 0.1 rad s−1, and a strain of 0.4% was used to ensure the linearity of dynamic viscoelasticity.
FTIR analysis
FTIR spectra of the organogel and microcrystals in DCM were collected on a Perkin-Elmer spectrophotometer by loading the samples into a KBr cuvette. The sample was prepared by using deuterium DCM as a solvent.
Wide angle X-ray diffraction
The X-ray diffraction patterns were obtained using a Shimadzu XRD-6000 diffractometer with a Ni filter and CuKα (λ = 1.54056 Å, voltage = 40 kV, and current = 40 mA). Each sample was scanned over the range from 5° to 30° with a step size of 0.026° and a count time of 5 seconds per step.
Molecular modelling
The ground-state geometry of the gelator was optimized using the Hartree–Fock Austin Model 1 (AM1) method. And the geometric optimization of four-molecule clusters of the gelator and its packing model were performed using the DREIDING force field as implemented in the molecular modelling programs (Accelrys Inc., San Diego, CA, USA).
Results and discussion
Design and synthesis
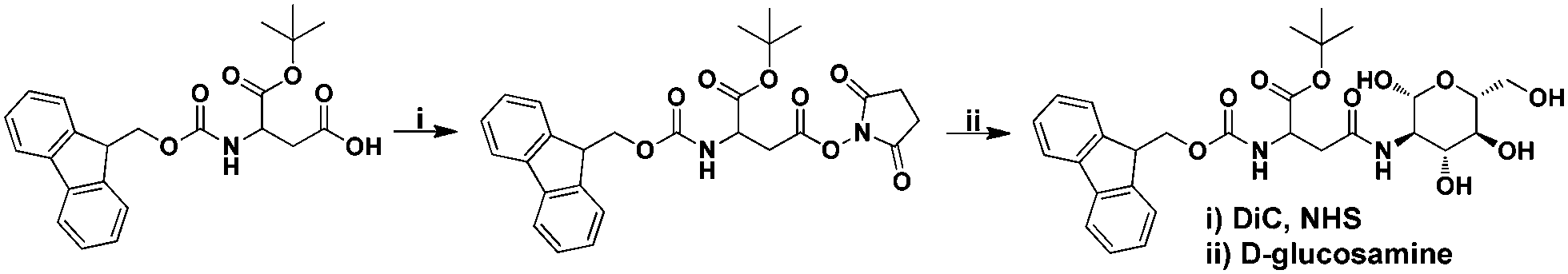
The formation of Fmoc-Asp(Glc)-OtBu was achieved by treating a solution of Fmoc-Asp-OtBu in N, N′-dimethylformamide (DMF) with N-hydroxysuccinimide (NHS) and N, N′-diisopropylcarbodiimide (DIC), followed by the dropwise addition of an aqueous solution of D-glucosamine (Scheme 2). In this process, the carboxylic acid group on the side chain of Fmoc-Asp-OtBu was firstly activated by NHS in the presence of DIC, and then coupled with the amine group of D-glucosamine to afford the final product. After the removal of reaction solvent, the product was purified using a flash column, and then characterized by NMR and MS, both of them gave satisfactory analysis data corresponding to its expected molecular structure (Fig. S1 and S2, ESI†). Overall, the integration of a partially protected amino acid and an amino sugar was proved to be an efficient method for the preparation of a glycoamino acid with regiospecific β-N-linkage.
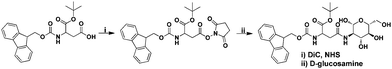 |
| | Scheme 2 Synthetic route for the preparation of Fmoc-Asp(Glc)-OtBu. | |
Gelation test
After obtaining this compound, we tested its gelation properties in various solvents such as, hexane, toluene, xylene, chlorobenzene, dichlorobenzene, ether, ethyl acetate, isobutanol, n-butanol, 2-propanol, ethanol, DCM, chloroform, methanol, water and DMF, ranging from protic to aprotic solvent and polar to apolar solvent. Typically, an appropriate amount of this compound was added to a sealed test tube containing one kind of organic solvent and heated gently until the sample was dissolved completely. After allowing the solution to cool to lower temperature (e.g., 25, 4, or −20 °C) for different periods of time, we examined the state of the sample by turning the tube upside down. If there was no gravitational flow, the sample was termed as a gel, and classified as G in Table 1. By following this procedure, we examined the gelation properties of Fmoc-Asp(Glc)-OtBu in different solvents, and the results are summarized in Table 1. The compound can gel efficiently in DCM, chloroform, chlorobenzene, m-dichlorobenzene and o-dichlorobenzene at room temperature by forming translucent organogels (Fig. 1A, B and Fig. S3, ESI†), while it afforded opaque gels in ethanol, 2-propanol and n-butanol, respectively, at much lower temperatures of around 4 °C or −20 °C in 24 h (Fig. 1C–E). Their critical gelation concentrations were 1.5, 1.5, 4.5, 4.5, 5.0, 5.5, 4.6 and 3.5 wt%, individually. In addition, it also exhibited the tendency to form opaque gels in the mixed solvents of water and ethanol, and the minimum gelation concentrations were highly dependent on the composition of mixed solvents (Table 1 and Fig. S3, ESI†). However, this compound was insoluble in hexane, ether, toluene, xylene, ethyl acetate and water even with gentle heating (Fig. S3 and S7, ESI†), due to the polarity of these solvents, but dissolved very well and formed homogenous solution in isobutanol, methanol and DMF. This study indicated that Fmoc-Asp(Glc)-OtBu can work as an effective gelator to form supramolecular gels in both protic and aprotic solvents, and its gelation properties were dependent on the polarity of solvent.
 |
| | Fig. 1 Optical images of the organogels formed in different solvents. (A) DCM, 1.5 wt%, 25 °C; (B) chloroform, 1.5 wt%, 25 °C; (C) ethanol, 5.5 wt%, 4 °C; (D) n-butanol, 3.5 wt%, −20 °C; (E) 2-propanol, 4.6 wt%, −20 °C. | |
Table 1 The gelation ability of Fmoc-Asp(Glc)-OtBu in various organic solvents
| Solvent |
Statea |
CGC wt% |
GTb at CGC (°C) |
|
G = gel, S = soluble, and I = insoluble.
GT = gelation temperature.
|
| DCM |
G |
1.5 |
25 |
| Chloroform |
G |
1.5 |
25 |
| Ethanol |
G |
5.5 |
4 |
| 2-Propanol |
G |
4.6 |
−20 |
|
n-Butanol |
G |
3.5 |
−20 |
| Chlorobenzene |
G |
4.5 |
25 |
|
o-Dichlorobenzene |
G |
4.5 |
25 |
|
m-Dichlorobenzene |
G |
5 |
25 |
H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (75:25) :ethanol (75:25) |
G |
2.5 |
25 |
| H2O:ethanol (60:40) |
G |
2.7 |
25 |
| H2O:ethanol (40:60) |
G |
3.7 |
25 |
| H2O:ethanol (25:75) |
G |
4.7 |
25 |
| Isobutanol |
S |
— |
— |
| Methanol |
S |
— |
— |
| DMF |
S |
— |
— |
| Hexane |
I |
— |
— |
| Toluene |
I |
— |
— |
| Xylene |
I |
— |
— |
| Ether |
I |
— |
— |
| Ethyl acetate |
I |
— |
— |
| H2O |
I |
— |
— |
Morphology
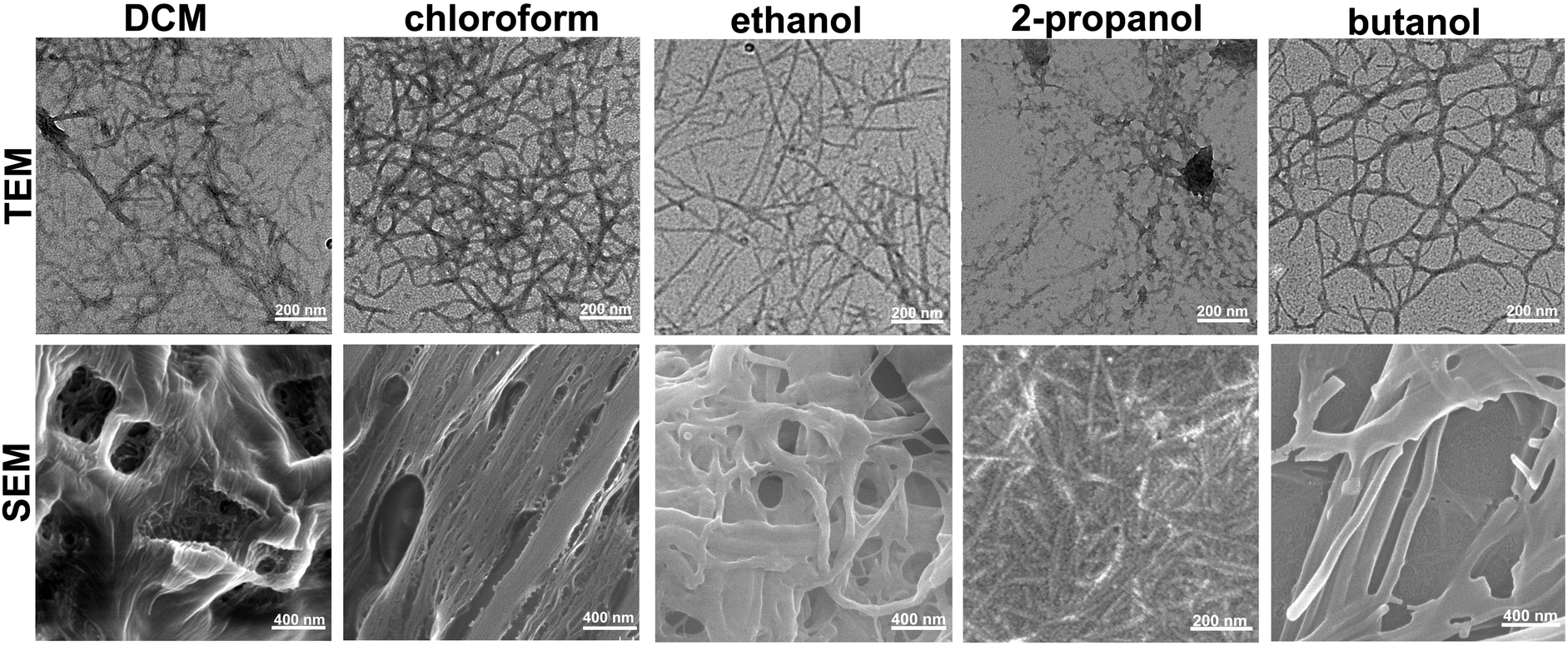
In order to obtain further insight into the solvent effect on the self-assembling properties of this gelator, we used transmission electron microscopy (TEM) and scanning electron microscopy (SEM) to investigate the nano- and micro-scale structures of these gels formed in different solvents. The TEM images shown in Fig. 2 revealed that the organogels in DCM and chloroform consisted of individual nanofibers which further aggregated together to form thick fiber bundles with diameters of 20 nm and 22 nm, respectively. However, the gel in ethanol displayed the formation of well-defined nanofibrous structures tens of micrometers in length and 18 nm in width. And we also identified the formation of nanofibrous-like structures in the organogels of 2-propanol, n-butanol, chlorobenzene, m-dichlorobenzene, o-dichlorobenzene and mixed solvents of water and ethanol (Fig. S4–S6 and S8–S11, ESI†). In addition, SEM analysis confirmed the morphological features of fibrous structures self-assembled from Fmoc-Asp(Glc)-OtBu, which physically cross-linked each other to form entangled bundles and immobilize organic solvents for the formation of organogels. However, structural differences among the gels in different solvents were obviously observed at the micro-scale. As evidenced by SEM images shown in Fig. 2, the gel in DCM revealed the formation of entangled fibres and bundles which worked as the main microstructures to support the formation of organogels. And the gel system in chloroform and ethanol showed predominantly interwoven fibrous structures which aggregated together to form the gel network. In addition, we can confirm the formation of microstructure networks in the organogels of 2-propanol and n-butanol, which supported the formation of organogels. This study demonstrated that solvent polarity played an important role in regulating the self-assembling behaviour of the gelator to form nanostructures in different morphologies.
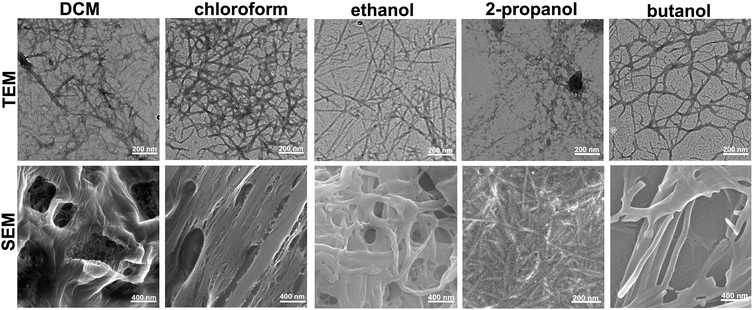 |
| | Fig. 2 TEM and SEM images of the organogels formed in DCM, chloroform, ethanol, 2-propanol and n-butanol shown in Fig. 1. | |
Rheological studies
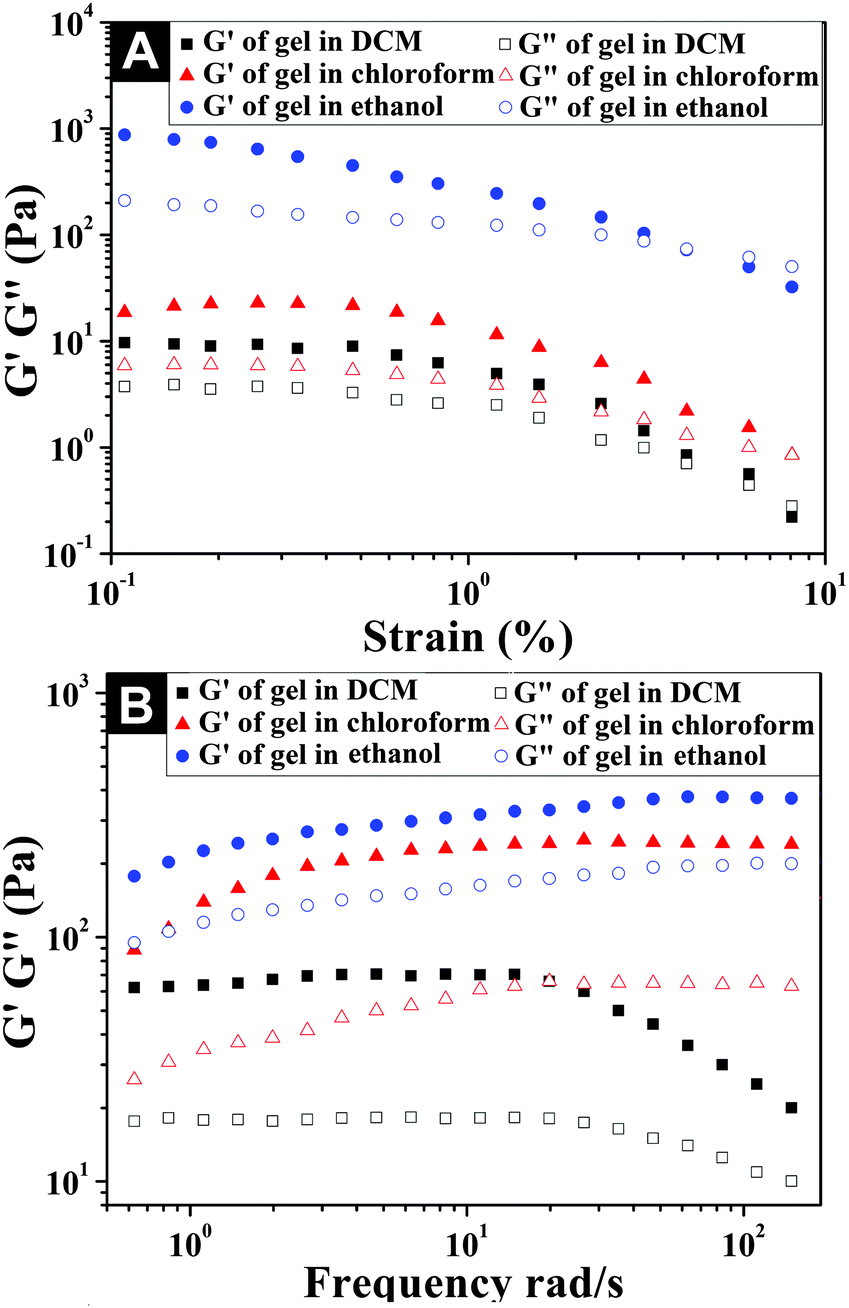
With the application of oscillatory rheology, we investigated the viscoelastic properties of organogels in different solvents. The viscoelastic behavior of a gel was determined by two key parameters, such as storage modulus (G′) and loss modulus (G′′), which represented the ability of a deformed gel to return to its original geometry and the tendency of a material to flow under exerted stress. For a gel material, G′ is greater than G′′, and G′ shows low dependence on oscillatory frequency, signifying the dominant elastic behavior of the gel. From the strain and frequency sweep profiles of the organogels in different solvents shown in Fig. 3, we found that the storage moduli (G′) of the organogels in chloroform and ethanol were much higher than their loss moduli (G′′) within the investigated oscillating strain (0.1–10%) and frequency limit (0.1–200 rad s−1), confirming the formation of stable organogels and their dominant elastic properties (Fig. S4–S6 and S8–S11, ESI†). However, the organogel formed in DCM was mechanically weaker than that formed in chloroform and ethanol, as exemplified by the low G′ value (61 Pa) and the high dependence of the G′ value on the oscillatory frequency from 20–200 rad s−1. Its abrupt decrease of storage moduli (G′) within 20–200 rad s−1 was due to the fact that the mechanical strength of the gel matrix in DCM was not high enough to resist the exerted shear stress, with the tendency to undergo gel–sol transition under high oscillatory frequency.46 The relatively high mechanical strengths of organogels in chloroform and ethanol could be due to the formation of compact microstructure networks observed in Fig. 2.
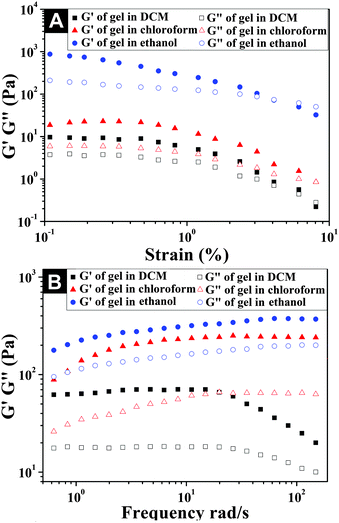 |
| | Fig. 3 Strain (A) and frequency (B) dependence of the dynamic storage moduli (G′) and the loss moduli (G′′) of the organogels formed in DCM, chloroform, and ethanol, respectively, which are shown in Fig. 1. | |
Phase transition
In addition, we also observed that the organogels in DCM, chloroform and ethanol were metastable, because they exhibited a tendency to transfer into microscopic crystals during storage. Typical gelation and phase transition processes of Fmoc-Asp(Glc)-OtBu in these solvents are shown in Fig. 4. In the system of DCM and chloroform, when the hot transparent solutions were cooled at room temperature, white precipitates firstly appeared at the top of the solutions in 5 min, which further aggregated together until the lower solution changed into a semi-transparent gel in 10 min, and the millimetre-sized aggregates were trapped inside the gels. From the SEM images shown in Fig. S12 and S14 (ESI†), we confirmed that these firstly formed precipitates belonged to irregular aggregates, not crystals. Afterwards, the surrounding part of the gels changed gradually from semi-transparent to translucent in 24 h. From microscopy and SEM images in Fig. 4 and Fig. S13 and S15 (ESI†), we confirmed the generation of a significant amount of micrometre-sized crystals within gels through gel to crystal transition, which should be responsible for the cloudiness of the gels. However, for the system in ethanol, when the solution was cooled at 4 °C, it gradually turned into suspension with the generation of precipitates deposited at the bottom of the tube (Fig. S16, ESI†). Later, the surrounding solution transformed to a translucent gel and an opaque gel gradually in 48 h, with the generation of microcrystalline fibres inside the gel (Fig. S17, ESI†). These results demonstrated that there existed a phase transition from the gel to the crystal after the formation of gels in DCM, chloroform and ethanol, and the morphologies of microcrystals formed in this process were dependent on the polarity of the solvents.
 |
| | Fig. 4 Photo- and microscopic images of the process for gel to crystal transition in DCM (1.5 wt%), chloroform (1.5 wt%) and ethanol (5.5 wt%), respectively. | |
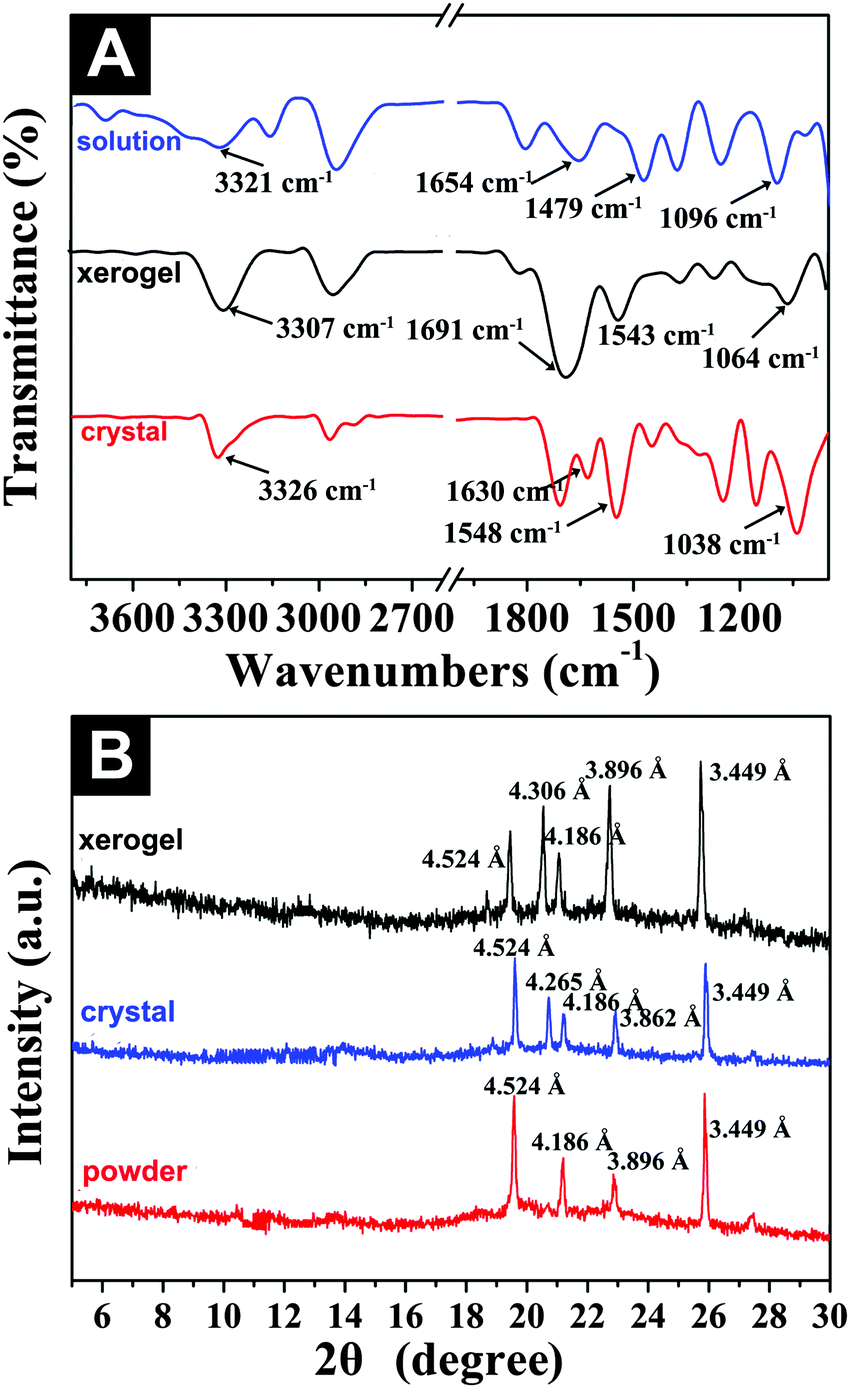
FTIR and XRD studies
With the purpose of elucidating the intermolecular interactions and molecular packing modes of the gelators in the gel and microcrystals, Fourier transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD) experiments were carried out by using the organogel formed in DCM as an example. Fig. 5A shows the FTIR spectra of Fmoc-Asp(Glc)-OtBu at different states (e.g., solution, xerogel and crystal). It was obvious that the spectrum of the xerogel was very similar to that of the crystal in DCM, indicating that the two forms existed in the same molecular interaction patterns. As shown in Fig. 5A, the three main bands at 3328, 1651, and 1536 cm−1 in DCM solution corresponded to the non-hydrogen-bonded NH band and the amide I and II bands frequencies,47 which shifted slightly to 3307, 1691, and 1543 cm−1 for the xerogel, and 3326, 1704, and 1548 cm−1 for the crystal, respectively, which indicated the presence of hydrogen bonding interactions between the carbonyl group and the amide NH group in the both gel and crystal samples. In addition, the stretching vibration of the glycosyl ring appeared at 1095 cm−1 in solution and shifted to 1064 cm−1 and 1038 cm−1, respectively, after the formation of the gel and the crystal, which implied that the hydroxyl groups also played an important role in the supramolecular self-assembly of the gelator via hydrogen-bonding interactions.48 A transmittance band at 2967 cm−1 was ascribed to the CH3 vibration of Fmoc-Asp(Glc)-OtBu in DCM solution, and shifted to 2953 and 2964 cm−1 after gelation and crystallization, due to the decreased vibration and enhanced organization of the tert-butyl groups via van der Waals interaction.49 Therefore, the observed spectroscopic data suggested the involvement of both hydrogen bonding and hydrophobic interaction for the formation of ordered structures.
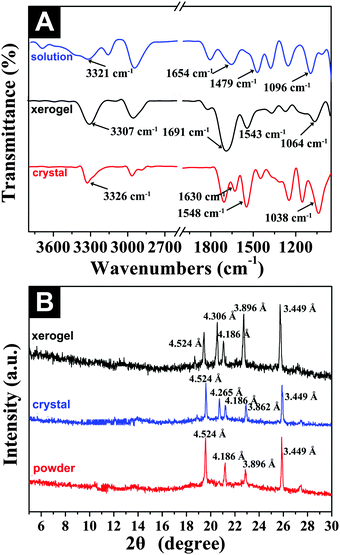 |
| | Fig. 5 (A) FTIR and (B) XRD analysis of the xerogel and microcrystals in DCM (1.5 wt%). | |
The molecular packing modes in different states (e.g., xerogel, crystal and powder) were further analyzed by XRD. The XRD pattern of the gel was almost identical to the result of the crystal, but different from that of the powder, confirming the similar molecular packing mode of the gelator in both gel and crystal states. As shown in Fig. 5B, the XRD traces of xerogel and crystal showed main peaks in the angle region (2θ values, 19.5°, 20.6°, 21.1°, 22.7°, and 25.7°), which corresponded to d values of 4.524 Å, 4.306 Å, 4.186 Å, 3.896 Å, and 3.449 Å, respectively.
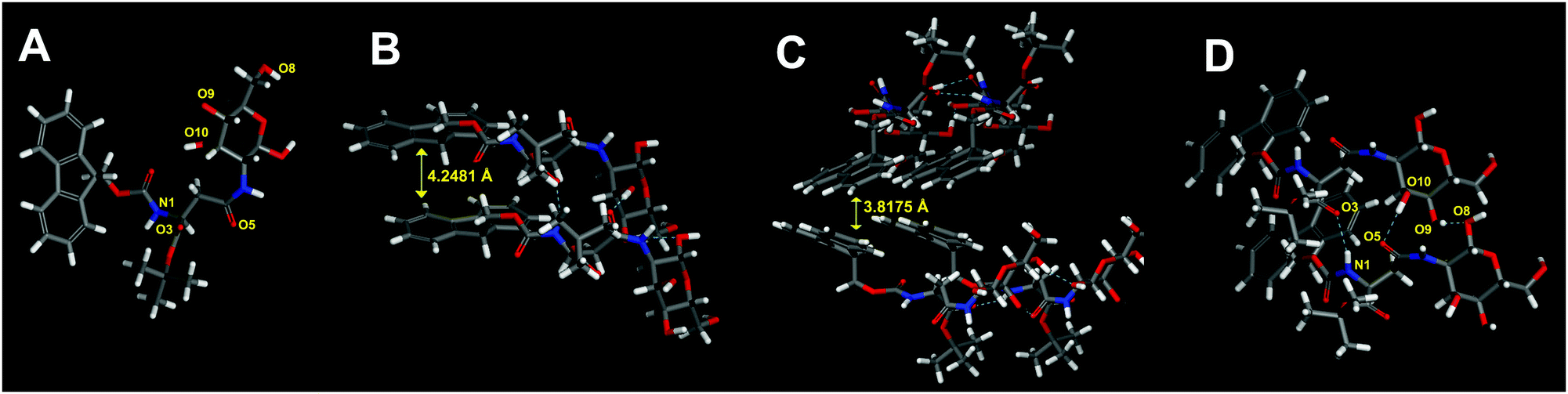
Molecular modeling
With reference to the molecular structure models illustrated in Fig. 6, the peak with a d spacing of 4.306 Å in XRD was assigned to a vertical spacing of two stacked aromatic rings for π–π stacking interaction in the gel state, and the distance was decreased to 4.265 Å after gel to crystal transition, while another peak at 22.9° with a d spacing value of 3.896 Å in the gel corresponded to a separation distance between two planar aromatic groups for edge-to-face interaction, and the distance was shortened to 3.86 Å in the crystal, indicating the occurrence of glide movement of the molecule during gel to crystal transition which was resulted from solvent and gelator interaction.50 These data indicated that aromatic–aromatic interactions worked as a significant driving force for supramolecular gelation and subsequent phase transition. The structural model was also characterized by the presence of intermolecular hydrogen bonds between the NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (N1H⋯O3, 2.17 Å), and between the hydroxyl groups of glucose residues (O10H⋯O5, 1.95 Å; and O9H⋯O8, 1.98 Å). On the basis of FTIR and XRD analysis, the organogel of Fmoc-Asp(Glc)-OtBu was found to be stabilized by a combination of π–π stacking and hydrogen bonding interactions.
O groups (N1H⋯O3, 2.17 Å), and between the hydroxyl groups of glucose residues (O10H⋯O5, 1.95 Å; and O9H⋯O8, 1.98 Å). On the basis of FTIR and XRD analysis, the organogel of Fmoc-Asp(Glc)-OtBu was found to be stabilized by a combination of π–π stacking and hydrogen bonding interactions.
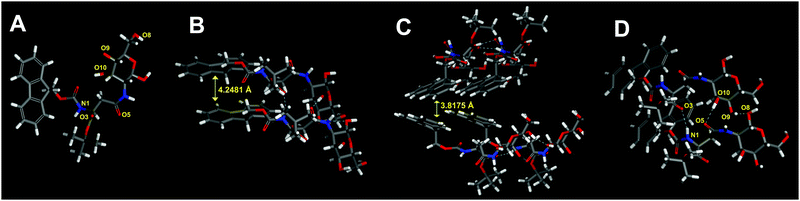 |
| | Fig. 6 (A) The molecular conformation of Fmoc-Asp(Glc)-OtBu with atom labelling for indicating the presence of hydrogen bonding interactions on these atoms. Optimized structures of molecular models of Fmoc-Asp(Glc)-OtBu showing the presence of (B) π–π stacking interaction with distance at 4.2481 Å, (C) edge-to-face interaction between two planar aromatic groups with distance at 3.8175 Å, and (D) the extended intermolecular hydrogen bond between glucose and amino acid (C: gray, H: white, N: blue, and O: red). The molecular models for the supramolecular interaction of Fmoc-Asp(Glc)-OtBu were obtained from molecular mechanical (MM) calculations. | |
Conclusions
In summary, we have designed and synthesized a new gelator from a glycosylated amino acid derivative, which contained three structural units, an aromatic group, a carbohydrate moiety and alkyl residues in a single molecule. These structural units can promote the supramolecular self-assembly of this gelator in both aprotic and protic solvents via π–π stacking, multiple hydrogen binding and van der Waals interactions. More importantly, for the systems in DCM, chloroform and ethanol, the organogels can undergo phase transition from the gel to the crystal, induced by coordinated solvent and gelator interaction.50 Although further studies are still required to elucidate the process of gel–crystal transition, we hope that this study introduces a new gelation system with phase transition properties, which would be helpful for investigating the relationship of supramolecular gelation and crystallization during the supramolecular self-assembly process of gelators.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (21344002 and 51402203), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Natural Science Foundation of Jiangsu Province (BK20151218 and BK20140326) to X. Li and Y. Pan, respectively; the National Science Council of the Republic of China (NSC 102-2113-M-009-006-MY2) and the “Aim for the Top University” program of the National Chiao Tung University and Ministry of Education, Taiwan, R.O.C. to H.-C. Lin; the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Key Lab of Health Chemistry and Molecular Diagnosis of Suzhou.
Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- Y.-S. Su, J.-W. Liu, Y. Jiang and C.-F. Chen, Chem. – Eur. J., 2011, 17, 2435–2441 CrossRef CAS PubMed.
- J.-W. Liu, J.-T. Ma and C.-F. Chen, Tetrahedron, 2011, 67, 85–91 CrossRef CAS.
- Y. Yan, Y. Lin, Y. Qiao and J. Huang, Soft Matter, 2011, 7, 6385–6398 RSC.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- A. R. Hirst and D. K. Smith, Chem. – Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed.
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC.
- K. Yabuuchi, Y. Tochigi, N. Mizoshita, K. Hanabusa and T. Kato, Tetrahedron, 2007, 63, 7358–7365 CrossRef CAS.
- J. T. van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Langmuir, 2013, 29, 8763–8767 CrossRef CAS PubMed.
- D. Das, A. Dasgupta, S. Roy, R. N. Mitra, S. Debnath and P. K. Das, Chem. – Eur. J., 2006, 12, 5068–5074 CrossRef CAS PubMed.
- A. Banerjee, G. Palui and A. Banerjee, Soft Matter, 2008, 4, 1430–1437 RSC.
- S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658–11664 RSC.
- O. Gronwald and S. Shinkai, Chem. – Eur. J., 2001, 7, 4328–4334 CrossRef CAS.
- J. H. Jung, G. John, K. Yoshida and T. Shimizu, J. Am. Chem. Soc., 2002, 124, 10674–10675 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425–3433 RSC.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- P. Duan, H. Cao, L. Zhang and M. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- S.-M. Hsu, Y.-C. Lin, J.-W. Chang, Y.-H. Liu and H.-C. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921–1927 CrossRef CAS PubMed.
- G. John, G. Zhu, J. Li and J. S. Dordick, Angew. Chem., Int. Ed., 2006, 45, 4772–4775 CrossRef CAS PubMed.
- G. Wang, S. Cheuk, H. Yang, N. Goyal, P. V. N. Reddy and B. Hopkinson, Langmuir, 2009, 25, 8696–8705 CrossRef CAS PubMed.
- Q. Chen, Y. Lv, D. Zhang, G. Zhang, C. Liu and D. Zhu, Langmuir, 2010, 26, 3165–3168 CrossRef CAS PubMed.
- N. Yan, G. He, H. Zhang, L. Ding and Y. Fang, Langmuir, 2010, 26, 5909–5917 CrossRef CAS PubMed.
- W. Wang, H. Wang, C. Ren, J. Wang, M. Tan, J. Shen, Z. Yang, P. G. Wang and L. Wang, Carbohydr. Res., 2011, 346, 1013–1017 CrossRef CAS PubMed.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- Z. Yang, G. Liang, M. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845 RSC.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS PubMed.
- X. Li, Y. Kuang and B. Xu, Soft Matter, 2012, 8, 2801–2806 RSC.
- G. Zhu and J. S. Dordick, Chem. Mater., 2006, 18, 5988–5995 CrossRef CAS.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154–5156 RSC.
- D. Braga, S. d'Agostino, E. D'Amen, F. Grepioni, D. Genovese, L. Prodi and M. Sgarzi, Dalton Trans., 2013, 42, 16949–16960 RSC.
- V. J. Anderson and H. N. W. Lekkerkerker, Nature, 2002, 416, 811–815 CrossRef CAS PubMed.
- N. M. Dixit and C. F. Zukoski, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67 Search PubMed.
- O. Lebel, M.-E. Perron, T. Maris, S. F. Zalzal, A. Nanci and J. D. Wuest, Chem. Mater., 2006, 18, 3616–3626 CrossRef CAS.
- P. Terech, N. M. Sangeetha and U. Maitra, J. Phys. Chem. B, 2006, 110, 15224–15233 CrossRef CAS PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem. – Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC.
- Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884–889 CAS.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- Y.-D. Huang, W. Tu, Y.-Q. Yuan and D.-L. Fan, Tetrahedron, 2014, 70, 1274–1282 CrossRef CAS.
- J. Cui, Z. Shen and X. Wan, Langmuir, 2010, 26, 97–103 CrossRef CAS PubMed.
- M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Langmuir, 2003, 19, 8622–8624 CrossRef CAS.
- C. Boettcher, B. Schade and J. H. Fuhrhop, Langmuir, 2001, 17, 873–877 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed reaction conditions and procedures for the synthesis of Fmoc-Asp(Glc)-OtBu and the corresponding NMR and MS spectra. See DOI: 10.1039/c5sm02111a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.