
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors†
Guo-Bo
Li
ab,
Martine I.
Abboud
 a,
Jürgen
Brem
a,
Hidenori
Someya
ac,
Christopher T.
Lohans
a,
Sheng-Yong
Yang
d,
James
Spencer
e,
David W.
Wareham
f,
Michael A.
McDonough
*a and
Christopher J.
Schofield
*a
a,
Jürgen
Brem
a,
Hidenori
Someya
ac,
Christopher T.
Lohans
a,
Sheng-Yong
Yang
d,
James
Spencer
e,
David W.
Wareham
f,
Michael A.
McDonough
*a and
Christopher J.
Schofield
*a
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk; michael.mcdonough@chem.ox.ac.uk
bKey Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu, 610041, China
cMedicinal Chemistry Research Laboratories, New Drug Research Division, Otsuka Pharmaceutical Co., Ltd., 463-10 Kagasuno, Kawauchi-cho, Tokushima 771-0192, Japan
dState Key Laboratory of Biotherapy/Collaborative Innovation Center for Biotherapy, West China Hospital, West China Medical School, Sichuan University, Sichuan 610041, China
eSchool of Cellular and Molecular Medicine, Biomedical Sciences Building, University of Bristol, Bristol BS8 1TD, UK
fAntimicrobial Research Group, Barts & The London School of Medicine and Dentistry, Queen Mary University of London, London, E1 2AT, UK
First published on 14th December 2016
Abstract
There are no clinically useful inhibitors of metallo-β-lactamases (MBLs), which are a growing problem because they hydrolyse almost all β-lactam antibacterials. Inhibition by most reported MBL inhibitors involves zinc ion chelation. A structure-based virtual screening approach combined with NMR filtering led to the identification of inhibitors of the clinically relevant Verona Integron-encoded MBL (VIM)-2. Crystallographic analyses reveal a new mode of MBL inhibition involving binding adjacent to the active site zinc ions, but which does not involve metal chelation. The results will aid efforts to develop new types of clinically useful inhibitors targeting MBLs/MBL-fold metallo-enzymes involved in antibacterial and anticancer drug resistance.
Introduction
One of the most important mechanisms of resistance to β-lactam antibacterials involves their hydrolysis as catalysed by serine-β-lactamases (SBLs) and metallo-β-lactamases (MBLs) (Fig. 1).1,2 Although clinically useful inhibitors of the SBLs are established, there are no such MBL inhibitors.3–7 Inhibitors of human MBL-fold proteins are also of medicinal interest, including for the human DNA cross-link repair enzymes SNM1-A/B, in order to combat resistance to major anti-cancer drugs such as cisplatin.8,9 To date, almost all reported MBL inhibitors work via zinc ion chelation (Fig. S1 and S2†).10–13 Development of new types of MBL-fold enzyme inhibitors that do not work via metal chelation is presently desirable, in part because this may enable improved selectivity than (readily) achievable with zinc ion chelation. Here we report how a virtual screening approach combined with NMR filtering, led to the identification of non-metal chelating inhibitors of the clinically relevant Verona Integron-encoded MBL (VIM)-2. As revealed by crystallographic, NMR, and biochemical studies, the new inhibitors bind via a mode that does not involve direct zinc chelation, but which may mimic interactions made by intact β-lactam substrates as they initially bind to VIM-2. VIM-2 is a clinically important representative of the class B1 MBLs (which also includes the imipenemase (IMP)-1, and New Delhi MBL (NDM)-1), that have a broad-spectrum substrate profile that includes penicillins, cephamycins, cephalosporins, oxacephamycins, and carbapenems.14 The B1 subfamily MBLs are di-Zn(II) utilizing enzymes, with both the Zn1 and Zn2 ions having crucial roles in catalysis, with respect to β-lactam substrate binding, and hydrolytic water activation.15–17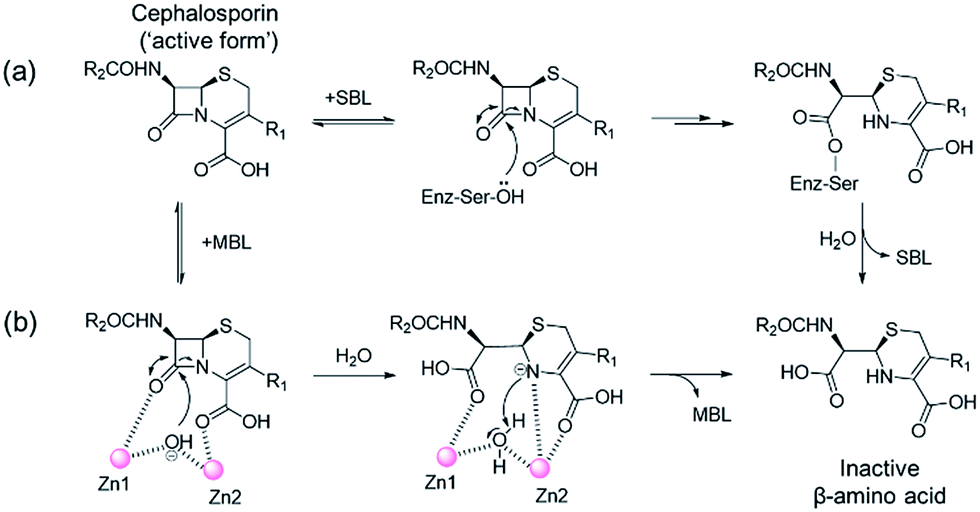 | ||
| Fig. 1 Outline mechanisms for (a) serine- (SBL) and (b) metallo-β-lactamase (MBL) catalysed hydrolysis. Note, in the case of the MBL variations of this mechanism are possible. | ||
We began by carrying out a virtual screen with VIM-2 for which several high-resolution (<1.5 Å) crystal structures are available.12,18–20 A customized virtual screening method, which combines molecular docking simulations with a molecular interaction fingerprints (IFPs)-based filtering approach, was used to identify compounds that are likely to interact with catalytically important active site residues including Arg228, Asn233, Phe61, Tyr67, and Asp120 (using the standard BBL numbering scheme for class B β-lactamases21) as well as zinc ions. Eight types of protein-ligand interactions (hydrogen-bonding donor, hydrogen-bonding acceptor, positively charged, negatively charged, face-to-face π–π stacking, edge-to-face π–π stacking, hydrophobic, and metal–ligand interactions) as defined in our previous work22,23 were used to generate the IFPs (for details of the virtual screening methods see ESI Experimental section SE. 1 and Fig. S3†). Although our strategy included the identification of potential zinc ion binding inhibitors, since we have found experimentally that metal ion chelation can serve to ‘template’ ligands to the active sites of metallo-enzymes,24–26 we were particularly interested in the identification of non-Zn chelating inhibitors. We subsequently screened selected compounds identified in the virtual screen for activity against VIM-2 using a fluorescence-based assay.27 We then used ligand-observe 1H Carr–Purcell–Meiboom–Gill (CPMG) NMR spectroscopy28 to test for binding to both the apo-VIM-2 and catalytically active di-Zn(II) VIM-2, with the aim of establishing whether the zinc ions are required for inhibitor binding.
Results
Application of the virtual screening method led to the identification of a number of fragment-sized compounds, mostly containing acidic groups, that are likely to interact with catalytically important active site features, replicating interactions involved in binding the carboxylate present in β-lactam antibacterials (i.e. with Arg228, Fig. S4–S6†). Of the 20 experimentally tested compounds, 15 exhibited inhibitory activity against VIM-2, and 8 compounds manifested IC50 values <400 μM, including compounds 6, 7, 12, 13, 16, 17, 18, and 20 (Table 1 and Fig. S7†). Using the same assay conditions, we observed that L-captopril inhibits VIM-2 with an IC50 value of 1.6 μM. As negative controls, we tested four compounds which did not pass the IFP score cut-off in the virtual screen at 2 mM against VIM-2 (44–47, Fig. S6, Table S4†); these compounds displayed substantially weaker inhibition than those with IFP scores above the cut-off.| Cpd IDa | Chemical structure | IC50 (μM)/pIC50/s.e. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50b IC50b |
|---|---|---|
| a Compounds 2, 4, 10, 11, 14, and 15 were tested as stereomeric mixtures. b The method for measuring IC50/pIC50 (n ≥ 3) values is described in ESI methods;27 IC50 curves are given in Fig. S7. | ||
| 1 |
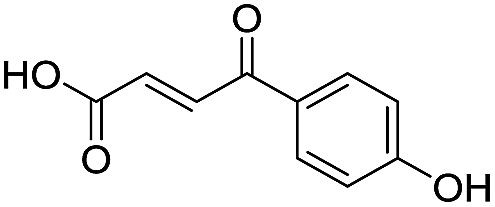
|
637/3.20/0.063 |
| 2 |
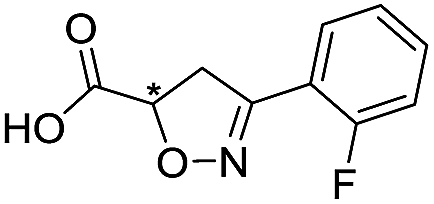
|
993/3.00/0.064 |
| 3 |
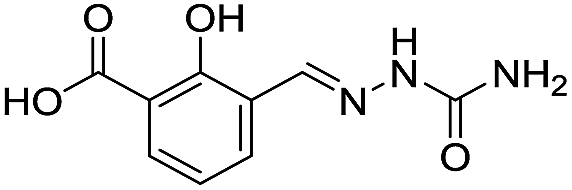
|
>1600/<2.8/— |
| 4 |
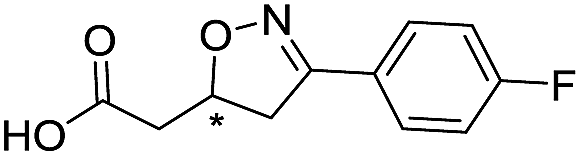
|
557/3.25/0.135 |
| 5 |
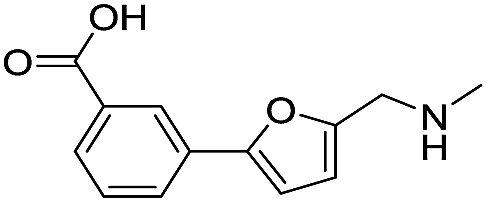
|
>1600/<2.8/— |
| 6 |
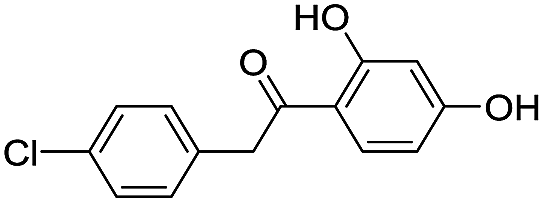
|
111/3.95/0.121 |
| 7 |
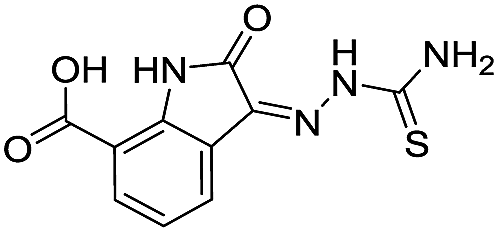
|
199/3.70/0.060 |
| 8 |
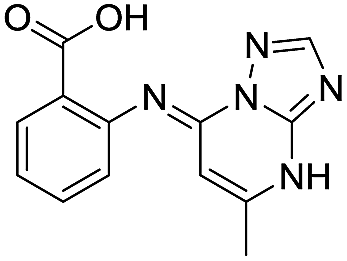
|
>1600/<2.8/— |
| 9 |
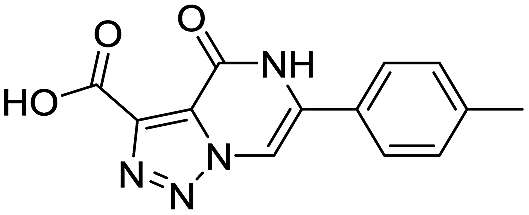
|
439/3.36/0.072 |
| 10 |
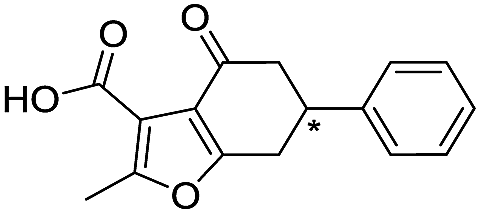
|
>1600/<2.8/— |
| 11 |
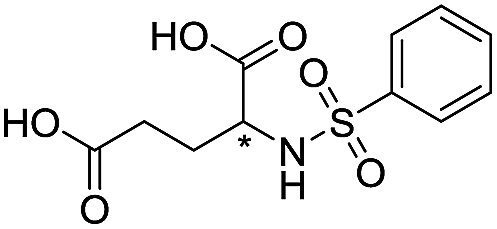
|
1110/2.96/0.043 |
| 12 |
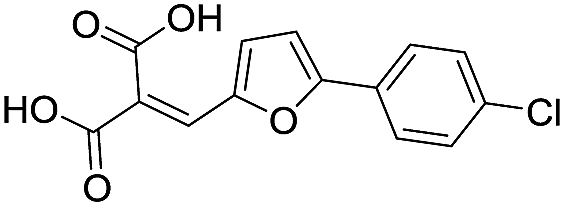
|
87/4.06/0.167 |
| 13 |
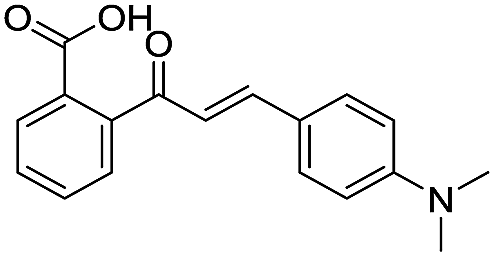
|
135/3.87/0.111 |
| 14 |
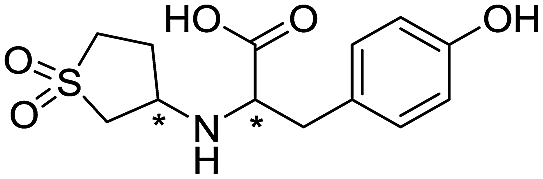
|
669/3.18/0.107 |
| 15 |
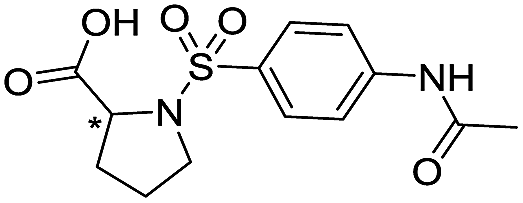
|
>1600/<2.8/— |
| 16 |
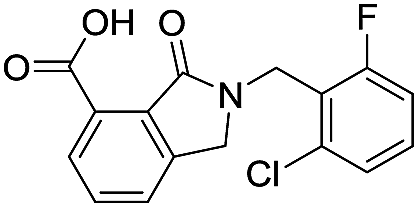
|
10.6/4.98/0.119 |
| 17 |
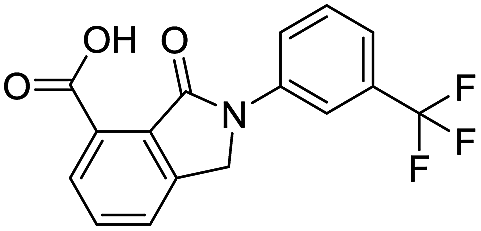
|
32.8/4.48/0.055 |
| 18 |
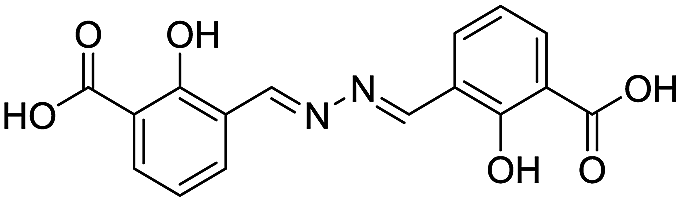
|
117/3.93/0.104 |
| 19 |
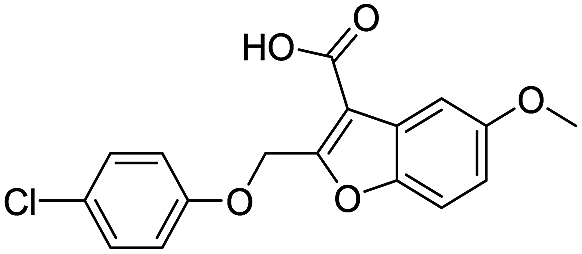
|
718/3.14/0.212 |
| 20 |
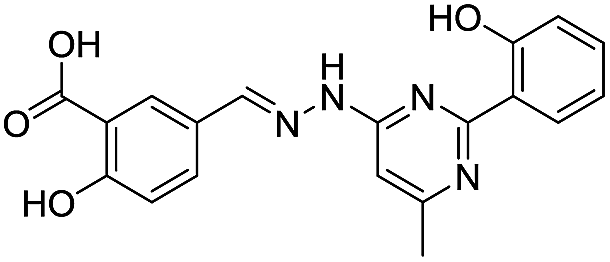
|
292/2.54/2.26 |
| L-Captopril |
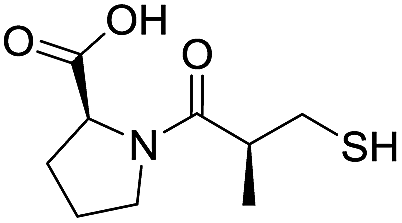
|
1.6/5.80/0.02 |
Since some of the compounds, e.g.6, 7, 12, 13, 16, 17, 18, and 20, possess potential metal-chelating motifs, further VIM-2 inhibition assays at three different concentrations of Zn(II) (0 μM, 1 μM, and 100 μM) were employed to investigate the potential for metal chelation in solution. With the exception of compound 18, no obvious differences between the inhibitory activity with or without excess Zn(II) were observed, suggesting that most of these compounds were not strong Zn(II) chelators in solution (Fig. S8†). L-Captopril was used as a positive control for active site/metal binding18 and showed IC50 values of 1.9 μM, 1.6 μM, and 1.4 μM at concentrations of 0 μM, 1 μM, and 100 μM Zn(II), respectively. Compound 18, however, likely causes inhibition, at least in part, by chelating Zn(II) in solution, so sequestering it from the enzyme.
With the aim of identifying compounds that bind (at least, substantially) without active site Zn(II) chelation, we used 1H CPMG NMR (ESI Experimental section SE. 4†) to investigate binding to catalytically active di-Zn(II) VIM-2 as well as to the apo-VIM-2 form for selected compounds from the virtual screen (including 6, 7, 12, 13, 16, 17, and 18). Notably, for 16 and 17, which displayed the most potent VIM-2 inhibition (IC50 values of 10.6 μM and 32.8 μM, respectively, Table 1), we observed binding to both the di-Zn(II) VIM-2 and apo-VIM-2 protein by 1H CPMG NMR analyses (Fig. 2a and b). For comparison, we selected a known inhibitor of the VIM-2 MBL, L-captopril, which works via an established binding mode that involves direct zinc chelation as revealed by crystallographic analyses;18,29 consistent with its reported mode of binding, we observed that L-captopril displays strong binding to di-Zn(II)-VIM-2, but only very weak binding to apo-VIM-2 (Fig. S9†). The reduction in signal intensity observations with 16 and 17 suggest that they bind more tightly to di-Zn(II) than apo-VIM-2 (Fig. 2), possibly reflecting the more ordered structure of the di-Zn(II) enzyme.
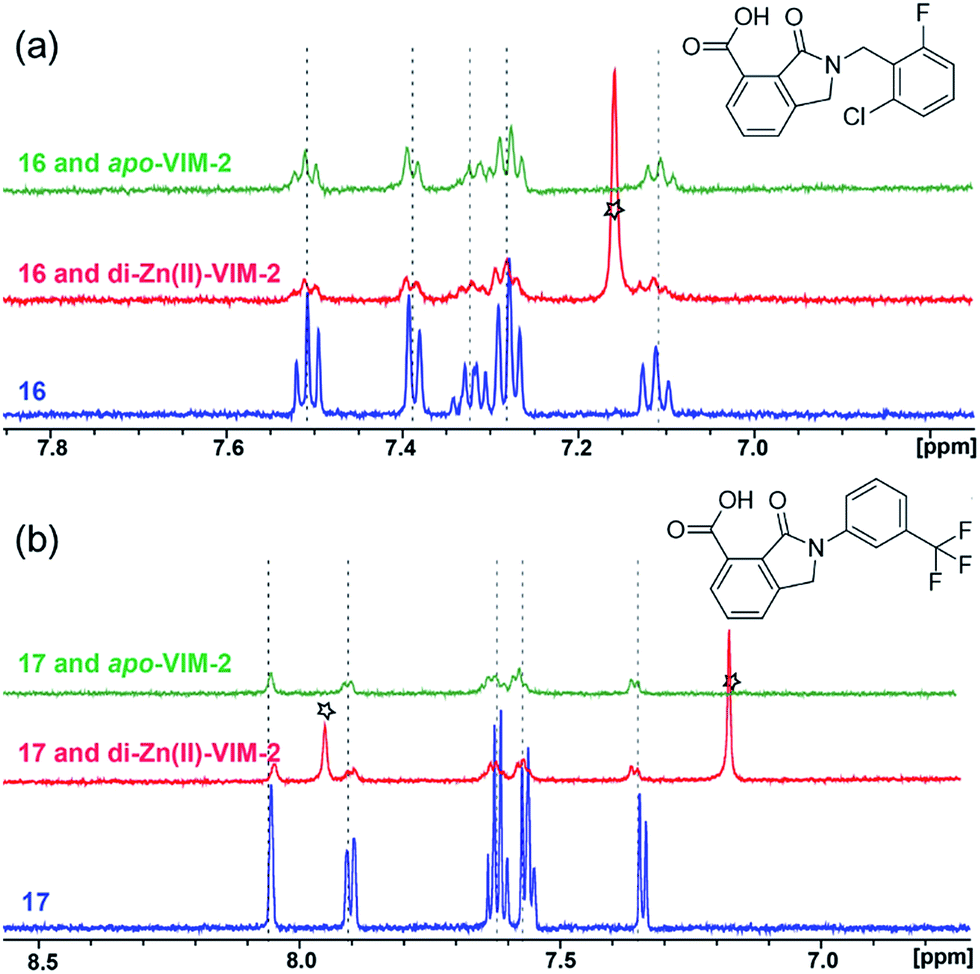 | ||
| Fig. 2 1H CPMG NMR analyses reveal that compounds 16 and 17 bind to both di-Zn(II) and apo-VIM-2 MBL. Binding studies of 16 (a) and 17 (b) to di-Zn(II)-VIM-2 and apo-VIM-2 by 1H CPMG NMR analyses. 16 and 17 bind to both di-Zn(II) and apo-VIM-2 as indicated by signal intensity reduction in the presence of VIM-2. Assay mixtures contained 50 μM enzyme (either di-Zn(II) VIM-2 in the presence of 50 μM Zn(II) or apo-VIM-2), and 50 μM of the compound of interest buffered with 50 mM Tris-D11, pH 7.5, in 90% H2O and 10% D2O. Black stars denote imidazole in the di-Zn(II)-VIM-2 buffer (Fig. S15†). Note, the 1H NMR spectra of 16 and 17 in DMSO-D6 (Fig. S16†) are different from those in 50 mM Tris-D11, pH 7.5, in 90% H2O and 10% D2O. | ||
Overall, these results show that 16 or 17 can bind to VIM-2 in the absence of zinc ions. However, the presence of zinc indirectly participates in the binding of these inhibitors to VIM-2, possibly through protein stabilisation, bridging interactions or a combination thereof. We also observed that 6 (IC50 = 111 μM) displays strong binding to both di-Zn(II) VIM-2 and apo-VIM-2 (Fig. S10†), indicating that, like 16 and 17, 6 may bind without metal chelation. In contrast, 12 (IC50 = 87 μM) and 13 (IC50 = 135 μM) displayed strong binding to di-Zn(II) VIM-2, but weak binding to the apo-VIM-2 protein (Fig. S11 and S12†), suggesting that these compounds inhibit VIM-2 by a mode directly involving zinc chelation. For 7 and 18, for which assays showed VIM-2 inhibition (IC50 values of 199 μM and 117 μM, respectively, Table 1), we observed binding to both di-Zn(II)- and apo-VIM-2 (Fig. S13 and S14†) by NMR. In contrast to 16 and 17 (see below), 12 and 13, which may be metal-chelating VIM-2 inhibitors as observed in the inhibition assays and NMR studies (Table 1, Fig. S11 and S12†), displayed broad-spectrum inhibition against almost all the tested class B MBLs (Fig. S21†).
In order to investigate the inhibitory mechanism of 16 and 17, we then sought to obtain crystal structures for complexes of VIM-2 with them (ESI Experimental section SE. 5 and Table S1† for details of crystallization and structure determinations). Co-crystallisation experiments yielded structures of VIM-2 in complex with 16 (1.40 Å) and 17 (1.93 Å) (Table S2†). VIM-2:16 and VIM-2:17 crystallized in the P21221 and P212121 space groups (Table S2†), respectively, with one molecule in the asymmetric unit (ASU); these space groups differ from those previously reported for VIM-2.12,18,19 In both structures, there was clear Fo − Fc density in the VIM-2 active site, into which 16 and 17 could be confidently modelled (Fig. S17†). The crystal structures reveal that 16 and 17 both bind adjacent to the zinc ions of VIM-2, but in a mode which does not involve direct zinc chelation (Fig. 3a–c), consistent with the NMR results (Fig. 2a and b). All of the atoms of the inhibitor are >4 Å from the zinc ions (Fig. 3c). The predicted binding modes of 16 and 17 are similar to those observed in the crystal structures (Fig. S18†), indicating that the virtual screening method, which searched for compounds likely to interact with catalytically important active site features, is a useful strategy for identification of new MBL-fold enzyme inhibitors.
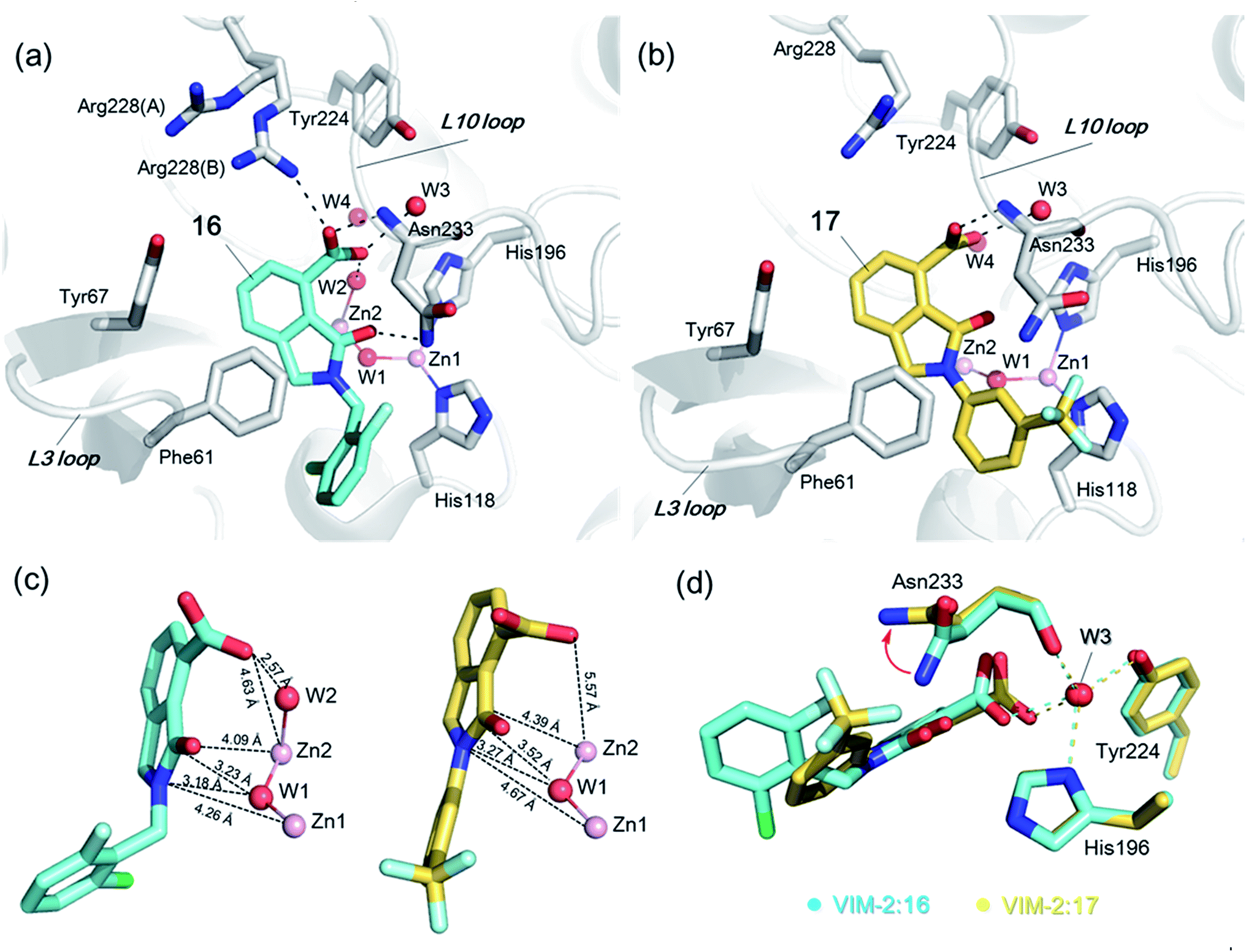 | ||
| Fig. 3 Crystallographic analyses reveals compound 16 and 17 binding modes to VIM-2. (a) View from a crystal structure of VIM-2 in complex with compound 16 (PDB ID 5LE1) reveals that the inhibitor binds to form hydrophobic and electrostatic interactions with residues on the L3 and L10 loops (e.g., π–π stacking interactions with Phe61, and hydrogen-bonding interactions with Asn233; see Fig. S19† for further details of binding interaction). (b) View from a crystal structure of VIM-2 in complex with 17 (PDB ID 5LCA) reveals that 17 binds via a similar mode with that of 16. (c) Comparison of complex structures of VIM-2:16 and VIM-2:17 shows that both bind adjacent to zinc ions of VIM-2, but in a mode which does not involve direct zinc chelation. Distances between the oxygen atoms of the carboxylate of 16 and 17 and Zn2 are 4.63 Å and 5.57 Å, respectively. (d) The water molecule W3 is positioned to form tight hydrogen-bonding interactions with His196, Tyr224, and Asn233, and is important for binding the carboxylate of 16 and 17. | ||
The crystal structures reveal that 16 and 17 have very similar binding modes, in which the 3-oxoisoindoline-4-carboxylate heterocyclic core is positioned to make hydrophobic and electrostatic interactions with the VIM-2 active site features, including via π–π stacking interactions with Phe61 on the L3 loop, hydrogen-bond interactions with Arg228 and Asn233 on the L10 loop and water molecules (such as W3) (Fig. 3a and b and S19†). 16 (IC50 = 10.6 μM) appears positioned to make stronger hydrogen-bonding interactions with Arg228 (2.9 Å), Asn233 (2.8 Å), and a water molecule W2 (2.7 Å) (Fig. 3a and b and S19†) than 17 (IC50 = 32.8 μM), which may, in part, explain why it inhibits more potently. As revealed by analysis of the Cambridge Structural Database, zinc–ligand coordination bond lengths are generally less than 2.5 Å,30 thus 16 and 17 both do not directly chelate the VIM-2 active site zinc ions. A structural water molecule, W3, also observed in other crystal structures of VIM-2,12,18–20 is positioned to form hydrogen-bonding interactions with His196 (2.9 Å), Tyr224 (2.7 Å), and Asn233 (2.7 Å) and appears to be important to the binding of the carboxylate groups of 16 and 17 (Fig. 3d).
We then tested 16 and 17 against; the subclass B1 enzymes VIM-5, VIM-1, NDM-1, SPM-1, and BcII; the subclass B2 enzyme Cph-A; the subclass B3 enzyme L1; and the class A serine β-lactamase TEM-1 using our previously established screening platform27 to investigate their selectivity profiles (see ESI Experimental section SE. 3 and Table S3†). Notably, 16, and to a greater extent, 17 have considerable selectivity for VIM-2 (Fig. 4a and b). Even for VIM-5, which is a close homolog of VIM-2 (sequence identity is 89.85%, Fig. S20a†),31,3216 and 17 showed lower inhibitory activities with IC50 values of 47.6 μM (10.6 μM for VIM-2) and >400 μM (32.8 μM for VIM-2), respectively (Fig. 3a and b and Table 2). As observed by crystallography, the active site features of VIM-2 (PDB 4BZ3)18 and VIM-5 (PDB 5A87)32 are highly similar with only three residues being different (Ile223VIM-2/Val223VIM-5, Tyr224VIM-2/Leu224VIM-5, and Glu225VIM-2/Ala225VIM-5) on the L10 loop (Fig. S20†). These substitutions may contribute to the small differences observed by crystallography in the conformations of the L10 loops of VIM-2 and VIM-5 (Fig. S20b†); however, differences in solution may be larger. Notably, a water molecule positioned similarly to W3 in VIM-2 (W3VIM-2), which we propose is important for binding of 16 and 17 (Fig. 3a–c), is apparent in the VIM-5 active site (W3VIM-5, Fig. S20b†); however, W3VIM-5 appears to be not so stable as W3VIM-2 (PDB 5A87). These differences may explain why 16 and 17 manifest substantial selectivity for VIM-2 over VIM-5 (Fig. 4).
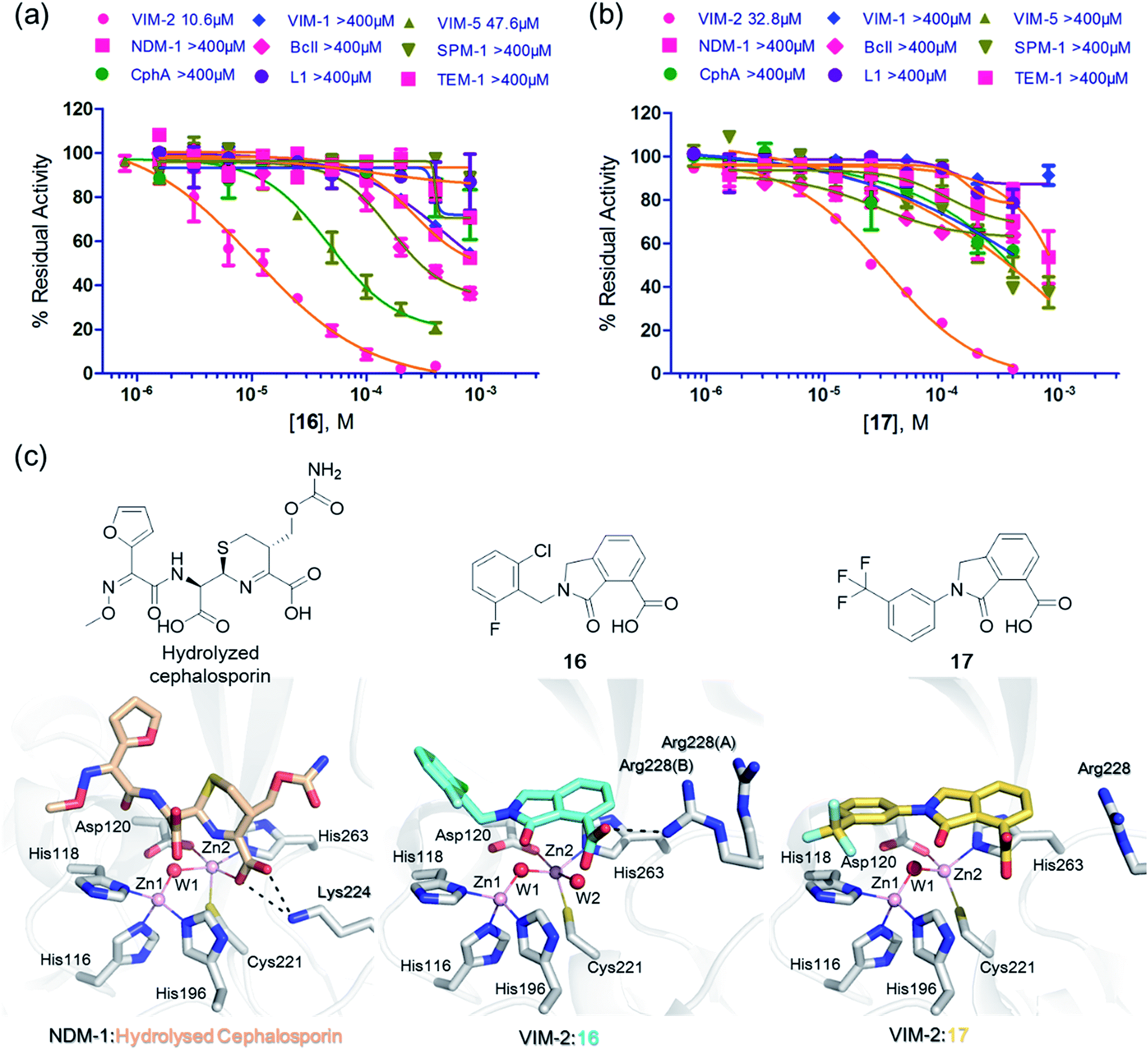 | ||
| Fig. 4 Compounds 16 and 17 mimic interactions made by substrates. Selectivity profiles of 16 (a) and 17 (b) for class B MBLs and the class A serine β-lactamase TEM-1 (for which no inhibition was observed). (c) Comparison of VIM2 structures in complex with 16 (PDB ID 5LE1) and 17 (PDB ID 5LCA) with that of a representative substrate intermediate of a hydrolysed cephalosporin in complex with NDM-1 (PDB ID 4RL0)33 indicates that 16 and 17 have related binding modes to the cephalosporin substrate. Docking studies imply the binding modes of 16 and 17 may mimic the binding mode of the substrate prior to β-lactam hydrolysis (Fig. S22†). | ||
|
|
|||||
|---|---|---|---|---|---|
| Cpd IDa | R1 | R2 | IC50 (μM)/pIC50/s.e. logIC50b |
||
| VIM-2 | VIM-5 | VIM-1 | |||
| a Compounds 28, 29, 38, 41, 42, and 43 were tested as racemic mixtures. b The method for measuring IC50/pIC50 (n ≥ 3) values is described in ESI methods.27 | |||||
| 16 |
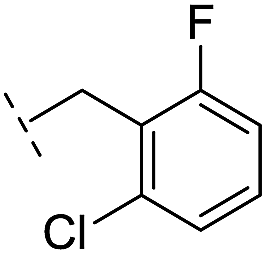
|
–H | 10.6/4.98/0.119 | 47.6/4.32/0.079 | >400/<3.4/— |
| 17 |
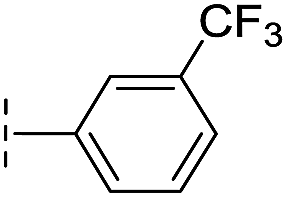
|
–H | 32.8/4.48/0.055 | >400/<3.4/— | >400/<3.4/— |
| 21 |
|
–H | 349/3.46/0.15 | >400/<3.4/— | >400/<3.4/— |
| 22 |

|
–H | >400/<3.4/— | >400/<3.4/— | >400/<3.4/— |
| 23 |
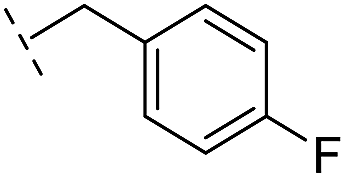
|
–H | 250/3.60/0.17 | >400/<3.4/— | >400/<3.4/— |
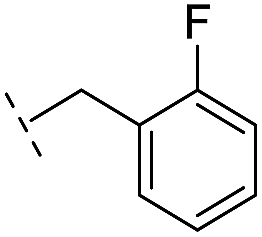
| 24 |
|
–H | 47.1/4.33/0.037 | 127/3.90/0.075 | >400/<3.4/— |
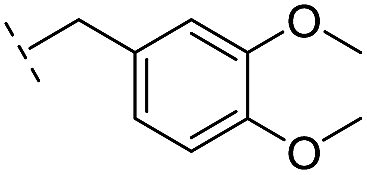
| 25 |
|
–H | 320/3.50/0.096 | >400/<3.4/— | >400/<3.4/— |
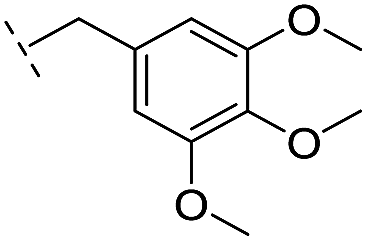
| 26 |
|
–H | >400/<3.4/— | >400/<3.4/— | >400/<3.4/— |
| 27 |
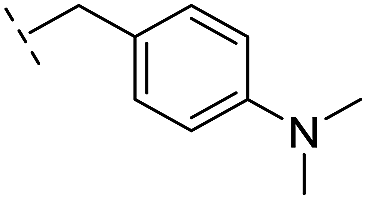
|
–H | 176/3.76/0.079 | >400/<3.4/— | >400/<3.4/— |
| 28 |
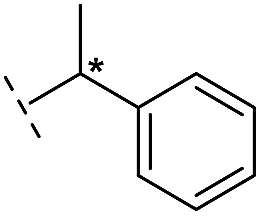
|
–H | 168/3.78/0.153 | >400/<3.4/— | >400/<3.4/— |
| 29 |
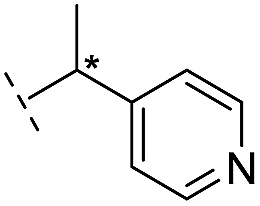
|
–H | 229/3.64/0.062 | >400/<3.4/— | >400/<3.4/— |
| 30 |
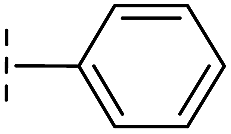
|
–H | 17.8/4.75/0.039 | >400/<3.4/— | >400/<3.4/— |
| 31 |
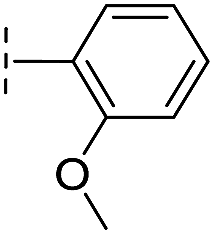
|
–H | 133/3.88/0.056 | >400/<3.4/— | >400/<3.4/— |
| 32 |
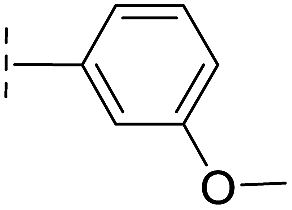
|
–H | 39.3/4.41/0.029 | >400/<3.4/— | >400/<3.4/— |
| 33 |
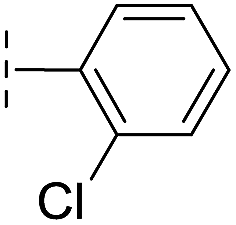
|
–H | 74.0/4.13/0.057 | >400/<3.4/— | >400/<3.4/— |
| 34 |
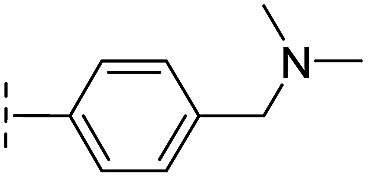
|
–H | 60.3/4.22/0.093 | >400/<3.4/— | >400/<3.4/— |
| 35 |
|
–H | 7.7/5.11/0.071 | >400/<3.4/— | >400/<3.4/— |
| 36 |
|
–H | 153/3.82/0.138 | 180/3.75/0.116 | >400/<3.4/— |
| 37 |
|
–H | 68.1/4.17/0.079 | >400/<3.4/— | >400/<3.4/— |
| 38 |
|
–H | >400/<3.4/— | >400/<3.4/— | >400/<3.4/— |
| 39 |
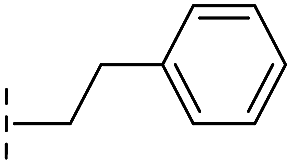
|
–H | 71.9/4.14/0.082 | >400/<3.4/— | >400/<3.4/— |
| 40 |
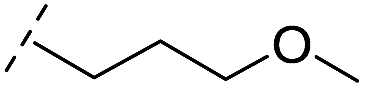
|
–H | >400/<3.4/— | >400/<3.4/— | >400/<3.4/— |
| 41 |
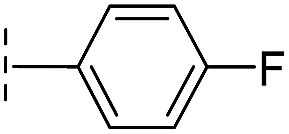
|
|
24.6/4.61/0.097 | 285/3.49/0.133 | >400/<3.4/— |
| 42 |
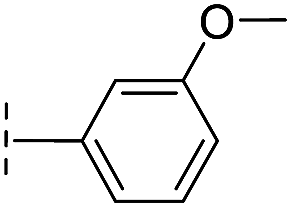
|
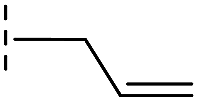
|
20.3/4.69/0.032 | 348/3.46/0.210 | >400/<3.4/— |
| 43 |
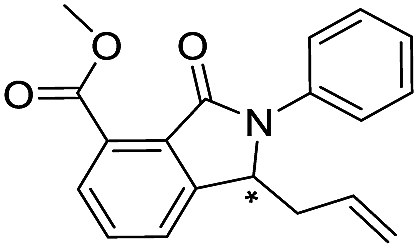
|
>400/<3.4/— | >400/<3.4/— | >400/<3.4/— | |
We were attracted by the resemblance of the 3-oxoisoindoline-4-carboxylate scaffold to that of the bicyclic β-lactam MBL substrates, e.g. cephalosporins (Fig. 1 and 4c). Using molecular docking analyses, we observed that the bicyclic core of a cephalosporin (a representative bicyclic β-lactam substrate) is likely to form hydrogen-bonding interactions with Asn233 and the water molecule W3 (Fig. S22a†), in a similar manner to the 3-oxoisoindoline-4-carboxylate scaffold of 16 and 17, as observed by our crystallographic analyses (Fig. S22b†). As shown in Fig. 4c, 16 and 17 also have a similar binding mode to that observed in a complex of NDM-1 with a ring-opened cephalosporin intermediate;33 the carboxylate groups of 16 and 17 are positioned to make electrostatic interactions with Arg228 in an analogous manner to that observed for the binding of the cephalosporin C-4 carboxylate to Lys224 (a structurally equivalent residue to Arg228) of NDM-1 (ref. 33) (Fig. 4c).
On superimposing the crystal structures of VIM-2:16, VIM-2:17, and di-Zn(II) VIM-2, we observe that the L3 loop of VIM-2 appears closer to the zinc ions when 16 and 17 are bound to VIM-2 (3.50 Å and 3.81 Å movements of the C-α atoms of Asp62, respectively, Fig. S23a†), whilst the L10 loop is positioned further away from the zinc ions (1.39 Å and 1.53 Å difference in the C-α atom of Asn233, respectively, Fig. S23†). These results support the proposal that, at least in some cases, these loops contribute to the highly efficient nature of MBL catalysis by capturing potential substrates and delivering them to the zinc ion containing active site for hydrolysis.
From the results above, we propose that the 3-oxoisoindoline-4-carboxylate scaffold of 16 and 17 is an important factor contributing to the inhibitory potency and the selectivity for VIM-2. We carried out structure–activity relationship (SAR) studies using commercially available and synthesised 3-oxoisoindoline-4-carboxylate derivatives with different indole-N-substituents (R1) and/or C-1 substituents (R2) (compounds 21–43 in Table 2). We synthesized 34 and 35via the routes in ESI methods SE. 2 and Scheme S1.† Compared to the hit compound 16 identified in initial screening (IC50 = 10.6 μM), which has a 2-chloro-6-fluorobenzyl moiety at the R1 position, compounds 21–29 with different substituted-benzyl moieties at the R1 position exhibited lower inhibitory activities against VIM-2. The results indicated that chloro- and fluoro-substituents at the ortho position of the N-benzyl group are important in binding. The VIM-2:16 complex structure implies that the fluorine and chlorine atoms of 16 are likely positioned to form halogen-bonding interactions with Asn233 (2.88 Å), and Trp87 (3.41 Å), and Asp120 (3.34 Å), respectively (Fig. 3a and S19a†). Compounds bearing substituted phenyl moieties at the R1 position (30–35) displayed better VIM-2 inhibitory activities than 21–29, some having comparable activity to 16 and 17. Compound 30 is more potent than 17 (IC50 = 32.8 μM) with an IC50 value of 17.8 μM, although its N-phenyl group is unsubstituted. As revealed by subsequent crystallographic analyses of VIM-2 complexes, 30 and 17 have similar binding modes (Fig. 5a and b and 3b). However, compared with 17, 30 is apparently better positioned to interact with the Asn233 side chain (the trifluoromethyl group of 17 hinders hydrogen bonding interactions between the Asn233 side chain and the 3-oxoisoindoline-4-carboxylate, Fig. 5b and S24†). Compound 35 (IC50 = 7.7 μM), which contains a 3-fluoro-4-hydroxyphenyl motif at its R1 position, was more potent against VIM-2 than compounds 16 and 17 (Table 2). As observed by crystallography, 35 has the same overall binding mode as 30 (Fig. 5c and d); compared with 30, 35 is positioned to make additional hydrogen-bonding interactions with Asp119 (3.03 Å), as well as halogen-bonding interactions with Asn233 (3.75 Å) (Fig. 5c and d and S25†). In addition to various substituted-benzyl/phenyl groups, compounds 36–40, containing thiophen-2-ylmethyl, cyclopentyl, tetrahydrofuran-2-yl, phenethyl, and 3-methoxypropyl at R1 position, respectively, were tested. These compounds displayed only moderate inhibitory potencies against VIM-2. 41 and 42, which both have an allyl group at the R2 position, showed relatively good inhibitory activities against VIM-2 with IC50 values of 24.6 μM and 20.3 μM, respectively (Table 2). As observed in a VIM-2:42 complex crystal structure, the allyl group of 42 is positioned to make hydrophobic interactions with Phe61 and Tyr67 (Fig. 5e and S26†).
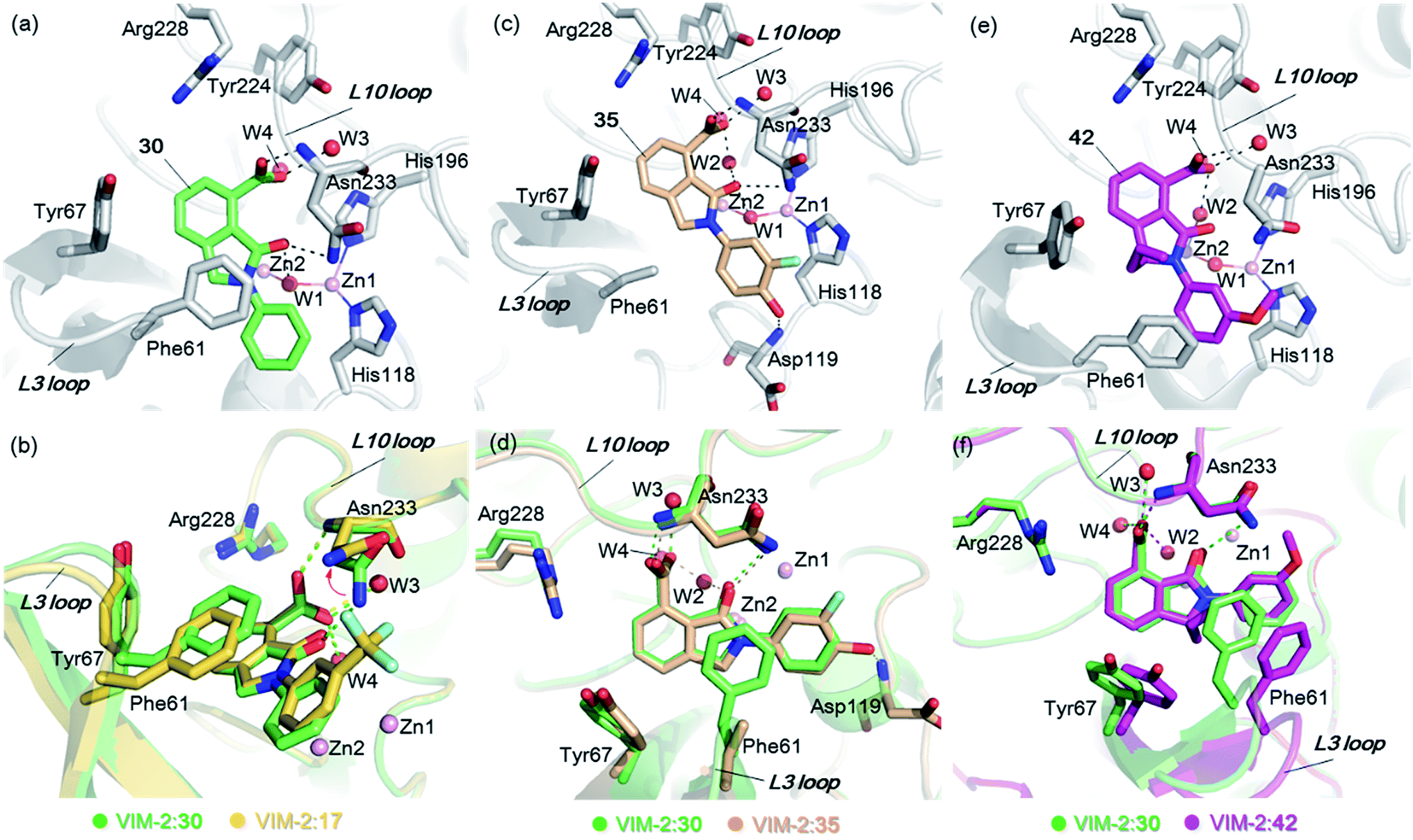 | ||
| Fig. 5 Crystallographic analyses reveals how the 3-oxoisoindoline-4-carboxylate derivatives bind to the VIM-2 MBL. (a) View from a crystal structure of VIM-2 in complex with 30 (PDB ID 5LCF). (b) Comparison of structures of VIM-2:17 and VIM-2:30 reveals the Asn233 side chain is unable to form hydrogen-bonds with the 3-oxoisoindoline-4-carboxylate of 17 due to its trifluoromethyl group, which may explain why 17 is less potent than 30 (Table 2). (c) View from a crystal structure of VIM-2 in complex with 35 (PDB ID 5LM6). (d) Comparison of VIM-2:30 and VIM-2:35 complex structures reveals 30 and 35 have the same binding mode. (e) View from a crystal structure of VIM-2 in complex with 42 (PDB ID 5LCH). (f) Comparison of structures of VIM-2:30 and VIM-2:42 reveals evidence for flexibility in the conformation of Tyr67 and Phe61 on the L3 loop, suggesting Tyr67 and Phe61 may play important roles in capturing substrates and delivering them to the zinc ions for hydrolysis. | ||
Notably, comparison of structures of VIM-2:30 and VIM-2:42 reveals evidence for flexibility in the conformation of the L3 loop, suggesting that Tyr67 and Phe61 may be important in substrate/inhibitor capture (Fig. 5f). 43 showed much lower inhibitory potency, supporting the proposed role of the 3-oxoisoindoline-4-carboxylate motif in binding/inhibition. For VIM-5 and VIM-1, almost all the tested 3-oxoisoindoline-4-carboxylate derivatives (except 16, 24, and 36) displayed low inhibitory activities, indicating that the 3-oxoisoindoline-4-carboxylate scaffold is an important factor (along with its precise substitution pattern) in determining selectivity for VIM-2. Together, these results led us to conclude that the 3-oxoisoindoline-4-carboxylate derivatives selectively inhibit the VIM-2 MBL via a mode which does not involve direct chelation with zinc ions.
Discussion
The overall results clearly reveal the potential of a customized virtual screening approach targeting specific active site features for the identification of hit compounds for MBL/MBL-fold/metallo-enzymes. We successfully used an NMR-based approach employing di-Zn(II)- and apo-VIM-2 to identify hits from virtual screening that bind to both the apo- and the di-Zn(II) containing enzymes, hence are less likely to inhibit solely via direct zinc ion coordination. Indeed, the approach led to some compounds which bind via direct zinc ion chelation and some which do not, e.g.16 and 17, as supported by subsequent crystallographic analyses.The combined biophysical analyses reveal 3-oxoisoindoline-4-carboxylate derivatives as a new class of MBL inhibitor, which do not, at least with the analysed compounds, strongly chelate the active site zinc ions. MBL selectivity profiling analyses showed 3-oxoisoindoline-4-carboxylate derivatives can be highly selective for VIM-2, even with respect to very closely related variants, e.g. VIM-5. The apparently narrow selectivity of the 3-oxoisoindoline-4-carboxylate derivatives may limit their use for potentiating β-lactam antibacterials, since clinically useful MBL inhibitors should be broad spectrum targeting multiple MBL types, at least the B1 subclass. However, the limited SAR results presented here suggest that increasing the potency and broadening the spectrum of the 3-oxoisoindoline-4-carboxylate inhibitors towards clinically relevant MBLs should be possible. Most importantly, the results reveal that non-metal-chelating inhibitors present a route to new types of selective MBL-fold enzyme inhibitors. This may be particularly useful in targeting human MBL-fold enzymes involved in cancer drug resistance,8,9e.g. where highly selective inhibition is desirable. When coupled with appropriate assays, such selective inhibitors may be useful in profiling clinically observed MBL variants.
From a mechanistic perspective, the biophysical analyses are of interest because the crystallographically observed binding modes for the 3-oxoisoindoline-4-carboxylate inhibitors, to some extent, mimic those of intact β-lactam substrates to MBLs. In this regard, the interactions of the 3-oxoisoindoline-4-carboxylate inhibitors with the L3/L10 loops are of particular interest, since it is possible, at least in some cases, that these loops may contribute to the highly efficient nature of MBL catalysis by capturing potential substrates and delivering them to the zinc ion containing active site for hydrolysis.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the Advanced Research Computing Centre, University of Oxford. We thank the staff at the Diamond Synchrotron Light source for access. This work was in part supported by the Wellcome Trust, the Medical Research Council (MRC) grant MR/L007665/1, the Medical Research Council (MRC)/Canadian Grant G1100135, the SWON alliance (C. J. Schofield), the Biochemical Society Krebs Memorial Award (M. I. Abboud), and the National Natural Science Foundation of China (81502989)/the China Postdoctoral Science Foundation Funded Project (2015M570789) (G.-B. Li).Notes and references
- K. Bush and G. A. Jacoby, Antimicrob. Agents Chemother., 2010, 54, 969–976 CrossRef CAS PubMed.
- M. W. Crowder, J. Spencer and A. J. Vila, Acc. Chem. Res., 2006, 39, 721–728 CrossRef CAS PubMed.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- C. Bebrone, P. Lassaux, L. Vercheval, J. Sohier, A. Jehaes, E. Sauvage and M. Galleni, Drugs, 2010, 70, 651–679 CrossRef CAS PubMed.
- K. A. Toussaint and J. C. Gallagher, Ann. Pharmacother., 2015, 49, 86–98 CrossRef PubMed.
- M. I. Abboud, C. Damblon, J. Brem, N. Smargiasso, P. Mercuri, B. Gilbert, A. M. Rydzik, T. D. Claridge, C. J. Schofield and J.-M. Frère, Antimicrob. Agents Chemother., 2016, 60, 5655–5662 CrossRef PubMed.
- I. Pettinati, J. Brem, S. Y. Lee, P. J. McHugh and C. J. Schofield, Trends Biochem. Sci., 2016, 41, 338–355 CrossRef CAS PubMed.
- S. Y. Lee, J. Brem, I. Pettinati, T. D. Claridge, O. Gileadi, C. J. Schofield and P. J. McHugh, Chem. Commun., 2016, 52, 6727–6730 RSC.
- P. Hinchliffe, M. M. González, M. F. Mojica, J. M. González, V. Castillo, C. Saiz, M. Kosmopoulou, C. L. Tooke, L. I. Llarrull and G. Mahler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3745–E3754 CrossRef CAS PubMed.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS PubMed.
- J. Brem, S. S. van Berkel, W. Aik, A. M. Rydzik, M. B. Avison, I. Pettinati, K.-D. Umland, A. Kawamura, J. Spencer and T. D. Claridge, Nat. Chem., 2014, 6, 1084–1090 CrossRef CAS PubMed.
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J.-C. Jimenez-Castellanos, M. B. Avison, J. Spencer, C. W. G. Fishwick and C. J. Schofield, Nat. Commun., 2016, 7, 12406 CrossRef CAS PubMed.
- L. Poirel, T. Naas, D. Nicolas, L. Collet, S. Bellais, J. D. Cavallo and P. Nordmann, Antibiotics, 2000, 44, 891–897 CAS.
- M. I. Page and A. Badarau, Bioinorg. Chem. Appl., 2008, 2008, 576297 Search PubMed.
- M. R. Meini, L. I. Llarrull and A. J. Vila, Antibiotics, 2014, 3, 285–316 CrossRef CAS PubMed.
- T. Palzkill, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104 CrossRef CAS PubMed.
- J. Brem, S. S. van Berkel, D. Zollman, S. Y. Lee, O. Gileadi, P. J. McHugh, T. R. Walsh, M. A. McDonough and C. J. Schofield, Antimicrob. Agents Chemother., 2016, 60, 142–150 CrossRef CAS PubMed.
- T. Christopeit, T. J. O. Carlsen, R. Helland and H.-K. S. Leiros, J. Med. Chem., 2015, 58, 8671–8682 CrossRef CAS PubMed.
- M. Aitha, A. R. Marts, A. Bergstrom, A. J. Moller, L. Moritz, L. Turner, J. C. Nix, R. A. Bonomo, R. C. Page, D. L. Tierney and M. W. Crowder, Biochemistry, 2014, 53, 7321–7331 CrossRef CAS PubMed.
- M. Galleni, J. Lamottebrasseur, G. M. Rossolini, J. Spencer, O. Dideberg and J.-M. Frère, Antimicrob. Agents Chemother., 2001, 45, 660–663 CrossRef CAS PubMed.
- G.-B. Li, L.-L. Yang, W.-J. Wang, L.-L. Li and S.-Y. Yang, J. Chem. Inf. Model., 2013, 53, 592–600 CrossRef CAS PubMed.
- G.-B. Li, S. Ji, L.-L. Yang, R.-J. Zhang, K. Chen, L. Zhong, S. Ma and S.-Y. Yang, Eur. J. Med. Chem., 2015, 93, 523–538 CrossRef CAS PubMed.
- M. Demetriades, I. K. Leung, R. Chowdhury, M. C. Chan, M. A. McDonough, K. K. Yeoh, Y. M. Tian, T. D. Claridge, P. J. Ratcliffe, E. C. Woon and C. J. Schofield, Angew. Chem., Int. Ed., 2012, 51, 6672–6675 CrossRef CAS PubMed.
- E. C. Y. Woon, M. Demetriades, E. A. L. Bagg, W. Aik, S. M. Krylova, J. H. Y. Ma, M. Chan, L. J. Walport, D. W. Wegman, K. N. Dack, M. A. McDonough, S. N. Krylov and C. J. Schofield, J. Med. Chem., 2012, 55, 2173–2184 CrossRef CAS PubMed.
- B. M. R. Liénard, R. Hüting, P. Lassaux, M. Galleni, J.-M. Frère and C. J. Schofield, J. Med. Chem., 2008, 51, 684–688 CrossRef PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- J. A. Aguilar, M. Nilsson, G. Bodenhausen and G. A. Morris, Chem. Commun., 2012, 48, 811–813 RSC.
- A. Badarau and M. I. Page, Biochemistry, 2006, 45, 10654–10666 CrossRef CAS PubMed.
- A. Nimmermark, L. Öhrström and J. Reedijk, Z. Kristallogr. - Cryst. Mater., 2013, 228, 311–317 CrossRef CAS.
- L. Poirel, Y. Yakupogullari, A. Kizirgil, M. Dogukan and P. Nordmann, Int. J. Antimicrob. Agents, 2009, 33, 287–294 CrossRef CAS PubMed.
- A. Makena, A. Ö. Düzgün, J. Brem, M. A. McDonough, A. M. Rydzik, M. I. Abboud, A. Saral, A. Ç. Çiçek, C. Sandalli and C. J. Schofield, Antimicrob. Agents Chemother., 2016, 60, 1377–1384 CrossRef CAS PubMed.
- H. Feng, J. Ding, D. Zhu, X. Liu, X. Xu, Y. Zhang, S. Zang, D. C. Wang and W. Liu, J. Am. Chem. Soc., 2014, 136, 14694–14697 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, crystallographic data collection and refinement statistics, details of chemical synthesis, additional figures and tables. See DOI: 10.1039/c6sc04524c |
| This journal is © The Royal Society of Chemistry 2017 |