
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective transition-metal-free vicinal cis-dihydroxylation of saturated hydrocarbons†
Luis
Bering
ab and
Andrey P.
Antonchick
*ab
aMax-Planck-Institut für molekulare Physiologie, Abteilung Chemische Biologie, Otto-Hahn-Straße 11, 44227 Dortmund, Germany. E-mail: Andrey.Antonchick@mpi-dortmund.mpg.de
bTechnische Universität Dortmund, Otto-Hahn-Straße 4a, 44227 Dortmund, Germany
First published on 22nd August 2016
Abstract
A transition-metal-free cis-dihydroxylation of saturated hydrocarbons under ambient reaction conditions has been developed. The described approach allows a direct and selective synthesis of vicinal diols. The new reaction thereby proceeds via radical iodination and a sequence of oxidation steps. A broad scope of one-pot dual C(sp3)–H bond functionalization for the selective synthesis of vicinal syn-diols was demonstrated.
Introduction
C–H bond functionalization of aliphatic hydrocarbons represents a long-standing goal in organic chemistry.1 The selective functionalization of the chemically inert and ubiquitous C(sp3)–H bond is a great challenge. The participation of hydrocarbons in chemical reactions often requires high temperatures at the expense of the controllability and economy of product formation.2 As well as aliphatic C–H bond halogenation, alkylation, aminations, dehydrogenation and borylation, direct C–H bond hydroxylation has gained significant interest.3 Alcohols and polyols are important synthetic precursors, structural motifs in natural products and bioactive molecules and are employed in numerous industrial processes.4 To date a variety of metal-based, cytochrome P450 inspired and metal-free methods for the hydroxylation of unactivated C–H bonds exist.5 Since 1983, Barton and co-workers pioneered the iron-mediated oxidation of alkanes under mild conditions (GIF chemistry) and more recently White et al. developed an iron-based catalyst that efficiently oxidizes one equivalent of cyclohexane to the corresponding ketone (Scheme 1A).6 In the absence of metal catalyst, the oxidation of hydrocarbons was realized using oxidants in the presence of strong acids or dimethyldioxirane and its analogues (Scheme 1B).7 Overoxidation, dehydration and low selectivity are common outcomes of the described methods. 1,2-Diols have high importance in organic synthesis and have found wide application. State of the art methods for diol synthesis are predominately based on the dihydroxylation of alkenes in a metal-catalyzed or metal-free manner.8 Despite the progress towards efficient and selective aliphatic C–H bond mono-functionalization, practical methods enabling the direct vicinal dihydroxylation of hydrocarbons are not known. Herein we report the first selective vicinal cis-dihydroxylation of saturated hydrocarbons (Scheme 1C). The developed method allows the synthesis of cis-diols under transition-metal-free and ambient reaction conditions.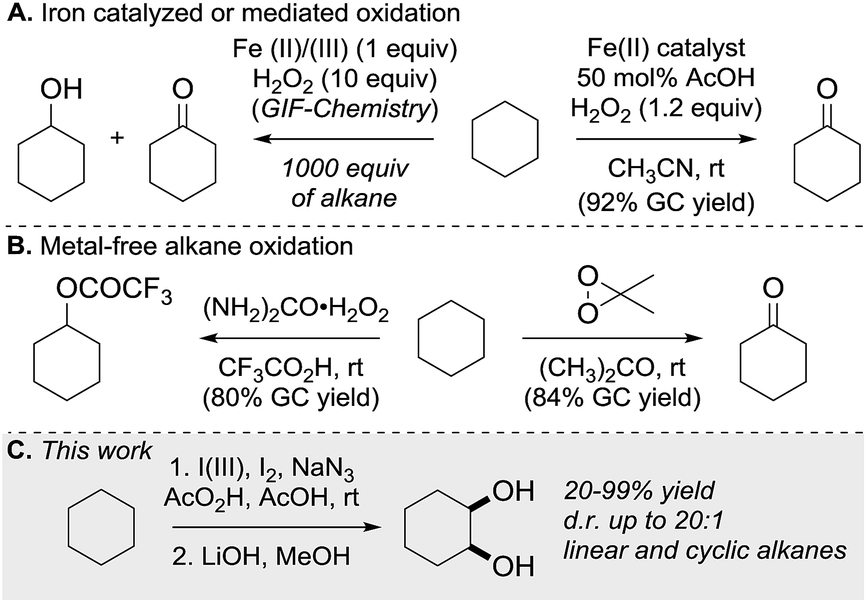 | ||
| Scheme 1 Direct C(sp3)–H bond oxidation of alkanes. | ||
Results and discussion
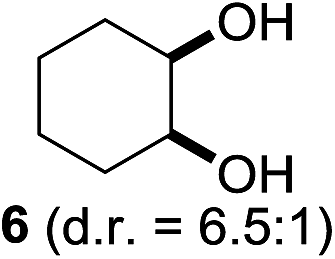
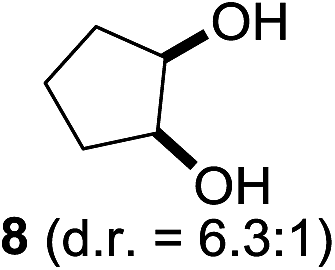
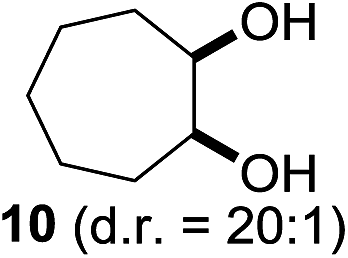
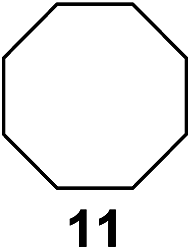
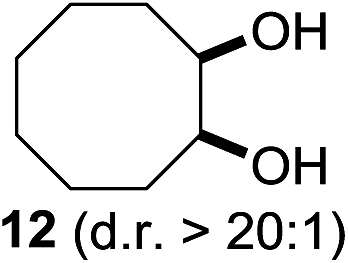
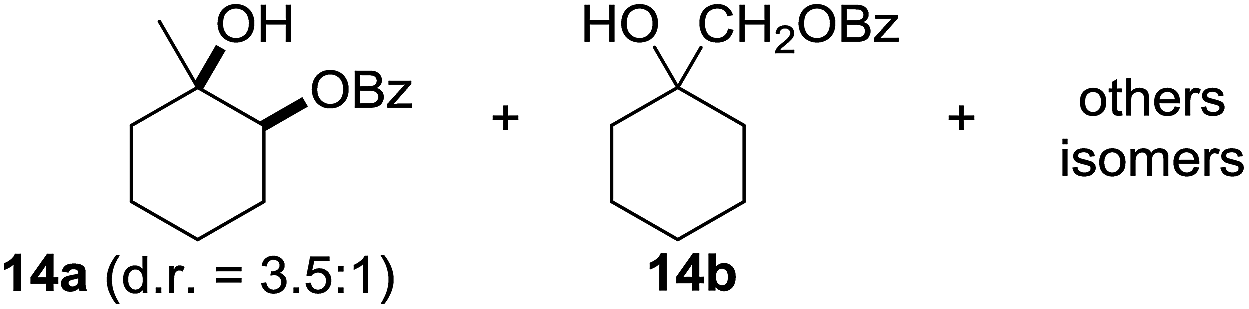
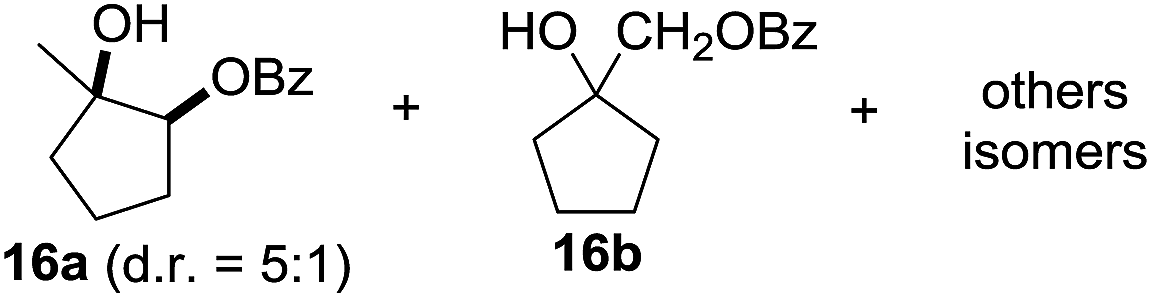
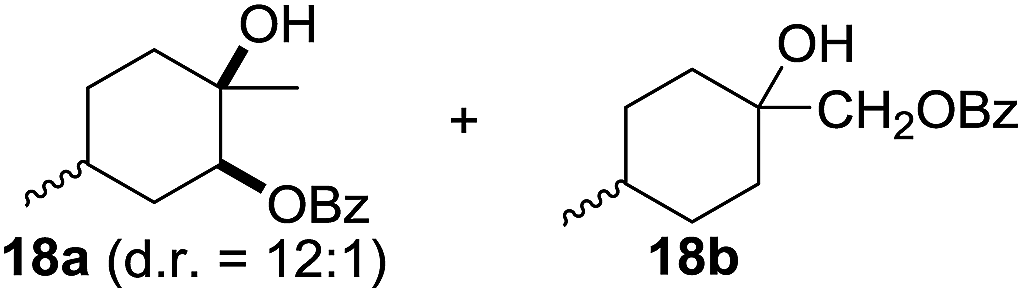
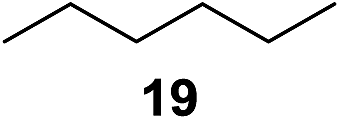
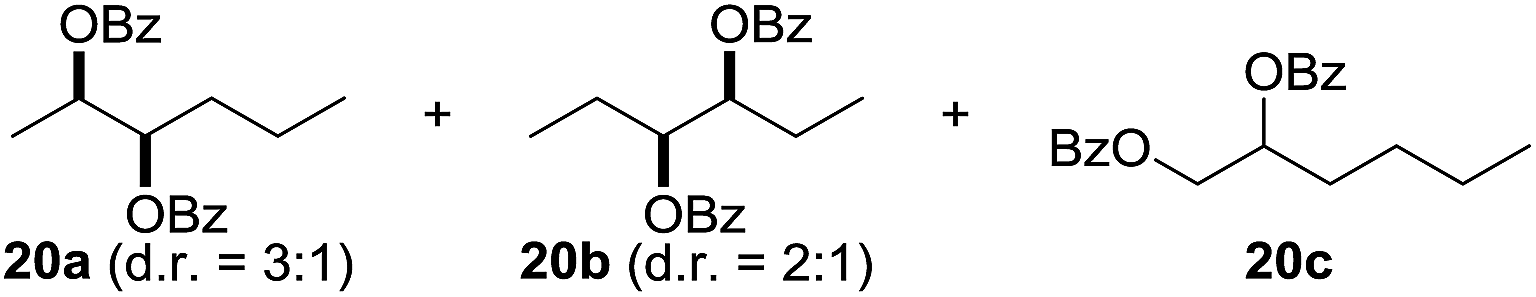
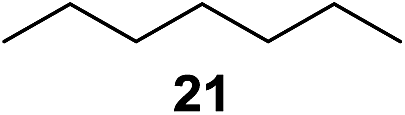
In 2002 Barluenga and co-workers reported the metal-free functionalization of cyclohexane (1) using phenyliodine diacetate (PIDA), I2 and tBuOH (Scheme 2).9 The reaction proceeds via the formation of iodocyclohexane (2), further oxidation and the trans-addition of acetylhypoiodide (AcOI) to give 2-iodohexyl acetate (4). Critical for the transformation is the generation of an alkene intermediate through the elimination of AcOI. The same course of reaction was proposed by Sudalai et al.10 Nevertheless, a high excess of alkane (200 equiv.) and elevated temperatures were required in both cases to achieve products. In the course of our studies exploring radical reactions and the functionalization of simple alkanes mediated by hypervalent iodine(III) reagents, we considered the transient formation of iodoalkanes and further oxidation in a cascade reaction fashion as a promising approach for the synthesis of diols from saturated hydrocarbons.11 During our primary screening we found that, beside known hypervalent iodine based methods, the PIDA-I2-NaN3 system enabled radical iodination.9,10,12 Furthermore, we hypothesized that by combining radical iodination with an excess of oxidant, such as peracetic acid, epoxidation of the generated alkene would take place and subsequent ring opening would lead to the formation of trans-2-hydroxycyclohexyl acetate. However, during the extensive screening, we found that the trans-addition of AcOI cannot be bypassed, but it is possible to accomplish an additional oxidation step of 2-iodocyclohexyl acetate (4) (Scheme 2). The formation of mono-acetylated diol 5 proceeds in this case via the oxidative nucleophilic substitution of iodine.13 We were pleased to find that the iodination and the sequence of oxidations could be performed as a one-pot reaction. Through hydrolysis of the crude product, cis-cyclohexane-1,2-diol (6) was obtained in 72% yield with a relative diastereoselectivity of 5.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). The systematic optimization was started by screening different solvents, but no improvement was found (Table 1, entries 2–6 and see the ESI†). The usage of AcOH was convenient, since AcO2H is a solution in AcOH. Among all of the tested oxidants, solely the use of peracetic acid yielded product 6 and an optimum was found when using 12.5 equiv. of oxidant (Table 1, entries 7–10 and see the ESI†). KI and NIS were examined as alternative iodine sources, but they yielded only traces of product (see the ESI†). Significant improvements were achieved during the screening of aryl iodides (Table 1, entries 11–16 and see the ESI†). 4-Iodotoluene yielded diol 6 in 99% yield with respect to the aryl iodide and slightly improved the diastereoselectivity (up to 6.5:1). Further screening of the additives revealed the substoichiometric use of iodine to be most advantageous (see the ESI†). High atom efficiency is a typical feature of iodination via radical activation.9,12 In contrast, the increased loading of I2 lowered product formation. We assume that unwanted radical scavenging disturbed the propagation of the reaction. Next, different azides were tested, but the initially used 2.5 equiv. of NaN3 yielded the highest product formation (see the ESI†). Finally, we stressed our novel system by reducing the amount of alkane as much as possible. Unchanged product formation was still observed when 8.5 equiv. of cyclohexane was added to the reaction (Table 1, entries 17–19 and see the ESI†). Nevertheless, the application of 8.5 equiv. of cyclohexane is one of the lowest reported loadings for the direct hydroxylation of simple alkanes. The obtained diol 6 is a valuable precursor for the production of adipic acid which is one of the most important chemical intermediates in industry.14 Having the optimized conditions in hand, we started to explore the scope of this novel method for transition-metal-free dihydroxylation alkanes by testing cyclic, linear and branched saturated hydrocarbons at a higher scale (Table 2). Gratifyingly, by varying the ring size to 5, 7 and 8 carbons the desired products were obtained in good to moderate yields. Comparable to cyclohexane-1,2-diol (6), cis-cyclopentane-1,2-diol (8) was formed in 86% yield and with a d.r. of 6.3:1. Significant improvement of diastereoselectivity was observed with the increasing ring size of the hydrocarbons. Only trace amounts of the trans-diastereomer of diol 10 were detected and cyclooctane (11) yielded exclusively the cis-isomer. Interestingly, the reaction conditions developed by Barluenga and co-workers were used to convert cycloheptane (9) into 2-iodo-1-methylcyclohexyl acetate, while our developed reaction conditions smoothly formed the desired diol without ring contraction.9,12 Furthermore, the results suggest that the relative stereochemistry is influenced by the neighboring group effect of the acetate group during the oxidative displacement of iodine.13c We assume that this effect becomes less dominant with increasing ring size. Next, tertiary (3°) carbon containing alkanes were tested. Although iodination predominantly occurred on the 3° position, minor functionalization of secondary (2°) positions occurred as well. The straightforward benzoylation of diols allowed the isolation of 14a and b and 16a and b in good yields (Table 2, entry 5 and 6) and revealed, that the functionalization of the 3° position proceeded with a high regioselectivity. A remarkable improvement regarding the selectivity of the radical iodination was observed when using 1,4-dimethylcyclohexane (17). Offering two 3° carbon atoms for iodination reduced the probability of the functionalization of the 2° C–H position and only traces of product 18b were formed, while 18a was isolated in good yields and excellent regioselectivity (Table 2, entry 7). The dihydroxylation of linear alkanes was realized in good yields as well (Table 2, entries 8 and 9). The benzoylation of 2° hydroxyl groups was applied to facilitate the handling of volatile compounds. Major products were identified as a result of the iodination of 2° carbon position and further transformation to give 20a and b and 22a and b. Although oxidation predominantly occurred at 2° carbon atoms, minor product formation as a result of 1° C–H bond iodination or elimination to a terminal double bond was observed (20c and 22c). Finally, branched alkanes containing 3° carbon atoms were tested in the reaction. 3-Methylpentane (23) yielded product 24 in 55% yield and demonstrated regioselectivity in a comparable manner to the previous examples. Elimination of oxidized iodine to the thermodynamically more stable double bond had a strong influence on the distribution of regioisomers. Interestingly, due to steric hindrance, product 26 revealed an opposite ratio of regioisomer formation, since dihydroxylation of a terminal double bond took preferential place. Although in the absence of 3° positions C–H functionalization via iodination resulted in the formation of regioisomers, it should be noted that among all possible oxidation products exclusively vicinal oxidation was observed and moreover overoxidation to ketones was not detected. Attempts to use aliphatic ethers, esters or carboxylic acids remained unsuccessful under the developed conditions.
:1 (Table 1, entry 1). The systematic optimization was started by screening different solvents, but no improvement was found (Table 1, entries 2–6 and see the ESI†). The usage of AcOH was convenient, since AcO2H is a solution in AcOH. Among all of the tested oxidants, solely the use of peracetic acid yielded product 6 and an optimum was found when using 12.5 equiv. of oxidant (Table 1, entries 7–10 and see the ESI†). KI and NIS were examined as alternative iodine sources, but they yielded only traces of product (see the ESI†). Significant improvements were achieved during the screening of aryl iodides (Table 1, entries 11–16 and see the ESI†). 4-Iodotoluene yielded diol 6 in 99% yield with respect to the aryl iodide and slightly improved the diastereoselectivity (up to 6.5:1). Further screening of the additives revealed the substoichiometric use of iodine to be most advantageous (see the ESI†). High atom efficiency is a typical feature of iodination via radical activation.9,12 In contrast, the increased loading of I2 lowered product formation. We assume that unwanted radical scavenging disturbed the propagation of the reaction. Next, different azides were tested, but the initially used 2.5 equiv. of NaN3 yielded the highest product formation (see the ESI†). Finally, we stressed our novel system by reducing the amount of alkane as much as possible. Unchanged product formation was still observed when 8.5 equiv. of cyclohexane was added to the reaction (Table 1, entries 17–19 and see the ESI†). Nevertheless, the application of 8.5 equiv. of cyclohexane is one of the lowest reported loadings for the direct hydroxylation of simple alkanes. The obtained diol 6 is a valuable precursor for the production of adipic acid which is one of the most important chemical intermediates in industry.14 Having the optimized conditions in hand, we started to explore the scope of this novel method for transition-metal-free dihydroxylation alkanes by testing cyclic, linear and branched saturated hydrocarbons at a higher scale (Table 2). Gratifyingly, by varying the ring size to 5, 7 and 8 carbons the desired products were obtained in good to moderate yields. Comparable to cyclohexane-1,2-diol (6), cis-cyclopentane-1,2-diol (8) was formed in 86% yield and with a d.r. of 6.3:1. Significant improvement of diastereoselectivity was observed with the increasing ring size of the hydrocarbons. Only trace amounts of the trans-diastereomer of diol 10 were detected and cyclooctane (11) yielded exclusively the cis-isomer. Interestingly, the reaction conditions developed by Barluenga and co-workers were used to convert cycloheptane (9) into 2-iodo-1-methylcyclohexyl acetate, while our developed reaction conditions smoothly formed the desired diol without ring contraction.9,12 Furthermore, the results suggest that the relative stereochemistry is influenced by the neighboring group effect of the acetate group during the oxidative displacement of iodine.13c We assume that this effect becomes less dominant with increasing ring size. Next, tertiary (3°) carbon containing alkanes were tested. Although iodination predominantly occurred on the 3° position, minor functionalization of secondary (2°) positions occurred as well. The straightforward benzoylation of diols allowed the isolation of 14a and b and 16a and b in good yields (Table 2, entry 5 and 6) and revealed, that the functionalization of the 3° position proceeded with a high regioselectivity. A remarkable improvement regarding the selectivity of the radical iodination was observed when using 1,4-dimethylcyclohexane (17). Offering two 3° carbon atoms for iodination reduced the probability of the functionalization of the 2° C–H position and only traces of product 18b were formed, while 18a was isolated in good yields and excellent regioselectivity (Table 2, entry 7). The dihydroxylation of linear alkanes was realized in good yields as well (Table 2, entries 8 and 9). The benzoylation of 2° hydroxyl groups was applied to facilitate the handling of volatile compounds. Major products were identified as a result of the iodination of 2° carbon position and further transformation to give 20a and b and 22a and b. Although oxidation predominantly occurred at 2° carbon atoms, minor product formation as a result of 1° C–H bond iodination or elimination to a terminal double bond was observed (20c and 22c). Finally, branched alkanes containing 3° carbon atoms were tested in the reaction. 3-Methylpentane (23) yielded product 24 in 55% yield and demonstrated regioselectivity in a comparable manner to the previous examples. Elimination of oxidized iodine to the thermodynamically more stable double bond had a strong influence on the distribution of regioisomers. Interestingly, due to steric hindrance, product 26 revealed an opposite ratio of regioisomer formation, since dihydroxylation of a terminal double bond took preferential place. Although in the absence of 3° positions C–H functionalization via iodination resulted in the formation of regioisomers, it should be noted that among all possible oxidation products exclusively vicinal oxidation was observed and moreover overoxidation to ketones was not detected. Attempts to use aliphatic ethers, esters or carboxylic acids remained unsuccessful under the developed conditions.
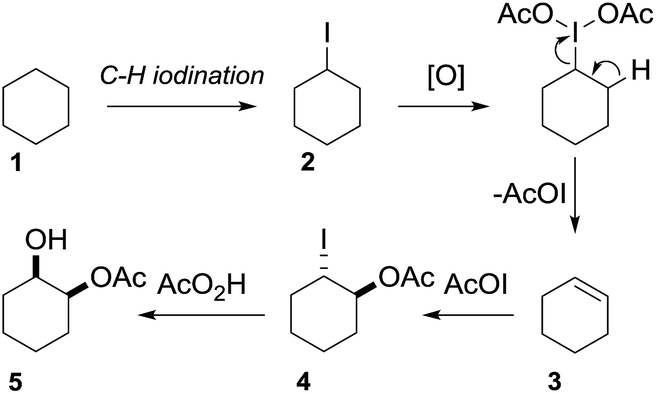 | ||
| Scheme 2 Proposal for the vicinal dihydroxylation. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (equiv.) | ArI or ArI(III) | Solvent | Yieldb (%) | d.r.c |
| a Reaction conditions: (1) ArI or ArI(III) (0.6 mmol, 1 equiv.), oxidant (see table), cyclohexane (12.5 equiv.), I2 (0.8 equiv.), NaN3 (2.5 equiv.), AcOH (0.1 M), rt, 24 h; (2) LiOH (2 equiv.), MeOH (0.2 M), rt. b Yields are given for isolated products after column chromatography. Calculated based on ArI or ArI(III). c Diastereomeric ratio (d.r.) according to 1H-NMR. d 8.5 equiv. e 6.25 equiv. of cyclohexane. f 1 equiv. of cyclohexane. Abbreviations: n.d. = not detected, w/o = without, HFIP = hexafluoroisopropanol, PIFA = phenyliodine bis(trifluoroacetate), DIBA = 2,2′-diiodo-4,4′,6,6′-tetra-methylbiphenyl, IBA = 2-iodobenzoic acid, n.c. = not calculated. | |||||
| 1 | AcO2H (16.5) | PIDA | AcOH | 72 | 5.3:1 |
| 2 | AcO2H (16.5) | PIDA | HCO2H | 32 | 7:1 |
| 3 | AcO2H (16.5) | PIDA | HFIP | 18 | 1.5:1 |
| 4 | AcO2H (16.5) | PIDA | CH3CN | 16 | 1.5:1 |
| 5 | AcO2H (16.5) | PIDA | CH2Cl2 | 20 | 1.5:1 |
| 6 | AcO2H (16.5) | PIDA | w/o | 17 | 4.3:1 |
| 7 | H2O2 (16.5) | PIDA | AcOH | n.d. | — |
| 8 | TBHP (16.5) | PIDA | AcOH | n.d. | — |
| 9 | mCPBA (16.5) | PIDA | AcOH | n.d. | — |
| 10 | Na2S2O8 (16.5) | PIDA | AcOH | Traces | — |
| 11 | AcO2H (12.5) | PIFA | AcOH | 53 | 5:1 |
| 12 | AcO2H (12.5) | DIBA | AcOH | 97 | 5:1 |
| 13 | AcO2H (12.5) | 4-MeC6H4I | AcOH | 99 | 6:1 |
| 14 | AcO2H (12.5) | 4-FC6H4I | AcOH | 79 | 6.3:1 |
| 15 | AcO2H (12.5) | 2-MeC6H4I | AcOH | 53 | 5:1 |
| 16 | AcO2H (12.5) | IBA | AcOH | 8 | 10:1 |
| 17d | AcO2H (12.5) | 4-MeC6H4I | AcOH | 99 | 6.5:1 |
| 18e | AcO2H (12.5) | 4-MeC6H4I | AcOH | 86 | 6:1 |
| 19f | AcO2H (12.5) | 4-MeC6H4I | AcOH | 8 | n.c. |
| Entry | Substrate | Productsb | r.r.c | Yieldd (%) |
|---|---|---|---|---|
| a Reaction conditions: (1) alkane (8.5 equiv.), 4-MeC6H4I (0.3 mmol, 1 equiv.), I2 (0.8 equiv.), NaN3 (2.5 equiv.) and AcO2H (12.5 equiv.) in AcOH (0.1 M), rt, 24 h; (2) LiOH (2 equiv.), MeOH (0.1 M), rt. b Diastereomeric ratio (d.r.) according to 1H-NMR. Major isomer is shown. c Regioisomeric ratio (r.r.) according to 1H-NMR. d Yields are given for isolated products. Calculated based on 4-MeC6H4I. e Scaled to 0.9 mmol of 4-MeC6H4I. f 3.5 equiv. of NaN3. g BzCl (2.5 equiv.) and DMAP (5 mol%) in DCM/Py (0.45 M). h Yield of isolated regioisomers 14a and 14b. i Yield of isolated regioisomers 16a and 16b. | ||||
| 1 |
|
|
— | 99 |
| 2 |
|
|
— | 86 |
| 3 |
|
|
— | 85 |
| 4 |
|
|
— | 37 |
| 5e,f,g |
|
|
21:1:7 |
90 (68h) |
| 6e,f,g |
|
|
18.5:1:5 |
91 (73i) |
| 7e,f,g |
|
|
>20:1 |
86 |
| 8e,g |
|
|
6.5:1.5:1 |
68 |
| 9e,g |
|
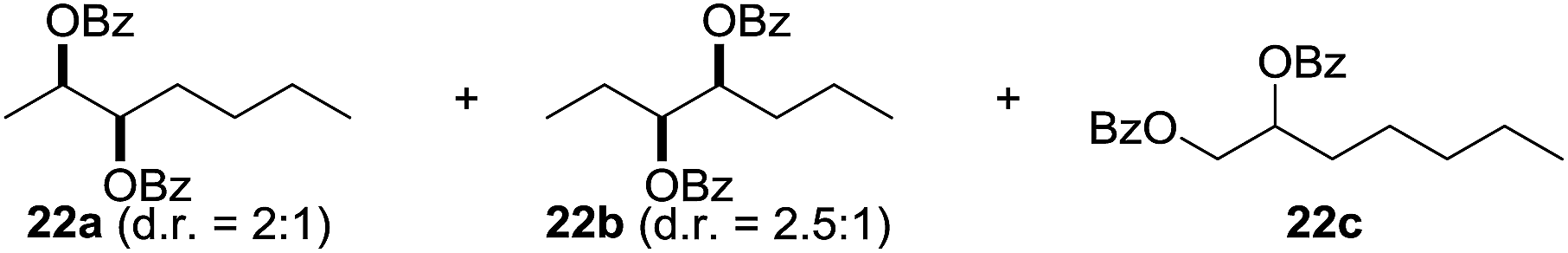
|
5.5:2:1 |
73 |
| 10e,f,g |
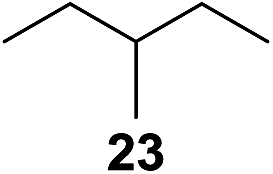
|
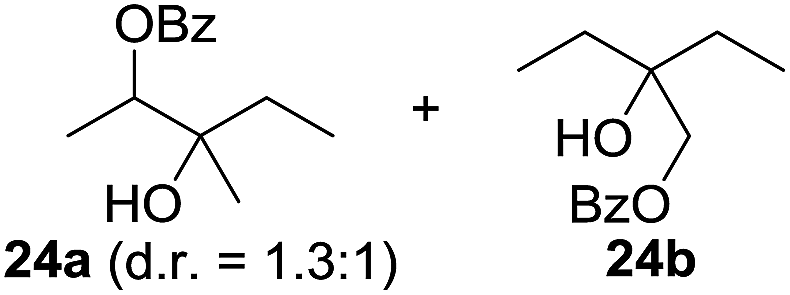
|
7.7:1 |
55 |
| 11e,f,g |
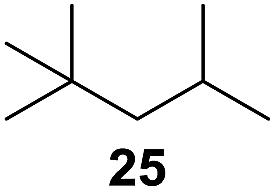
|
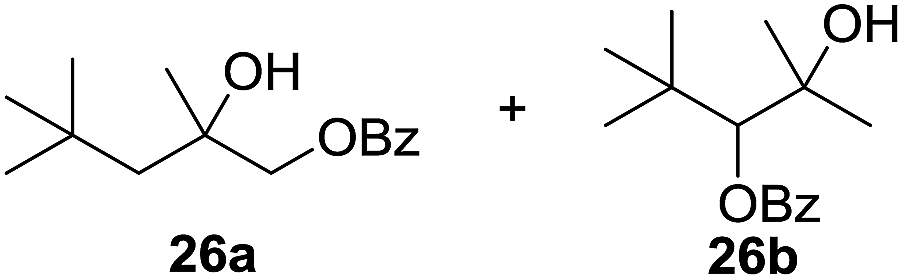
|
8:1 |
20 |
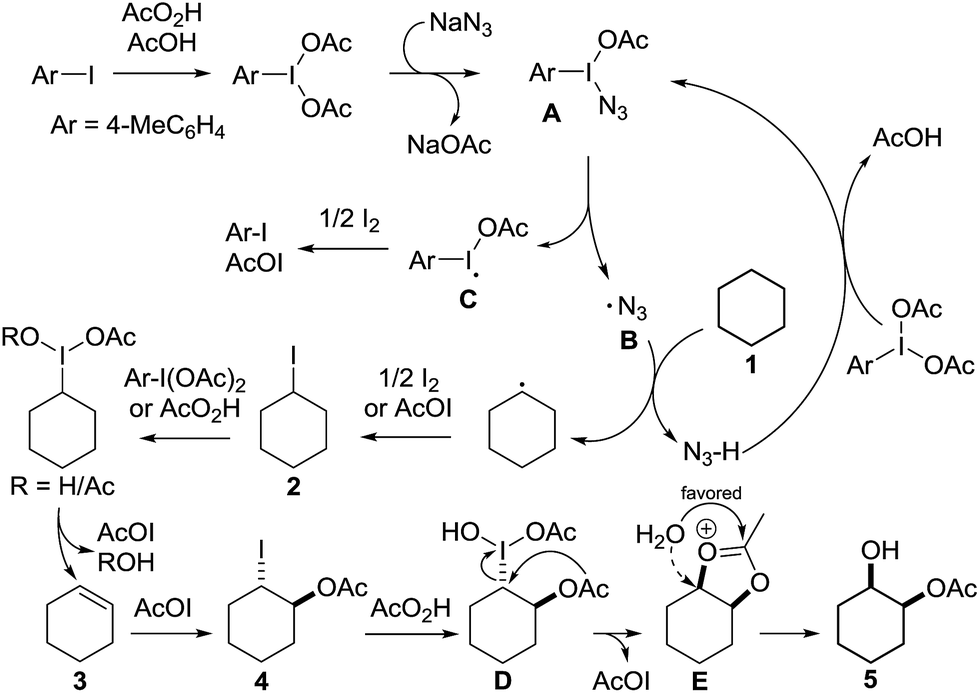
Having established the scope of the vicinal dihydroxylation using saturated hydrocarbons, the reaction mechanism was studied. By omitting the aryl iodide, iodine or sodium azide no product formation took place. The application of iodocyclohexane instead of cyclohexane smoothly yielded the expected product which was isolated in 98% yield (see ESI†). Additionally, we were able to study the reaction profile by means of GC-MS. The monitoring of the important intermediates 2 and 4 and product 5 formation was possible (see the ESI† and Scheme 3). A significant kinetic deuterium isotope effect was observed (KIE = 7.4), when using d12-cyclohexane and 1 (see the ESI†). Consequently, the abstraction of hydrogen from cyclohexane is the rate-limiting step. A proposed reaction mechanism is outlined in Scheme 3. Initially 4-iodotoluene is oxidized by peracetic acid in the presence of acetic acid. Ligand exchange with NaN3 leads to intermediate A, which undergoes thermolysis at ambient temperature to give an azide radical (B) and an iodine centred radical (C).11aC is scavenged by iodine whereupon AcOI and 4-iodotoluene are formed. The azide radical reacts with 1 providing a cyclohexyl radical. This transformation is the rate limiting step of the developed cascade reaction. In the following step, the cyclohexyl radical is trapped by iodine or AcOI to provide iodocyclohexane (2). The principal difference compared to the reaction conditions developed by Barluenga and co-workers9 is the application of sodium azide instead of tBuOH.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
The use of azide radicals allows a dramatically reduced loading of alkanes and enables iodination at ambient temperature, which makes the formation of iodoalkanes more efficient. The formation of IN3 as an intermediate10 was excluded, since the formation of 1-azido-2-iodocyclohexane or 2-azidocyclohexyl acetate was not observed under the developed reaction conditions. In control experiments iodocyclohexane (2) was converted to product 5 under the developed reaction conditions (see the ESI†). Next, iodocyclohexane (2) is oxidized and the reductive elimination of AcOI leads to cyclohexene (3). The subsequent trans-addition of AcOI to cyclohexene (3) provides 4. Further oxidation of 4 by peracetic acid and nucleophilic substitution gives product 5.13c The stereochemical outcome can be reasoned through a competition between a Woodward- and Prévost-type reaction.13c,15 The Woodward-type reaction was favoured for cyclic alkanes. It is important to note that under the developed reaction conditions epoxidation of cyclohexene by peracetic acid (3) does not occur. A control experiment revealed the selective formation of trans-cyclohexane-1,2-diol after hydrolysis upon oxidation of cyclohexene (3) with peracetic acid (see the ESI†). This finding highlights the importance of AcOI which is formed in situ.
The application of C–H bond functionalization for the selective derivatization of complex molecules has gained great interest and offers unique advantages in terms of efficiency and atom economy.16 However, the required excess of starting material in this protocol negotiates these advantages. Based on the results obtained during the studies on the reaction mechanism, we envisioned that the use of iodoalkanes offers the opportunity to selectively introduce vicinal diols into more complex substrates by mimicking the developed reaction conditions. In this process radical iodination is excluded and the iodine atom in alkyl iodide plays the role of a traceless directing group for metal-free dihydroxylation. According to our proposal, ester 27 yielded the dioxygenated product 28 in a good yield using 1 equiv. of starting material (Scheme 4). Furthermore, the functionalization of complex molecules was tackled by using cholestane derivative 29. The dihydroxylated products were isolated with good cis selectivity after hydrolysis in 40% yield (Scheme 4). It is notable that 29 contains 7 weak tertiary C–H bonds, which were untouched under the applied reaction conditions. It must be noted, that common reaction routes include multiple reaction steps and the use of expensive and toxic osmium catalysts or are mediated by metal reagents.17
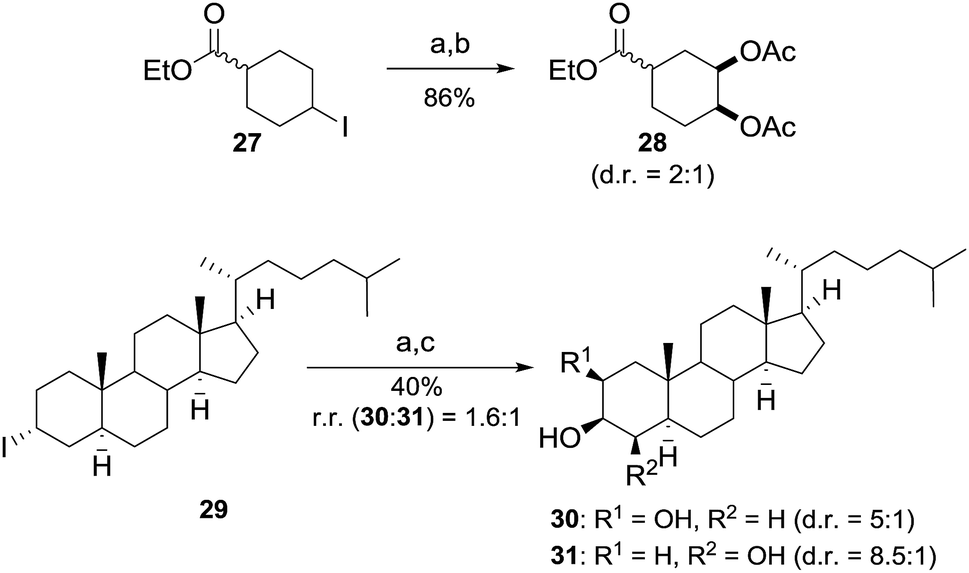 | ||
| Scheme 4 Dihydroxylation of iodoalkanes. Reaction conditions: a iodoalkane (1 equiv., 0.5 mmol), 4-MeC6H4I (0.6 equiv.), AcO2H (7.5 equiv.) in AcOH (0.3 M), rt, 24 h; b Ac2O (3 equiv.), DMAP (5 mol%) in DCM/Py (0.45 M); c LiOH (2 equiv.), MeOH (0.1 M), rt. | ||
Conclusions
We have developed the first efficient and scalable method for the transition-metal-free double C(sp3)–H bond functionalization of saturated hydrocarbons. Cyclic, linear and branched alkanes were converted selectively to vicinal diols under ambient reaction conditions. Furthermore, we demonstrated for the first time the use of the iodine atom from alkyl iodides as a traceless directing group for metal-free dihydroxylation. Our discovery is grounded in an unprecedented cascade reaction.Experimental
Full synthetic characterization, general experimental procedures and detailed optimization and mechanistic studies are provided in the ESI.†Acknowledgements
We gratefully acknowledge Prof. Dr H. Waldmann for his generous support. This work was supported by the Verband der Chemischen Industrie (FCI Fellowship to L. Bering).Notes and references
- (a) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed; (c) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (d) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- (a) M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712 CrossRef CAS PubMed; (b) G. C. Fortman, N. C. Boaz, D. Munz, M. M. Konnick, R. A. Periana, J. T. Groves and T. B. Gunnoe, J. Am. Chem. Soc., 2014, 136, 8393 CrossRef CAS PubMed; (c) R. A. Periana, G. Bhalla, W. J. Tenn Iii, K. J. H. Young, X. Y. Liu, O. Mironov, C. J. Jones and V. R. Ziatdinov, J. Mol. Catal. A: Chem., 2004, 220, 7 CrossRef CAS; (d) V. N. Cavaliere, B. F. Wicker and D. J. Mindiola, in Adv. Organomet. Chem., Academic Press, 2012, vol. 60 Search PubMed; (e) J. A. Labinger, J. Mol. Catal. A: Chem., 2004, 220, 27 CrossRef CAS; (f) S. A. Vladimir and I. V. Vladimir, Russ. Chem. Rev., 1991, 60, 1384 CrossRef.
- (a) G. A. Russell and H. C. Brown, J. Am. Chem. Soc., 1955, 77, 4578 CrossRef CAS; (b) A. Demonceau, A. F. Noels, A. J. Anciaux, A. J. Hubert and P. Teyssié, Bull. Soc. Chim. Belg., 1984, 93, 949 CrossRef CAS; (c) R. H. Crabtree, J. M. Mihelcic and J. M. Quirk, J. Am. Chem. Soc., 1979, 101, 7738 CrossRef CAS; (d) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS; (e) J. T. Groves, T. E. Nemo and R. S. Myers, J. Am. Chem. Soc., 1979, 101, 1032 CrossRef CAS.
- (a) J. E. Bäckvall, in Modern Oxidation Methods, John Wiley & Sons, 2011 Search PubMed; (b) H. C. Kolb and K. B. Sharpless, in Transition Metals for Organic Synthesis, Wiley-VCH Verlag GmbH, 2008 Search PubMed; (c) A. B. Zaitsev and H. Adolfsson, Synthesis, 2006, 11, 1725 Search PubMed; (d) U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1 CrossRef CAS.
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (b) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS PubMed; (c) A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352 CrossRef CAS; (d) J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3723 CrossRef CAS; (e) B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391 CrossRef CAS PubMed; (f) B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS; (g) W. A. Duetz, J. B. Van Beilen and B. Witholt, Curr. Opin. Biotechnol., 2001, 12, 419 CrossRef CAS PubMed; (h) J. Buddrus and H. Plettenberg, Angew. Chem., Int. Ed., 1976, 15, 436 CrossRef; (i) M. M. Konnick, B. G. Hashiguchi, D. Devarajan, N. C. Boaz, T. B. Gunnoe, J. T. Groves, N. Gunsalus, D. H. Ess and R. A. Periana, Angew. Chem., Int. Ed., 2014, 53, 10490 CrossRef CAS PubMed; (j) B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232 CrossRef CAS PubMed.
- (a) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052 CrossRef CAS PubMed; (b) M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216 CrossRef CAS PubMed; (c) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed; (d) D. H. R. Barton, Tetrahedron, 1998, 54, 5805 CrossRef CAS; (e) D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504 CrossRef CAS; (f) D. H. R. Barton, M. J. Gastiger and W. B. Motherwell, Chem. Commun., 1983, 41 RSC.
- (a) R. Mello, M. Fiorentino, C. Fusco and R. Curci, J. Am. Chem. Soc., 1989, 111, 6749 CrossRef CAS; (b) C. J. Moody and J. L. O'Connell, Chem. Commun., 2000, 1311 RSC; (c) R. Curci, L. D'Accolti and C. Fusco, Acc. Chem. Res., 2006, 39, 1 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. S. Van Nieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (c) S. Haubenreisser, T. H. Wöste, C. Martínez, K. Ishihara and K. Muñiz, Angew. Chem., Int. Ed., 2016, 55, 413 CrossRef CAS PubMed; (d) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS PubMed; (e) N. Ghavtadze, F. S. Melkonyan, A. V. Gulevich, C. Huang and V. Gevorgyan, Nat. Chem., 2014, 6, 122 CrossRef CAS PubMed; (f) Y. Usui, K. Sato and M. Tanaka, Angew. Chem., Int. Ed., 2003, 42, 5623 CrossRef CAS PubMed; (g) Y.-B. Kang and L. H. Gade, J. Am. Chem. Soc., 2011, 133, 3658 CrossRef CAS PubMed; (h) B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320 CrossRef CAS PubMed; (i) L. Emmanuvel, T. M. A. Shaikh and A. Sudalai, Org. Lett., 2005, 7, 5071 CrossRef CAS PubMed; (j) S. Picon, M. Rawling, M. Campbell and N. C. O. Tomkinson, Org. Lett., 2012, 14, 6250 CrossRef CAS PubMed.
- J. Barluenga, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2002, 41, 2556 CrossRef CAS.
- P. V. Chouthaiwale, G. Suryavanshi and A. Sudalai, Tetrahedron Lett., 2008, 49, 6401 CrossRef CAS.
- (a) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed; (b) K. Matcha, R. Narayan and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 7985 CrossRef CAS PubMed; (c) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS PubMed; (d) R. Narayan and A. P. Antonchick, Chem. Eur. J., 2014, 20, 4568 CrossRef CAS PubMed.
- (a) D. D. Tanner and G. C. Gidley, J. Am. Chem. Soc., 1968, 90, 808 CrossRef CAS; (b) P. R. Schreiner, O. Lauenstein, E. D. Butova and A. A. Fokin, Angew. Chem., Int. Ed., 1999, 38, 2786 CrossRef CAS; (c) A. A. Orlinkov, S. Vitt and A. Chistyakov, Tetrahedron Lett., 2002, 43, 1333 CrossRef; (d) R. Montoro and T. Wirth, Org. Lett., 2003, 5, 4729 CrossRef CAS PubMed; (e) J. Barluenga, E. Campos-Gómez, D. Rodríguez, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2005, 44, 5851 CrossRef CAS PubMed; (f) R. Montoro and T. Wirth, Synthesis, 2005, 9, 1473 Search PubMed.
- (a) N. S. Zefirov, V. V. Zhdankin, G. V. Makhon'kova, Y. V. Dan'kov and A. S. Koz'min, J. Org. Chem., 1985, 50, 1872 CrossRef CAS; (b) R. I. Davidson and P. J. Kropp, J. Org. Chem., 1982, 47, 1904 CrossRef CAS; (c) R. C. Cambie, D. Chambers, B. G. Lindsay, P. S. Rutledge and P. D. Woodgate, J. Chem. Soc., Perkin Trans. 1, 1980, 822 RSC; (d) T. L. Macdonald, N. Narasimhan and L. T. Burka, J. Am. Chem. Soc., 1980, 102, 7760 CrossRef CAS; (e) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed.
- K. C. H. Wang and A. Sagadevan, Science, 2014, 346, 1495 CrossRef PubMed.
- T. H. Wöste and K. Muñiz, Synthesis, 2016, 48, 816 CrossRef.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (c) J. Yamaguchi, K. Itami and A. D. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- (a) M. M. C. Silva, S. S. Riva and M. L. Sá e Melo, Tetrahedron, 2005, 61, 3065 CrossRef; (b) F. D'Onofrio and A. Scettri, Synthesis, 1985, 12, 1159 CrossRef; (c) G. A. G. Santos, A. P. Murray, C. A. Pujol, E. B. Damonte and M. S. Maier, Steroids, 2003, 68, 125 CrossRef CAS PubMed; (d) B. S. Jursic, S. K. Upadhyay, C. C. Creech and D. M. Neumann, Bioorg. Med. Chem. Lett., 2010, 20, 7372 CrossRef CAS PubMed; (e) V. Sepe, R. Ummarino, M. V. D'Auria, G. Lauro, G. Bifulco, C. D'Amore, B. Renga, S. Fiorucci and A. Zampella, Org. Biomol. Chem., 2012, 10, 6350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03055f |
| This journal is © The Royal Society of Chemistry 2017 |