
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multicomponent approach for the preparation of homoallylic alcohols†‡
Jian-Siang
Poh§
 ,
Shing-Hing
Lau§
,
Iain G.
Dykes
,
Duc N.
Tran
,
Claudio
Battilocchio
and
Steven V.
Ley
*
,
Shing-Hing
Lau§
,
Iain G.
Dykes
,
Duc N.
Tran
,
Claudio
Battilocchio
and
Steven V.
Ley
*
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk
First published on 8th July 2016
Abstract
Here we report the in situ generation of transient allylic boronic species, by reacting TMSCHN2 and E-vinyl boronic acids, followed by their subsequent trapping with aldehydes as electrophiles to yield homoallylic alcohols. This metal-free reaction was initially discovered by the use of a flow chemistry approach to generate a variety of homoallylic alcohols in a straightforward fashion and then transferred to a batch protocol.
Introduction
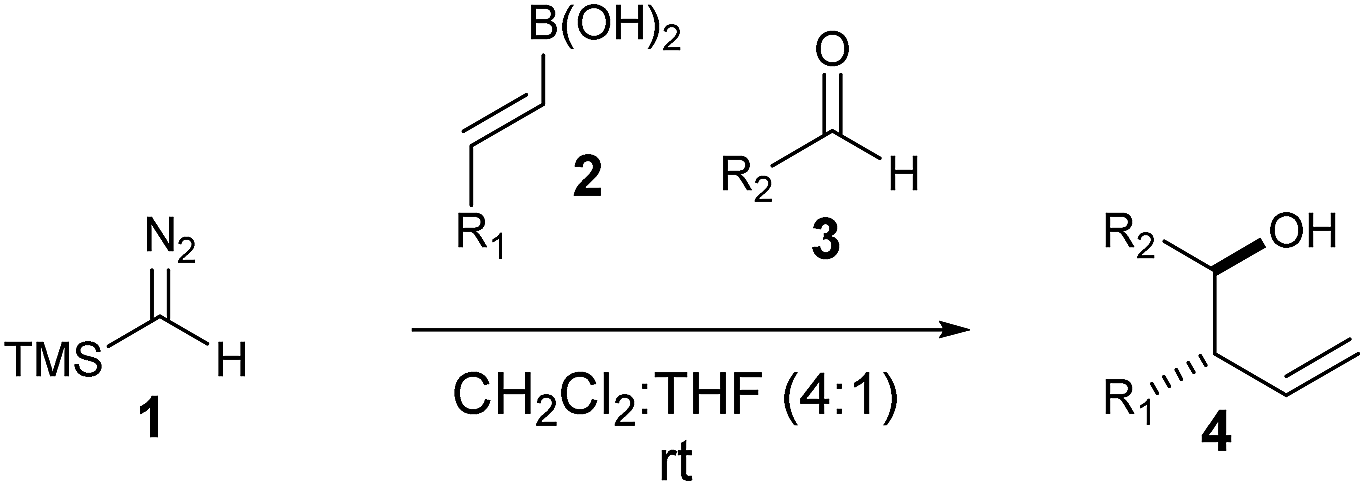
Homoallylic alcohols are valuable synthetic building blocks, in particular, for the construction of polyketide natural products.1–4 Whilst they can be accessed in a metal-free fashion by crotylation/allylation methods of Roush5–10 and Brown,11–19 the instability or availability of the appropriate allyl boronic coupling partner is an important limitation.20–22 Furthermore, installation of side chain groups other than methyl is not always straightforward using these methods. Other procedures are known but tend to lack generality.23–38 In a continuation of our work in the area (Fig. 1),39–44 we report here a new multi-component coupling to prepare homoallylic alcohols which was first discovered using flow chemistry45 methods and later transferred to a batch protocol, involving reactions of vinyl boronic acids with trimethylsilyl diazomethane (TMSCHN2)46–49 to generate the allylic boronic species in situ which are then intercepted with aldehydes to form new C–C bonds.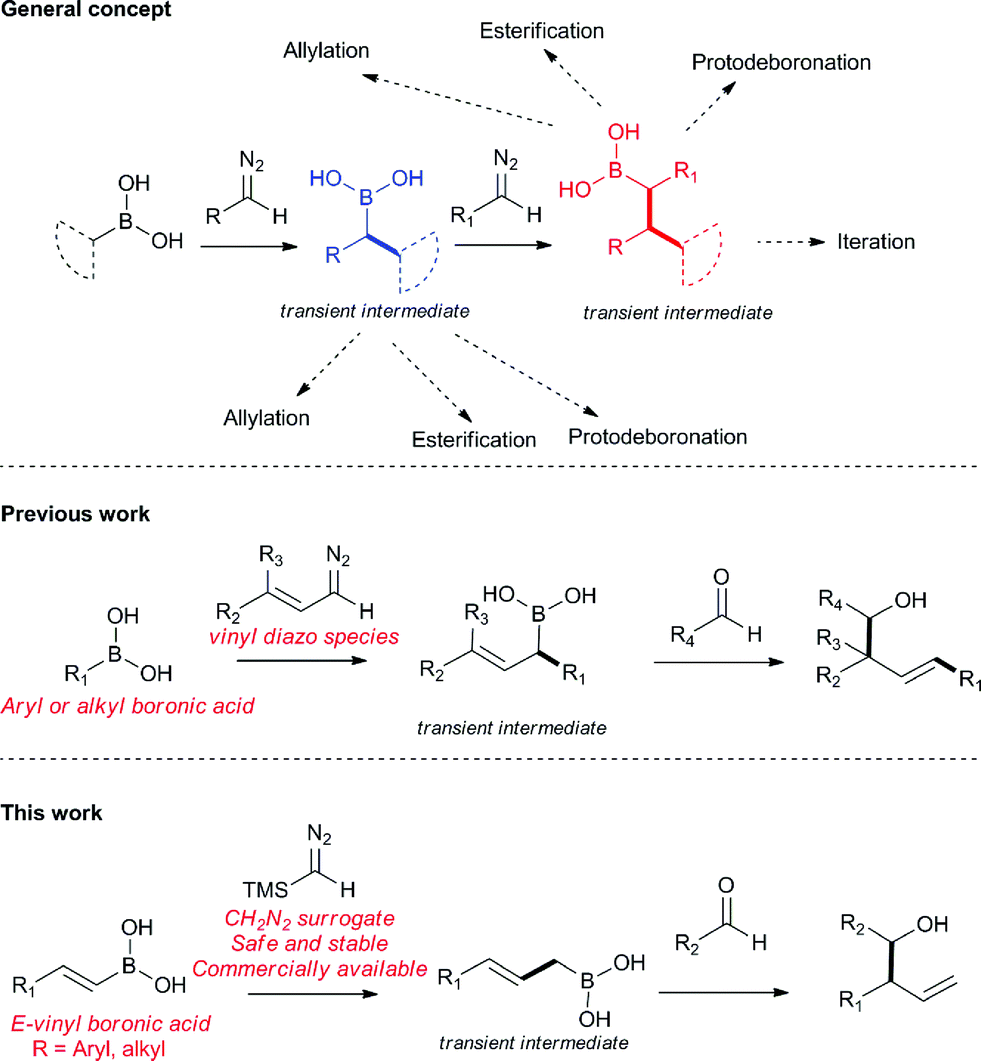 | ||
| Fig. 1 General concept for the reaction of diazo compounds and boronic acid species for the iterative C–C bond formation strategy, previous work on allylation of transient boronic acids, and current work using TMSCHN2 as commercially available diazo compounds. | ||
Results and discussion
The reaction was initially optimised with available flow equipment using TMS-diazomethane 1 (TMSCHN2), E-4-methylphenyl vinyl boronic acid 2a and 4-bromobenzaldehyde 3a as substrates (Scheme 1). Interestingly, early investigations showed the product to be a homoallylic alcohol (4a), which was obtained in good yield. We therefore initiated a rapid evaluation of the reaction parameters in order to obtain a robust system. The first attempt to define these reaction parameters employed a flow strategy whereby a solution containing the vinyl boronic acid 2a (0.04 M, in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF, 4:1 v/v) and a solution of TMSCHN21 and aldehyde 3a (0.066 M and 0.033 M, respectively, in CH2Cl2) were combined at a T-piece and reacted in a 20 mL perfluoroalkoxy alkanes (PFA) reactor coil (τ = 25 min). The exiting stream of borylated intermediate was then quenched with MeOH (2 mL), in a batch-fed integrated fashion, to yield the final product 4a. Following this approach, we were able to rapidly identify suitable conditions, with compound 4a being generated (0.29 mmol scale, 91% yield) at 60 °C. The choice of solvent was crucial as solvent systems that are able to strongly coordinate with the boronic acid partner (e.g. neat THF) can favour the formation of by-products. We were soon able to identify that a mixture of CH2Cl2 and THF (4:1 v/v) was the optimal solvent system to carry out the reaction continuously without causing any undesirable rate or selectivity issues on the reaction.
:THF, 4:1 v/v) and a solution of TMSCHN21 and aldehyde 3a (0.066 M and 0.033 M, respectively, in CH2Cl2) were combined at a T-piece and reacted in a 20 mL perfluoroalkoxy alkanes (PFA) reactor coil (τ = 25 min). The exiting stream of borylated intermediate was then quenched with MeOH (2 mL), in a batch-fed integrated fashion, to yield the final product 4a. Following this approach, we were able to rapidly identify suitable conditions, with compound 4a being generated (0.29 mmol scale, 91% yield) at 60 °C. The choice of solvent was crucial as solvent systems that are able to strongly coordinate with the boronic acid partner (e.g. neat THF) can favour the formation of by-products. We were soon able to identify that a mixture of CH2Cl2 and THF (4:1 v/v) was the optimal solvent system to carry out the reaction continuously without causing any undesirable rate or selectivity issues on the reaction.
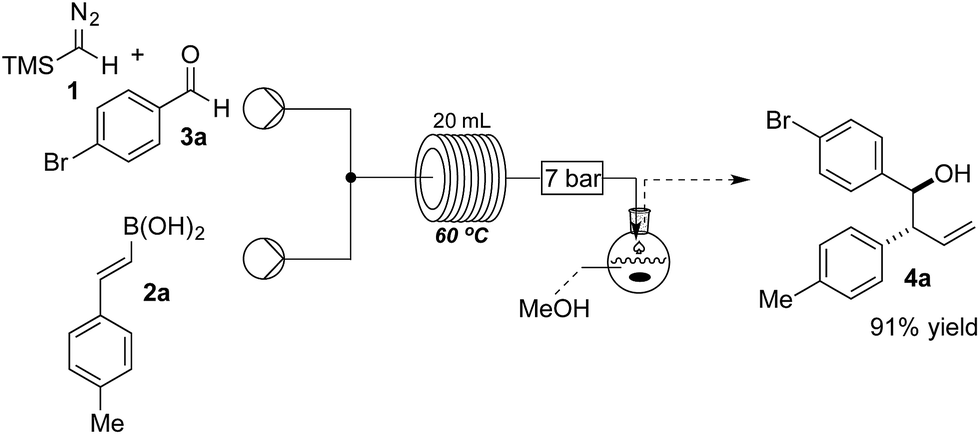 | ||
| Scheme 1 Flow set up for the continuous synthesis of homoallylic alcohols 4. | ||
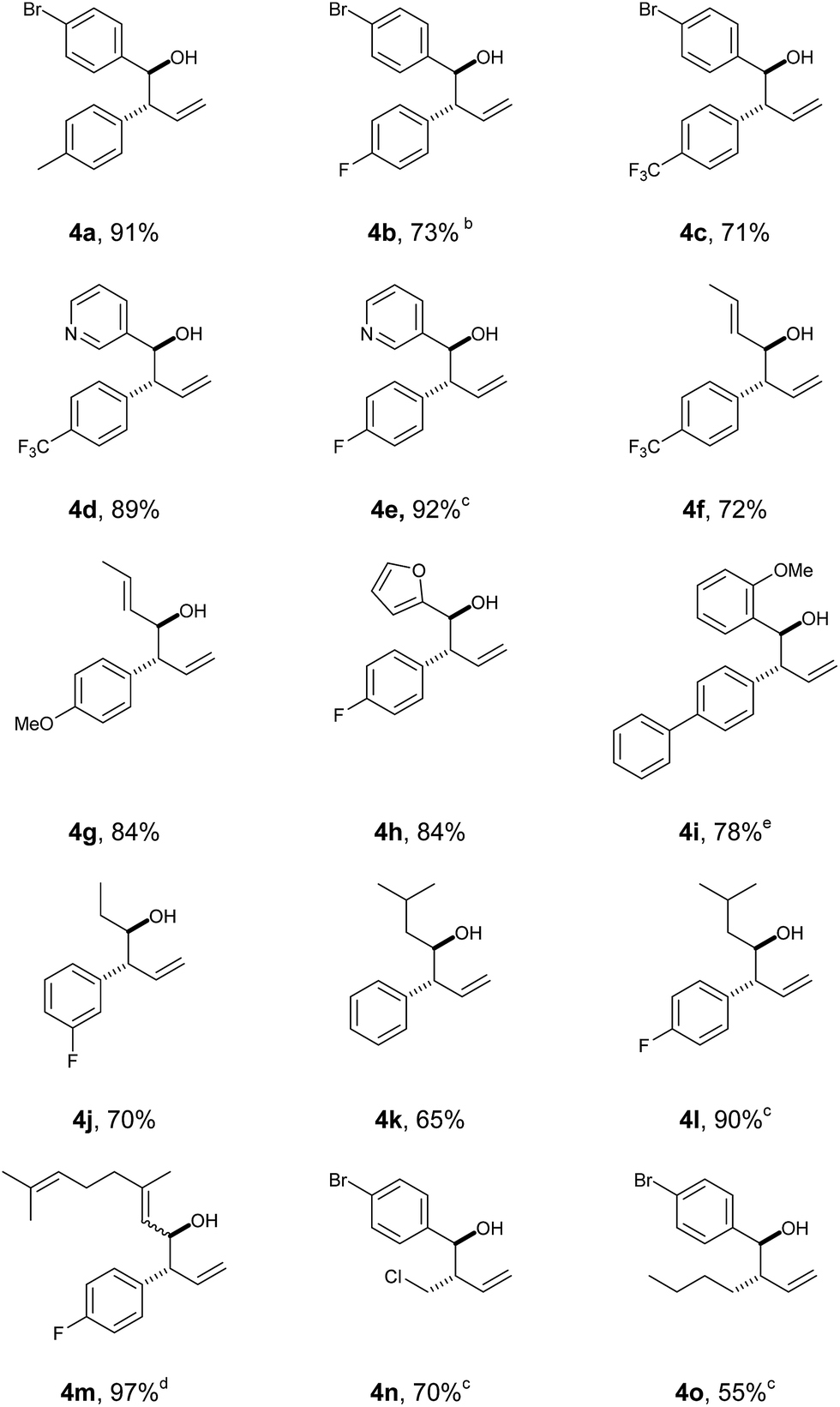
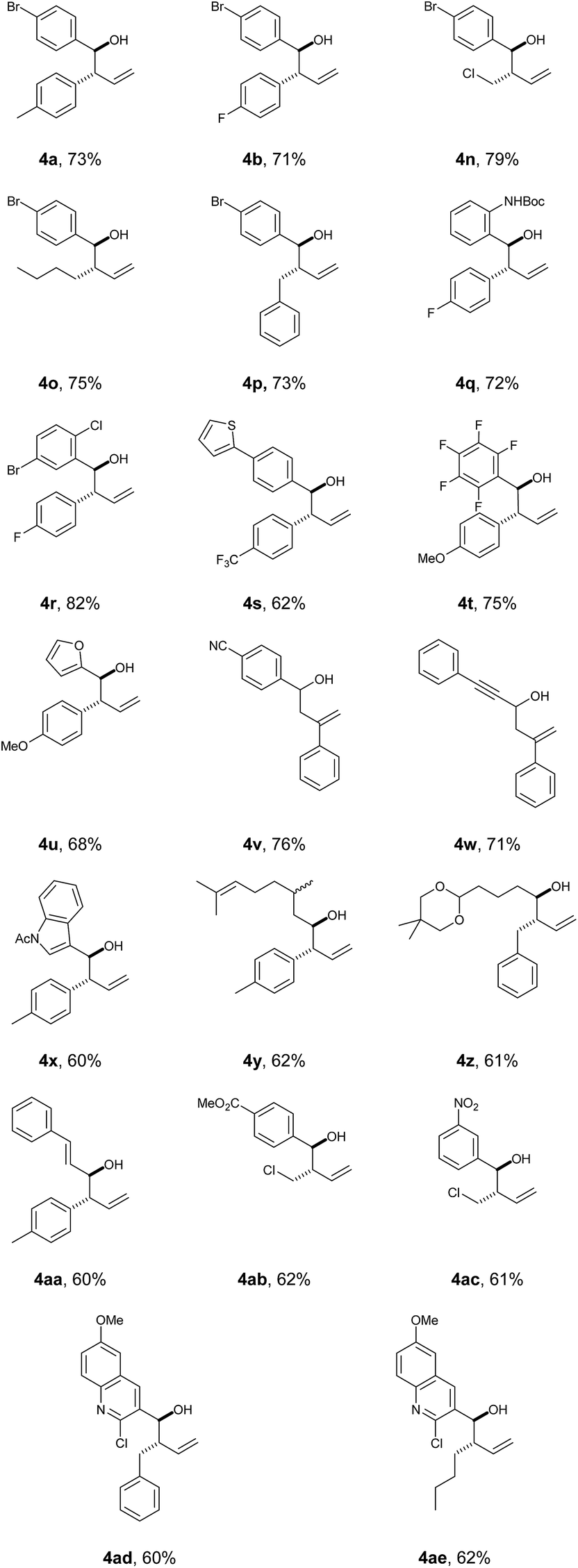
With this protocol in place, we were able to rapidly access a range of homoallylic alcohols by using various vinyl boronic acids 2 and aldehydes 3 (Table 1).
|
|
|---|
|
a Standard reaction conditions: solution of TMSCHN21 and aldehyde 3 in CH2Cl2 (0.066 M and 0.033 M, respectively), solution of vinyl boronic acid 2 in CH2Cl2:THF (4:1 v/v, 0.04 M), 60 °C, residence time 23 min (isolated yield); for more detail see ESI.
b Reaction performed on 10 mmol scale (analytic sample was collected at steady state for 340 min).
c Yield was measured using a 1H-NMR standard (1,3,5-trimethoxybenzene).
d
E/Z ratio 2:1.
e Solvent mixture used for the vinyl boronic acid was CH2Cl2:THF (2:1 v/v, 0.04 M).
|
|
Both electron withdrawing and electron-donating substituents on the vinyl boronic partner were associated with particularly good yields (including aryl and alkyl groups). Screening of both aromatic and aliphatic aldehyde traps successfully provided the products with good-to-excellent yields in all cases. In all the examples, high degree of diastereoselectivity was observed (confirmed by X-ray crystallography data, see ESI‡).
It is worth mentioning that reacting Z-vinyl boronic acids under the standard conditions led to a complex mixture, suggesting that the molecular geometry of the boronic species is mechanistically relevant.
The robustness of the protocol was quickly assessed by performing a larger-scale reaction (10 mmol) which gave product 4b in a consistent yield of 73%.
In order to facilitate downstream processing and improve the practicality of the method, any residual aldehyde was scavenged using a polymer-supported tosylhydrazine resin.50,51
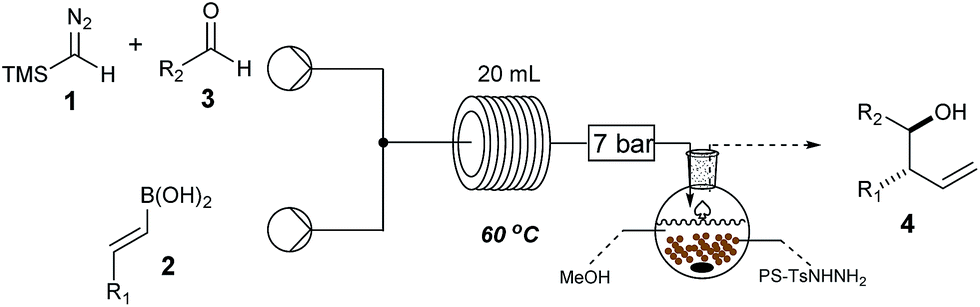
Wishing to broaden the practical aspect of this multicomponent metal-free reaction, we decided to transfer the method directly into a batch process (Scheme 2), having taken care to safely handle the TMSCHN2 reagent. The optimised batch protocol was developed at room temperature with reaction times being within the range from 2 h to 16 h for similar reaction scale (0.3 mmol). The scope of the reaction under batch conditions was expanded and demonstrated the generality of the method (Table 2).
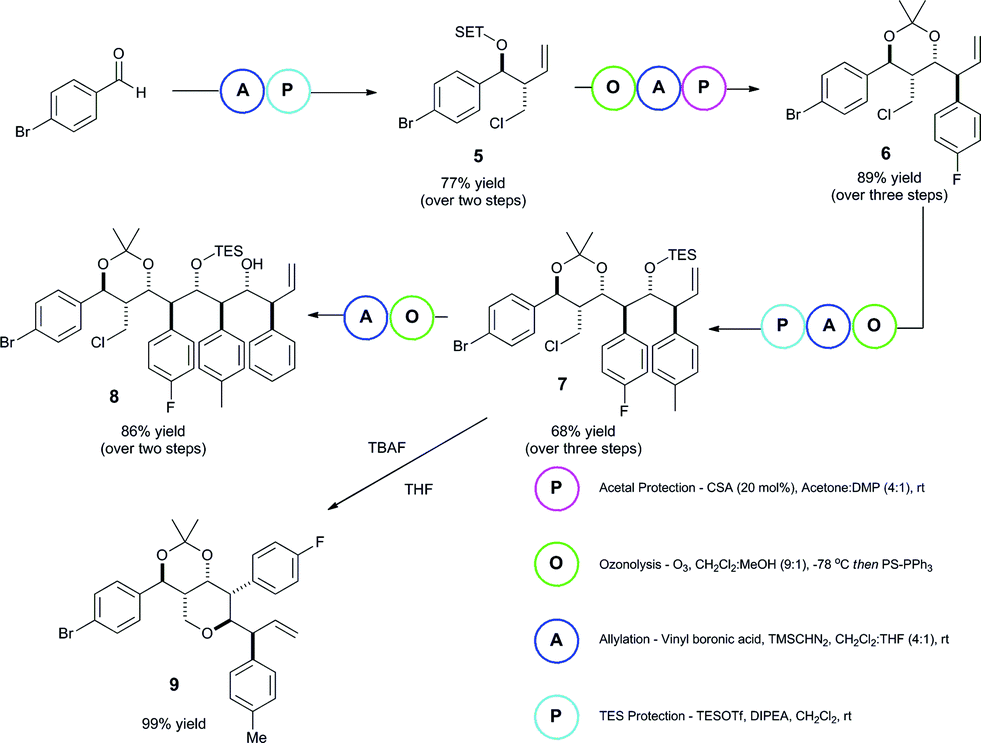 | ||
| Scheme 2 Iterative process to polyol 8 and tetrahydropyran derivative 9. | ||
|
|
|---|
|
a Standard reaction conditions: 0.6 mmol of TMSCHN21, 0.36 mmol of vinyl boronic acid 2, 0.3 mmol of aldehyde 3, in CH2Cl2:THF (4:1 v/v, 4 mL) at rt, 2 h to 16 h; for more detail see ESI.
|
|
The iterative importance of this methodology was then showcased in the generation of polyol backbone materials (Scheme 2). The iteration process consisted of a sequence of allylation processes. In between each allylation, the homoallylic alcohol intermediate underwent an alcohol protection stage, followed by ozonolysis of the olefin to regenerate the aldehyde component for further reaction. The sequence was repeated four times to rapidly provide a complex derivative 8 (see ESI‡ for a detailed reaction description). Analysis of the 1H/13C shifts of the acetonide for the 2nd iteration product 6 confirms a twist-boat structure, indicating Felkin–Anh selectivity.52–55
In addition to this, we were able to selectively elaborate intermediate 7 into the corresponding tetrahydropyran derivative 9, showing the versatility of such a method for generation of rather underrepresented molecular entities.
From a mechanistic viewpoint, we speculate that the absence of the trimethylsilyl group in the product may be due to the instability of TMSCHN2, in the presence of E-vinyl boronic acids.56–58 Efforts to elucidate the mechanism confirmed that the addition of a base (e.g. TEA) shuts down the reaction. Similarly, the treatment of the vinyl boronic acid solution with a drying reagent (e.g. molecular sieves) followed by addition of aldehyde and TMSCHN2, afforded a very complex mixture and only traces of product. This suggests that the acidity of the vinyl boronic acid is important in respect to the desired reaction outcome.59 Another piece of information showed that when catechol borane 10 was employed as the vinyl boronic ester partner under the same reaction conditions, vinyl derivative 11 was obtained (19% yield) along with recovered starting material. Additionally, 1H-NMR studies indicate that after addition of TMSCHN2 to the boronic acid partner, the main intermediate in the reaction mixture is the corresponding allylic boronic acid, in which no silylated intermediate is observed.60 We therefore propose a mechanism where TMSCHN2 is first protonated by the boronic acid; the boronate intermediate then reacts with the trimethylsilane group in order to generate a highly reactive diazomethane partner; this diazomethane reacts with the vinyl boronic acid to provide the corresponding allylic boronic acid (Scheme 3). This intermediate then reacts with the aldehyde electrophile to yield the final anti product after deborylation.
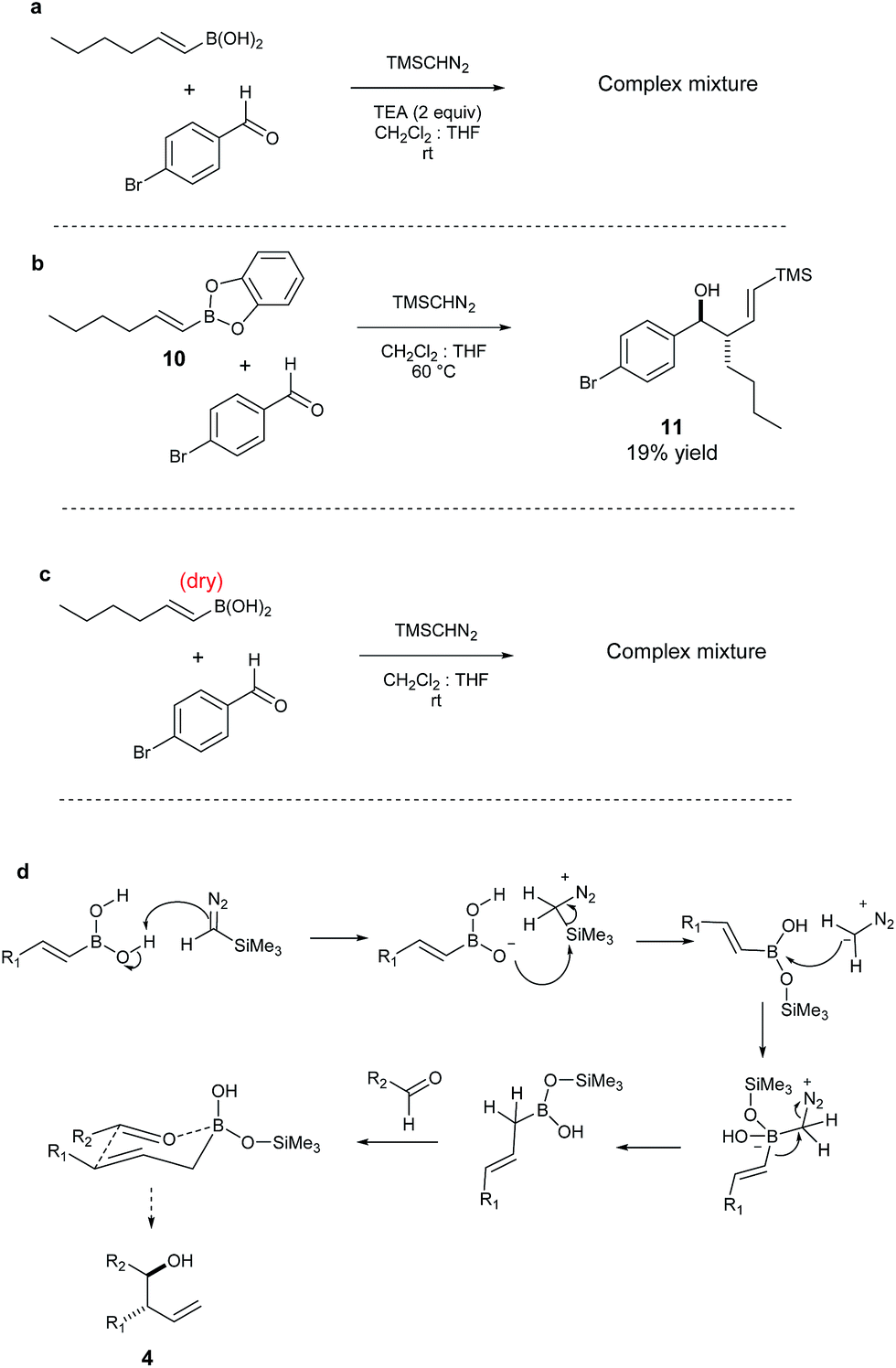 | ||
| Scheme 3 (a) Reaction run in the presence of a base; (b) reaction run with vinyl boronic catechol ester; (c) reaction run with “dry” boronic acid; (d) proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a robust and scalable metal-free multicomponent approach towards the synthesis of homoallylic alcohols, using TMSCHN2, E-vinyl boronic acids and aldehydes. The method was first developed using a flow-based route and subsequently transferred to a batch procedure in order to demonstrate the generality of the chemistry. The power of this method was further exemplified in an iterative fashion to build novel and potentially useful oligomeric materials. Current investigations are directed towards the development of an asymmetric version of this method.Acknowledgements
We are grateful to the Cambridge Home and European Scholarship Scheme (J. S. P.), Croucher Foundation (S. H. L.), the Swiss National Science Foundation (D. N. T.), Pfizer Worldwide Research & Development (C. B.), and the Engineering and Physical Sciences Research Council (S. V. L. grant no. EP/K0099494/1 and EP/K039520/1) for financial support. We would also like to thank Dr Richard Turner for providing assistance for the project. The X-ray crystal structure in this paper was determined by Dr Andrew D. Bond (University of Cambridge).Notes and references
- E. Srinivas, P. Dutta, B. Ganganna, A. A. Alghamdi and J. S. Yadav, Synthesis, 2016, 48, 1561–1567 CrossRef CAS.
- G. Helmchen, R. Hoffmann, J. Mulzer and E. Schaumann, in Stereoselective Synthesis, Methods of Organic Chemistry (Houben-Weyl), ed. G. Helmchen, R. Hoffmann, J. Mulzer and E. Schaumann, Thieme Stuttgart, New York, 1996, vol. 3, pp. 1357–1602 Search PubMed.
- S. R. Chemler and W. R. Roush, in Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, ch. 11 Search PubMed.
- S. E. Denmark and N. G. Almstead, in Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, ch. 10 Search PubMed.
- W. R. Roush, A. E. Walts and L. K. Hoong, J. Am. Chem. Soc., 1985, 107, 8186–8190 CrossRef CAS.
- W. R. Roush and R. L. Halterman, J. Am. Chem. Soc., 1986, 108, 294–296 CrossRef CAS.
- W. R. Roush, A. D. Palkowitz and M. J. Palmer, J. Org. Chem., 1987, 52, 316–318 CrossRef CAS.
- W. R. Roush, K. Ando, D. B. Powers, R. L. Halterman and A. D. Palkowitz, Tetrahedron Lett., 1988, 29, 5579–5582 CrossRef CAS.
- W. R. Roush, K. Ando, D. B. Powers, A. D. Palkowitz and R. L. Halterman, J. Am. Chem. Soc., 1990, 112, 6339–6348 CrossRef CAS.
- W. R. Roush, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 2, pp. 1–53 Search PubMed.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS.
- P. K. Jadhav, K. S. Bhat, P. T. Perumal and H. C. Brown, J. Org. Chem., 1986, 51, 432–439 CrossRef CAS.
- H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 5919–5923 CrossRef CAS PubMed.
- H. C. Brown, K. S. Bhat and R. S. Randad, J. Org. Chem., 1987, 52, 319–320 CrossRef CAS.
- H. C. Brown, P. K. Jadhav and K. S. Bhat, J. Am. Chem. Soc., 1988, 110, 1535–1538 CrossRef CAS.
- H. C. Brown, K. S. Bhat and R. S. Randad, J. Org. Chem., 1989, 54, 1570–1576 CrossRef CAS.
- H. C. Brown, R. S. Randad, K. S. Bhat, M. Zaidlewicz and U. S. Racheria, J. Am. Chem. Soc., 1990, 112, 2389–2392 CrossRef CAS.
- U. S. Racheria and H. C. Brown, J. Org. Chem., 1991, 56, 401–404 CrossRef.
- M. Srebnik and P. V. Ramachandran, Aldrichimica Acta, 1987, 20, 9–24 CAS.
- S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed.
- Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS.
- T. R. Wu, L. Shen and J. M. Chong, Org. Lett., 2004, 6, 2701–2704 CrossRef CAS PubMed.
- G. E. Keck, K. H. Tarbet and L. S. Geraci, J. Am. Chem. Soc., 1993, 115, 8467–8468 CrossRef CAS.
- M. J. Zacuto and J. L. Leighton, J. Am. Chem. Soc., 2000, 122, 8587–8588 CrossRef CAS.
- J. W. A. Kinnaird, P. Y. Ng, K. Kubota, X. Wang and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7920–7921 CrossRef CAS PubMed.
- K. Kubota and J. L. Leighton, Angew. Chem., Int. Ed., 2003, 42, 946–948 CrossRef CAS PubMed.
- B. M. Hackman, P. J. Lombardi and J. L. Leighton, Org. Lett., 2004, 6, 4375–4377 CrossRef CAS PubMed.
- J. L. Leighton, Aldrichimica Acta, 2010, 43, 3–14 CAS.
- W. A. Chalifoux, S. K. Reznik and J. L. Leighton, Nature, 2012, 487, 86–89 CAS.
- I. S. Kim, M.-Y. Ngai and J. M. Krische, J. Am. Chem. Soc., 2008, 130, 6340–6341 CrossRef CAS PubMed.
- I. S. Kim, M.-Y. Nagi and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891–14899 CrossRef CAS PubMed.
- M. Wadamoto and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 14556–14557 CrossRef CAS PubMed.
- T. Miura, Y. Nishida, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2013, 135, 11497–11500 CrossRef CAS PubMed.
- H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376–5377 CrossRef CAS PubMed.
- K. R. Fandrick, D. R. Fandrick, J. J. Gao, J. T. Reeves, Z. Tan, W. Li, J. J. Song, B. Lu, N. K. Yee and C. H. Senanayake, Org. Lett., 2010, 12, 3748–3751 CrossRef CAS PubMed.
- S. Yamasaki, K. Fujii, R. Wada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 6536–6537 CrossRef CAS PubMed.
- U. Schneider, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13824–13825 CrossRef CAS PubMed.
- B. Dutta, N. Gilboa and I. Marek, J. Am. Chem. Soc., 2010, 132, 5588–5589 CrossRef CAS PubMed.
- C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed.
- J. S. Poh, D. N. Tran, C. Battilocchio, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 7920–7923 CrossRef CAS PubMed.
- N. M. Roda-Monsalvez, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 Search PubMed.
- D. N. Tran, C. Battilocchio, S. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC.
- J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618–3621 CrossRef CAS PubMed.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016 10.1039/c5cs00902b.
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed.
- D. Seyferth, A. W. Dow, H. Menzel and T. C. Flood, J. Am. Chem. Soc., 1968, 90, 1080–1082 CrossRef CAS.
- D. Seyferth, H. Menzel, A. W. Dow and T. C. Flood, J. Organomet. Chem., 1972, 44, 279–290 CrossRef CAS.
- T. Shioiri, T. Aoyama and S. Mori, Org. Synth., 1990, 68, 1–7 CrossRef CAS.
- N. G. Murphy, S. M. Varney, J. M. Tallon, J. R. Thompson and P. D. Blanc, Clin. Toxicol., 2009, 47, 712 Search PubMed.
- M. Baumann, I. R. Baxendale, L. J. Martin and S. V. Ley, Tetrahedron, 2009, 65, 6611–6625 CrossRef CAS.
- L. J. Martin, A. L. Marzinzik, S. V. Ley and I. R. Baxendale, Org. Lett., 2011, 13, 320–323 CrossRef CAS PubMed.
- D. A. Evans, D. L. Rieger and J. R. Gage, Tetrahedron Lett., 1990, 49, 7099–7100 CrossRef.
- M. Chérest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 9, 2199–2204 CrossRef.
- N. T. Anh and O. Eisenstein, Nouv. J. Chim., 1977, 1, 61–70 Search PubMed.
- N. T. Anh, O. Eisenstein, J. M. Lefour and M. E. Tran-Huu-Dau, J. Am. Chem. Soc., 1973, 95, 6146–6147 CrossRef.
- M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2012, 51, 5953–5957 CrossRef PubMed.
- For the related reactions of diazo compounds with organoboranes, see: (a) J. Hooz and S. Linke, J. Am. Chem. Soc., 1968, 90, 5936–5937 CrossRef CAS; (b) H. C. Brown, M. M. Midland and A. B. Levy, J. Am. Chem. Soc., 1972, 94, 3662–3664 CrossRef CAS.
- For the related reactions of diazo compounds with aryl boronic acids, see: (a) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed; (b) O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed; (c) G. Wu, Y. Deng, C. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 10510–10514 CrossRef CAS PubMed; (d) G. A. Molander and D. Ryu, Angew. Chem., Int. Ed., 2014, 53, 14181–14185 CrossRef CAS PubMed.
- E. Kühnel, D. D. P. Laffan, G. C. Lloyd-Jones, T. Martínez del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef PubMed.
- M. Shimizu, H. Kitagawa, T. Kurahashi and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 4283–4286 CrossRef CAS.
Footnotes |
| † Raw spectra can be found at DOI: 10.17863/CAM.591 |
| ‡ Electronic supplementary information (ESI) available. CCDC 1483699. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02581a |
| § J-SP and S-HL contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |