
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atmospheric chemistry of bioaerosols: heterogeneous and multiphase reactions with atmospheric oxidants and other trace gases
Armando D.
Estillore
a,
Jonathan V.
Trueblood
a and
Vicki H.
Grassian
*ab
aDepartment of Chemistry & Biochemistry, University of California San Diego, La Jolla, California 92093, USA. E-mail: vhgrassian@ucsd.edu; Fax: +1-858-534-6255; Tel: +1-858-534-2499
bScripps Institution of Oceanography and Department of Nanoengineering, University of California San Diego, La Jolla, California 92093, USA
First published on 28th July 2016
Abstract
Advances in analytical techniques and instrumentation have now established methods for detecting, quantifying, and identifying the chemical and microbial constituents of particulate matter in the atmosphere. For example, recent cryo-TEM studies of sea spray have identified whole bacteria and viruses ejected from ocean seawater into air. A focal point of this perspective is directed towards the reactivity of aerosol particles of biological origin with oxidants (OH, NO3, and O3) present in the atmosphere. Complementary information on the reactivity of aerosol particles is obtained from field investigations and laboratory studies. Laboratory studies of different types of biologically-derived particles offer important information related to their impacts on the local and global environment. These studies can also unravel a range of different chemistries and reactivity afforded by the complexity and diversity of the chemical make-up of these particles. Laboratory experiments as the ones reviewed herein can elucidate the chemistry of biological aerosols.
 Armando D. Estillore | Armando D. Estillore obtained his B.S. in chemistry from MSU-IIT, Iligan City, Philippines and his Ph.D. in physical chemistry from Wayne State University under Prof. Arthur G. Suits in 2012. His doctoral work on the reaction dynamics of radicals with polyatomic hydrocarbons using crossed-beam ion imaging techniques earned the Dan Trivich Memorial Award for research in physical chemistry. He was a Camille and Henry Dreyfus Postdoctoral Fellow in Environmental Chemistry at UC Berkeley prior to joining the group of Prof. Vicki H. Grassian at the University of Iowa and the University of California, San Diego where he is currently a postdoctoral researcher. |
 Jonathan V. Trueblood | Jonathan V. Trueblood received his B.A. in physics from Dordt College in Sioux Center, Iowa in 2011 and his M.S. in Atmospheric Science from North Carolina State University in Raleigh, North Carolina. He is currently pursuing his Ph.D. in chemistry under the direction of Prof. Vicki H. Grassian at the University of California, San Diego. His research is focused on the heterogeneous reactions of sea spray aerosols with trace atmospheric gases and probing the impacts of these reactions on aerosols' physical, chemical, and climate-relevant properties. |
 Vicki H. Grassian | Vicki H. Grassian is a Distinguished Professor at the University of California San Diego with appointments in Chemistry & Biochemistry, Nanoengineering and Scripps Institution of Oceanography. She is also Distinguished Chair of Physical Chemistry and currently serves as co-director for the Center for Aerosol Impacts on Climate and the Environment. Her research focuses on environmental molecular surface science, heterogeneous and multiphase chemistry of atmospheric aerosol particles, the impact of atmospheric aerosol on air quality and climate, and environmental and health aspects of nanoscience and nanotechnology. |
Introduction
Generated from various sources, aerosols are ubiquitous in the atmosphere and play a crucial role in global climate, urban visibility, and human health. These airborne particles, ranging in size from a few nanometers (nm) to tens of micrometers (μm), can be released from primary sources such as industrial processes, biomass burning, volcanic emissions, or sea spray.1–5 In addition, aerosol particles can nucleate from atmospheric gases to form secondary organic aerosol (SOA).6 SOA is composed of a myriad of complex chemical compounds exhibiting a wide range of molecular structures and phase states which influences their physicochemical properties and reactivities.7,8 Atmospheric aerosols are strongly related to a plethora of health concerns as they are respiratory irritants, can cause cardiovascular problems, and are linked to increased mortality.9–13 Furthermore, depending on their chemical composition, aerosols can absorb or scatter solar and terrestrial radiation and can act as cloud condensation and ice nuclei which in turn plays a major role in global temperatures and precipitation.14A major source of primary aerosol organic matter, ranging from nm to μm in size, in the atmosphere is primary biological organic aerosol particles (PBAP) that are emitted directly from the biosphere to the atmosphere.15 These particles are comprised of dead and live cells, cell fragments, pollen (>10 μm), bacteria (∼1 μm), fungi, algae, moss and fern spores (∼10 μm), viruses (<0.1 μm), and other fragments of animals and plants.15–19 Studies on exposure to these particles are associated with a variety of health issues with major public health impacts, including infectious diseases, acute toxic effects, allergies and cancer.20,21 Collective efforts are actively pursued to decode the intricate details of the interaction of the coexisting population of microbiomes and nanobiomes of human, plants, the earth, the ocean and the atmosphere from molecular to global scale.22,23 As an example, the diversity of the atmospheric microbiome is severely affected by dust events24 and tropical storms.25 The relative abundance of desert soil-associated bacteria has been shown to increase during dust events, while the relative abundance of anthropogenic-influenced taxa decreases.24
In addition, biologically-derived compounds are known to be present and are incorporated in other primary aerosol particles such as sea spray aerosol (SSA) and dust particles that contribute to aerosol mass.26–30 For example, amino acids in both free and combined forms (i.e., peptides and proteins) are an important component in SSA31–33 as well as a range of terrestrial bioaerosol.34 They are the most abundant group of pollen allergens35 and are found in both coarse biological particles such as pollen grains (>10 μm) as well as the fine fraction of air particulate matter (<2.5 μm). In addition to being an important component of particulate aerosol, free and combined forms of amino acids have been found in hydrometeors36 and rain.37
Recent studies showed that oceans, which cover >70% of earth's surface, are a major source of bacteria, viruses and biologically-derived compounds to the atmosphere. The interaction of ocean-atmosphere is schematically illustrated in Fig. 1. In a study conducted using a unique ocean-atmosphere facility containing 3400 gallons of natural seawater, Wang et al.38 generated two successive phytoplankton blooms producing SSA with different composition and properties. In the first bloom, aliphatic-rich organics were present in submicron SSA that followed the abundance of phytoplankton measured by chlorophyll-a concentrations. During the second bloom, no organic enrichment was found in the particles collected. The studies explained the key link between biological, chemical, and physical processes occurring in the ocean and the resulting physicochemical properties of SSA particles.38 Furthermore, using cryogenic transmission electron microscopy (cryo-TEM) Patterson et al.,27 recently reported the detection of whole hydrated bacteria, diatoms, virus particles, marine vesicles from SSA samples as shown in Fig. 2.
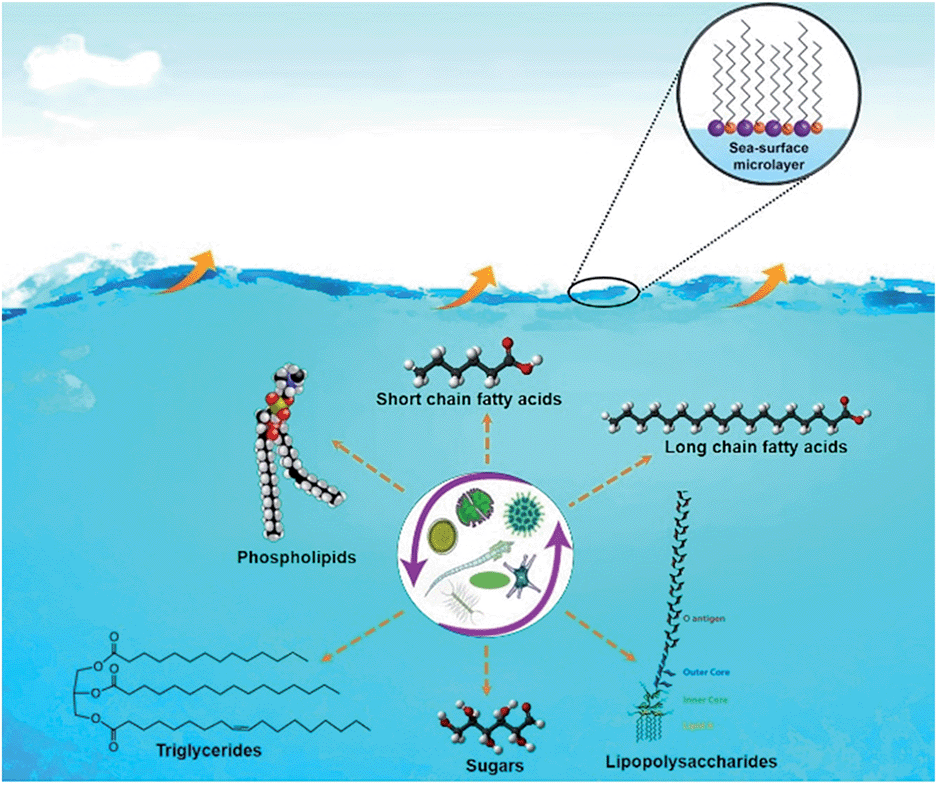 | ||
| Fig. 1 Oceans, covering >70% of Earth's surface, are a major source of biologically-derived compounds in the atmosphere. The biological, chemical, and physical processes in oceans affects the major chemical composition of sea-spray aerosol emitted into the atmosphere. | ||
 | ||
| Fig. 2 Bright field TEM images of SSA prepared by cryo-TEM showing (a) a whole bacterium inside a wet SSA droplet, (b) a diatom, (c) a virus particle, and (d) marine membrane vesicles. Yellow arrows indicate the edge of the SSA, the red arrows indicate contamination from the cryo-TEM preparation, and the green arrows indicate the biological particles. Adapted with permission from Patterson et al., ACS Cent. Sci., 2016, 2, 40–47. | ||
Overall, it is believed that primary biological aerosol particles make up a sizable fraction of the aerosol budget, with 1000 Tg per year emitted in comparison to 3300 Tg per year for sea salt and 2000 Tg per year for mineral dust.34 Interest in biological aerosol particle research is rapidly growing due in part to the continued progress and availability of sophisticated analytical instrumentation. Primary biological organic aerosol has been identified, characterized and quantified in several field studies.15,39,40 Bioaerosols are detected using several analytical techniques which includes electron impact ionization aerosol mass spectrometry,41,42 fluorescence spectroscopy,43,44 and Raman spectroscopy,45,46 among others. For additional information on bioaerosol detection, we refer the readers to several excellent reviews recently published.15,47–49
Aerosols evolve away from their native states as they undergo heterogeneous and multiphase reactions that modify gas and particle phases,50–55 affecting both atmospheric composition and air quality.50 Tremendous progress has been made in the fundamental understanding of heterogeneous chemical reactions involving primary aerosols such as mineral dust56,57 and sea-spray aerosol.33,58 Similarly, great strides have been made through the many studies on secondary organic aerosol formation.8,54,59–61 Information gathered from these studies has made huge impacts in understanding the chemistry and properties of atmospheric aerosols. However equivalent investigations on biological aerosol and how they are chemically transformed in the atmosphere remains limited. The importance of studying the reactivity of PBAP are several fold, both practical as well as elementary in nature. These studies cover many areas of chemistry, biology, clinical medicine, and even biodefense. These biologically derived particles are fundamentally different from inorganic aerosols; potentially more interesting and certainly much more complicated as these particles come in varying size and shape, carry different molecular functional groups and consist of different conformations. Because of their complexity, PBAP respond differently to abrupt changes of environmental conditions in the atmosphere such as relative humidity (RH), temperature, and reactive trace gases. Furthermore, studies have shown that these particles play an important role as ice and cloud condensation nuclei.38,62–68
The focus of this article is to review some processes related to the reactivity of bioaerosols with emphasis on select oxidants; OH, O3, and NO3. We do not attempt to comprehensively review the entire field of bioaerosol reactivity, as this is a continuously evolving area of research. For simplicity, we use the term “bioaerosols” to refer to primary biological and biologically-derived aerosol particles. This review covers several sections on the reactivity with oxidants in the atmosphere including OH, O3 and NO3 as well as a section on the reactivity of other trace gases, new reaction pathways and photochemistry.
Bioaerosol reactivity – oxidant chemistry and other reaction pathways
A. OH reaction with bioaerosols and biologically derived compounds
The hydroxyl radical is one of the most reactive atmospheric oxidants and is known to be one of the reactive oxygen species (ROS) in atmospheric processes. Its interactions with gas-phase organic species and the underlying processes by such interactions have been investigated extensively.59,69–71 Biologically-derived molecules lofted into the atmosphere can be chemically and physically altered through reactive encounters with OH radicals in the daytime when their concentration is highest. For example, OH radicals can reactively modify saturated and unsaturated molecular moieties by causing H atom abstraction of the former and both H-atom abstraction from the alkyl chain and addition to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– of the latter.72,73 Damage to biological systems can be induced by direct attack of ROS such as OH radicals.74 Investigations on the interactions of proteins and amino acids with OH radicals reveal H atom abstraction from C–H bonds, either from the protein backbone or from an amino acid side chain. This H atom abstraction step represents the first step in the protein oxidation pathway.75–77 In an in situ mass spectrometric study of the interaction of OH radicals with glutathione (GSH), an antioxidant which is a cysteine-containing tripeptide in epithelial lining fluids (ELF), Enami et al., showed that GSH are oxidized into sulfenic acid GSOH−, sulfinic acid GSO2H−, and sulfonic acid GSO3H−. This reaction oxidizes the GSH when inhaled particulates generate OH radical in the ELF via the Fenton-type chemistry.78
C– of the latter.72,73 Damage to biological systems can be induced by direct attack of ROS such as OH radicals.74 Investigations on the interactions of proteins and amino acids with OH radicals reveal H atom abstraction from C–H bonds, either from the protein backbone or from an amino acid side chain. This H atom abstraction step represents the first step in the protein oxidation pathway.75–77 In an in situ mass spectrometric study of the interaction of OH radicals with glutathione (GSH), an antioxidant which is a cysteine-containing tripeptide in epithelial lining fluids (ELF), Enami et al., showed that GSH are oxidized into sulfenic acid GSOH−, sulfinic acid GSO2H−, and sulfonic acid GSO3H−. This reaction oxidizes the GSH when inhaled particulates generate OH radical in the ELF via the Fenton-type chemistry.78
Another class of molecules prone to reactive encounters in the atmosphere with OH radicals is phospholipids. Phospholipids are found at the sea surface microlayer (SSML), in the top most region of the ocean surface and in the atmosphere as an organic coating in sea-spray aerosol.29,79–81 Moreover, phospholipids in both the saturated and unsaturated forms are a major component in cell membranes.82 Finlayson-Pitts and co-workers performed experiments on the reaction of OH radical with 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) as a proxy for saturated and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) as proxy for unsaturated phospholipids on a surface of sodium chloride using diffuse reflection infrared Fourier transform spectrometry (DRIFTS) and matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF-MS) methods.83,84Fig. 3 shows the structure of different phospholipids on the surface of NaCl as depicted in ref. 82, showing an “inverted micelle” where the hydrocarbon chains are exposed to the oxidizing environment.85 DPPC is a saturated phospholipid and thus only abstraction from the alkyl chain is possible, while both H atom abstraction and OH addition to the carbon–carbon double bond from the alkyl chains will occur in OPPC and its structural isomer POPC. In this “top down” experiment, the OH radical was generated in the gas phase over the phospholipid via photolysis of isopropyl nitrite in a mixture of N2 and air as shown in reactions (1)–(3):
| (CH3)2CHONO + hν → (CH3)2CHO˙ + NO | (R1) |
| (CH3)2CHO˙ + O2 → CH3COCH3 + HO2 | (R2) |
| HO2 + NO → OH + NO2 | (R3) |
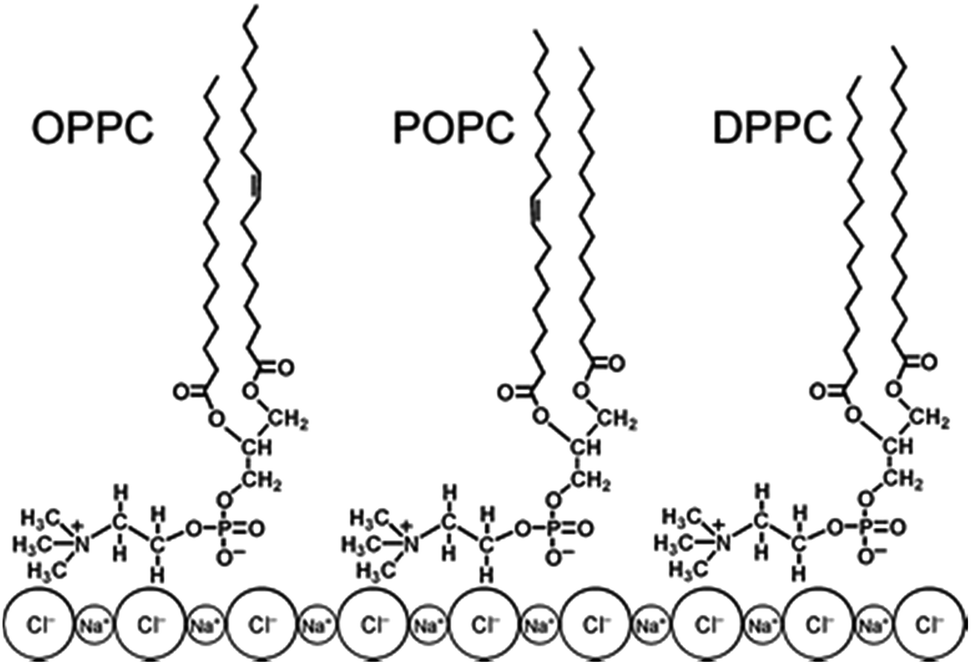 | ||
| Fig. 3 Structures of OPPC, POPC and DPPC, shown on the surface of solid NaCl. Adapted with permission from Dilbeck et al., Phys. Chem. Chem. Phys., 2013, 15, 9833–9844. | ||
The results of this experiment identified aldehydes, ketones, organic nitrates and nitrite ions as products of the reaction. The rate constant (k1) for the OH addition to the double bond in POPC was measured to be k1 > (3 ± 1) × 10−13 cm3 per molecule per s corresponding to a reaction probability, γ, for the addition reaction greater than (4 ± 1) × 10−3 while the rate constant (k2) for the DPPC–NaCl + OH is k2 = (7 ± 1) × 10−14 cm3 per molecule per s corresponding to γ of (8 ± 1) × 10−4 for the abstraction reaction.
Interestingly, unsaturated phospholipids such as OPPC can also be oxidized from the “bottom up” by the OH generated during the photolysis of nitrite ion in a NaNO2/NaCl salt mixture upon which OPPC is adsorbed, as shown in (R4). The presence of nitrite in the substrate is made possible by the displacement of chloride ion in SSA particles with nitrate, which in turn can photolyze readily to yield nitrite which then undergoes further reactions that yield OH radical as shown below.
| NO2− + hν (290–400 nm) → NO + O− + H2O → OH + OH− | (R4) |
Irradiation of the OPPC/NaNO2/NaCl mixture leads to the formation of organic nitrates and carbonyl compounds similar to the “top down” approach. In the absence of water vapor, carboxylate ions are also formed.84,86 These two different oxidation mechanisms of unsaturated phospholipids have important implications on the lifetime of the lipids. Freshly emitted SSA particles with phospholipid coating will experience top down oxidation. The onset of bottom up oxidation will soon set in when the SSA particles are exposed to species such as NO2− and NO3− that can be photolyzed to generate the OH oxidants. The organic nitrates generated as oxidation products of the two mechanisms can serve as reservoirs for oxides of nitrogen that can decompose to release NOx back to the atmosphere.87
B. NO3 reaction with bioaerosols and biologically derived compounds
Proteins are present in most biological systems at particularly high concentrations. Thus they are one of the most targeted components for oxidation in bioaerosols. To better access the activities of these aerosols, the valence electronic property of bioaerosols play a determinant role. The electronic properties of amino acid aerosols have been characterized which underlines the recent development on the instrumentation for characterizing bioaerosols as well as the advanced understanding on the properties of such types of bioaerosols. For example, Wilson et al. applied the VUV photoelectron imaging to study the electronic properties of glycine (Gly) and phenylalanine-glycine-glycine (Phe-Gly-Gly) in the dehydrated aerosol particle phase and determined the ionization energies and the molecular polarizabilities.88Protein molecules that have been nitrated by polluted urban air can promote allergies upon their inhalation and deposition in the human respiratory tract.89,90 Several research investigations have shown that phenols and other aromatic compounds can be nitrated by gaseous air pollutants such as NO2 and NO3.91–94 Additionally, previous studies have shown that within biological systems, proteins can undergo a nitration reaction leading to the formation of 3-nitrotyrosine residues.95,96 In a key study done by Goschnick and Schuricht, it was found that the nitrogen content of pollen surfaces increased upon exposure to NO2.97 Working from this evidence, Franze et al.89,98 first showed that the proteinaceous component of bioaerosols could also be nitrated when exposed to polluted urban outdoor air (e.g. vehicle exhaust) as well as synthetic gas mixtures containing H2O, NO2, and O3 at atmospherically relevant concentrations. The nitration reaction occurs by the addition of nitro groups to the aromatic ring of tyrosine residues in the polypeptide chain, resulting in the formation of 3-nitrotyrosine. Importantly, the extent of nitration was reduced under dry conditions, indicating that the hydration state of the aerosols could influence reaction pathways and kinetics. This process then can boost the power of existing allergens or make benign proteins allergenic. In their study, Franze and coworkers89 also collected samples of urban dust and found that up to 0.1% of the proteins in dust had been nitrated by traffic smog. Samples of allergenic proteins from birch pollen exposed to road junction in Munich, Germany found 10% of the proteins were nitrated and that number rose to 20% when exposed to smog in the laboratory. Interestingly, this study of pollutant–allergen interactions provided a molecular rationale for the medical reports which found an increase in allergic diseases in area with high traffic-related air pollution concentrations in combination with high NO2 and O3.89,98 The results were further corroborated by immunological experiments by Gruijthuijsen et al. who reported that allergenic potential of proteins are enhanced upon nitration.99 In a similar study, Rudich and co-workers100 conducted outdoor and laboratory exposure studies on the allergenicity of the allergenic mold Aspergillus fumigatus. The outdoor exposure experiments were performed in Modi'in, Israel. Their study demonstrated a dual effect of NO2/O3 pollution on the allergenic activity of A. fumigatus spore. Results showed that initial exposure of fungal spores leads to a 2–5 fold increase in allergenicity due to nitration compared to the control for short exposure times of up to 12 hours. At higher exposure times, the increase of allergenicity was less pronounced, which the authors ascribed to protein deamidation.100
From these initial studies on protein nitration of bioaerosols in the atmosphere, several investigations then focused on the underlying kinetics and reaction mechanisms. Aiding such studies was the development of simple, fast, efficient, and inexpensive methods for the quantification of nitrotyrosine residues in protein molecules.101–103 Shiraiwa et al.104 used a new kinetic modeling approach to show that the nitration of proteins with O3 and NO3 occurs through a two-step process involving the formation of long-lived reactive oxygen intermediates (ROIs) followed by nitration of the ROI via exposure to NO2. Shiraiwa et al.105 then further studied the kinetics and reaction mechanism of protein nitration using an aerosol flow tube and the short lived radioactive tracer 13N in addition to a kinetic multilayer model. Their results further proved that the reactions proceed through the two-step mechanism as previously proposed by Shiraiwa104et al. and as summarized in Fig. 4. In addition, it was found that the reaction of proteins on the surface of aerosol particles were rapidly nitrated, with the rate of nitration of the proteins in the interior of the particle being controlled by diffusion. In the absence of NO2, the ROIs were found to react with each other forming protein dimers. The efficiency and specificity of the protein nitration strongly depends on the nitrating agent and the reaction conditions. In another nitration study of the major birch pollen allergen Bet v 1, tetranitromethane was found to be the most efficient nitrating reagent, yielding nitration degree (ND) values of up to 70% over peroxynitrite (ONOO−) and O3/NO2 which yielded ND values up to ∼50 and ∼20%, respectively, with substantial amounts of side products from protein oxidation and degradation. Nitration rates were found to be higher for aqueous protein solutions (∼20% per day) than for solid or semisolid protein samples (∼2% per day).106
 | ||
| Fig. 4 Schematic of a reaction pathway of residues within the protein BSA following exposure to ozone and followed by nitrogen dioxide. Adapted with permission from Shiraiwa et al., Environ. Sci. Technol., 2012, 46, 6672–6680. | ||
While it has been shown by Shiraiwa et al. that the principal reaction is a two-step process, the actual reaction mechanism and pathways still remained unclear as there are many reactive sites in tyrosine other than the phenolic site in which the first oxidation step could take place. Using quantum chemical methods, Sandhiya107 showed that the initial oxidation of tyrosine by O3 proceeds by H atom abstraction and addition reactions, leading to the formation of six different intermediates. The subsequent nitration reaction was then studied for all six intermediates in gas, aqueous, and lipid media. Their results showed that the nitration could affect both the side chain and aromatic ring of tyrosine, with some other resulting products formed from other reaction pathways.
Studies conducted by Wille et al.108–110 on the reaction of nitrate radicals with aromatic amino acids demonstrated that the reaction leads to irreversible oxidative functionalization at the β-position or at the aromatic ring of the amino acids.108,109 In the case of aromatic dipeptides, in addition to oxidation at the ring, the oxidative damage of dipeptide linkage occurs in the aromatic N- and C-protected dipeptides.110 The results strongly suggest that this important atmospheric oxidant could potentially cause damage to peptides lining the respiratory tract and may contribute to pollution-derived diseases.
C. O3 reaction with bioaerosols and biologically-derived compounds
The ozonolysis of unsaturated volatile organic compounds in the troposphere has been known to proceed via the Criegee intermediate (CI)111 and has been well studied in laboratory and theoretical investigations. The details of this reaction have been recently compiled in an excellent review article.112 Additionally, the interaction of ozone with organic aerosols, especially those containing unsaturated bonds, has been extensively studied.55,113–117Karagulian and coworkers performed studies on the oxidation of OPPC adsorbed on NaCl as a model for lipids on sea salt using DRIFTS. Product identification was confirmed by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry and Auger electron spectroscopy.118 Following exposure to O3, loss of the CC functionality was observed with the subsequent formation of a stable secondary ozonide (1,2,4-trioxolane, SOZ) known to be formed in alkene–ozone reaction and confirmed by the strong band at ∼1110 cm−1. MALDI-TOF also confirmed the formation of SOZ at m/z = 809 and 831 attributed to SOZ adducts with H+ and Na+. The effect of water vapor on the ozonolysis reaction was also conducted at relative humidity (RH) ranging from 2 to 25%. These are relatively low RH values in order to maintain the phase state of NaCl as a solid to preserve the sample integrity for DRIFTS analysis. With increasing RH, the band at 1110 cm−1 that is associated with SOZ was dramatically decreased. This decrease of SOZ with hydration was more pronounced at the lower O3 concentration, which suggests competition of SOZ formation with the reaction of Criegee intermediate (CI) with water vapor. Vesna et al. suggested that, for the ozonolysis of arachidonic acid under humid conditions, the reaction of water with CI might open a pathway for the formation of smaller acids that lead to more significant changes in hygroscopicity.119 The authors also showed the increase of product yields of the ozonolysis of oleic acid with increasing RH.120
Dilbeck et al.,121 reported the ozone oxidation of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) adsorbed on salt mixtures using DRIFTS. Ozone reacts with the –CC– backbone in the POPC via the Criegee mechanism. The experiments conducted at room temperature yielded secondary ozonide and a phospholipid aldehyde and carboxylic acid. The presence of water vapor was found to affect the accessibility of the double bond for reaction as well as the reaction probability (γ = 6 × 10−7). Several studies by Finlayson-Pitts and co-workers have shown that unsaturated phospholipids are readily oxidized by O3 from the “top down” to generate secondary ozonides and a number of products.122–124 The group of Ye125,126 probed the structure and stability of phospholipids in low ozone concentration by a combination of π–A isotherms, vibrational spectroscopy, and force microscopy. Their results demonstrate that the CC moieties in the unsaturated lipids are selectively oxidized by a trace amount of ozone in the ambient environment. The oxidation process, however, is partially inhibited by mixing with the saturated lipids in the monolayers. This information is useful to understand the stability and functionality of cell membranes under similar conditions.
Ozone is also known to be a notorious air pollutant. The US Environmental Protection Agency (EPA) concluded that ozone pollution poses serious health threats ranging from productive and development harm to early death.127,128 Chemical interactions of ozone with protein are shown to induce post-translational modifications, similar to nitration, rendering the altered protein more allergenic than the original.99 A number of studies investigated the general mechanisms and kinetics of protein exposure to O3 as well as protein nitration.104,105,129 Protein exposure to ozone leads to formation of dimers105 and oligomers130 which may also proceed through the tyrosyl radicals as shown in Fig. 5. The chemical mechanism for the dimerization of protein such as bovine serum albumin (BSA), a globular protein with molecular mass of 67 kDa and 21 tyrosine residues per molecule, by O3 proceeds through a two-step mechanism, as suggested in previous studies.105 The first step involves the formation of long-lived reactive oxygen intermediates (ROI-1) from the reaction of a Tyr residue of the protein backbone with O3. In the second step, the ROI-1 reacts with each other to form protein dimers. A dimer itself can react further with O3 forming tyrosyl radicals, yielding a second type of reactive oxygen intermediate (ROI-2), which may react with ROI-1 to form a trimer (R5)–(R11).130
| O3 + BSA → c1ROI-1 + (1 − c1) oxidized monomer | (R5) |
| ROI-1 + ROI-1 → dimer | (R6) |
| Dimer + O3 → c2ROI-2 + (1 − c2) oxidized dimer | (R7) |
| ROI-2 + ROI-1 → trimer | (R8) |
| Trimer + O3 + BSA → c3ROI-3 + (1 − c3) oxidized trimer | (R9) |
| ROI-2 + ROI-2 → tetramer | (R10) |
| ROI-1 + ROI-3 → tetramer | (R11) |
 | ||
| Fig. 5 Schematic overview of the most relevant reactions and intermediates for protein oligomerization upon exposure of protein to environmentally relevant O3 concentrations. Reprinted from Kampf et al., Environ. Sci. Technol., 2015, 49, 10859–10886. | ||
Geddes and coworkers studied the transformation of dipeptides in an unsaturated organic matrix (i.e., oleic acid) upon exposure to ozone. Importantly, their results showed that peptide groups that do not feature side groups susceptible to direct ozonolysis may still undergo chemical transformations within the particle as a result of reactions with the ozonolysis products of the unsaturated components. Using photoelectron resonance capture ionization aerosol mass spectrometry (PERCI-AMS), they found that upon exposure to ozone, mixed particles containing a dipeptide and oleic acid resulted in the loss of peptide ions and carrier matrix and the appearance of ion signals corresponding to high molecular weight imides and amides. This type of reactivity is hypothesized to be of importance for aerosols containing cell derived materials including proteinaceous matter and unsaturated components such as fatty acids, phospholipids, and other unsaturated lipids.134
It is known that the viability of bacteria on surfaces is directly affected upon exposure to ozone.135 Furthermore, it was found that the absorption and fluorescence of aromatic amino acids in solution136,137 and in animal tissues138 changed upon ozone exposure. However, the effects of ozone on the fluorescence properties of the bacterial component of bioaerosols have not been extensively studied. Studies on the fluorescence spectra of un-aerosolized pollens treated with ozone were conducted by Roshchina.139,140 Effects of ozone and relative humidity on fluorescence spectra of bioaerosol particles have been investigated in a number of studies to simulate atmospheric processing. Santarpia et al.141 exposed samples of aerosolized Yersinia rohdei and MS2 bacteriophage-in-Escherichia coli lysate to atmospheric concentrations of ozone and found that the fluorescence peak at 330 nm decreased in intensity and became slightly blue shifted. Using a rotating drum chamber, Ratnesar-Shumate and coworkers142 conducted aging experiments by subjecting bioaerosols, Bacillus thuringiensis Al Hakam (BtAH) spores and MS2 bacteriophages to O3 and varying RH. The experiments yielded a significant decrease in the fluorescence for both BtAH and MS2 at high ozone concentration and high RH when excited at 263 nm excitation. The decreases in 263 nm excited fluorescence are indicative of hydrolysis and oxidation of tryptophan in the aerosols. Similarly, Pan et al. studied the properties of single octapeptide bioaerosol particles when exposed to O3 and RH.143 The results of these studies indicate that the physical and biological properties of bioaerosols change significantly after exposure to ozone and water vapor. The exact mechanisms for the observed changes were not reported and require future study.
Finally, ozone can dramatically change the structure and functionality of many biomolecules.144,145 Studying the effect of ozone exposure at the air–water interface, Hemming and coworkers146 found structural damage to the lung surfactant protein B. In addition, they found out that the presence of phospholipid, DPPC does not prevent oxidation of the peptide at the conditions in which the study was conducted. Similarly, ozone exposure caused degradation and rearrangement of lung surfactant phospholipid, POPC.147 Novel mass spectrometric approaches have enabled researchers to enquire the interaction of O3 with biomolecules at an air–liquid interface. Under conditions relevant to the interaction of ambient O3 with lung surfactant, these phase-boundary experiments confirm that cysteine (Cys) residues are the preferential sites of oxidation in peptides and proteins.148–151 Cys often serves as the redox active site in peptides and proteins. Recently, Su et al.152 reported the valence electronic properties and pH-dependent ionization energies of Cys aqueous bioaerosols using the aerosol VUV photoelectron spectroscopy. Ionization energy determines how readily the electron can be taken away from the donor. Their results show altered molecular orbital character of Cys when its form changes with pH.152 Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are also prone to the oxidizing potential of ozone.145,153,154
D. Other trace gases, new reaction pathways and photochemistry
Another important trace gas in the atmosphere is sulfur dioxide (SO2). Although SO2 is known to react with surfaces in mineral dust,155–157 its reactive collisions with biological and organic aerosol have not been well explored. Shang et al.158 investigated the uptake of SO2 on oleic acid (OA), which is a monounsaturated long fatty-acid found in plant membranes that has been detected in atmospheric aerosols.159 Their experiments on the heterogeneous uptake of SO2 on OA showed a decreasing uptake coefficient with increasing SO2 concentration, suggesting a Langmuir–Hinshelwood type mechanism characterized by the adsorption of the oxidant followed by a chemical reaction on the surface or within the bulk. In the presence of water vapor, the uptake coefficient of the reaction revealed an upward trend with increasing RH. Monitoring the products of the heterogeneous reaction of OA with SO2 using high resolution mass spectrometry (HRMS) revealed that, devoid of any other oxidants, sulphur-containing compounds were produced, leading the authors to conclude the formation of organosulfates.158 The direct SO2 addition may be relevant in highly populated megacities such as those in China where sulphate levels are high.Another type of reaction in the atmosphere is the acid–base reaction. It has been very well documented in aerosol studies, in both atmospheric and laboratory settings, that atmospheric nitric acid (HNO3) can displace chloride in the SSA particles rendering the aerosol deficient in chloride ion content.51,58 Single particle analysis on substrate deposited particles collected from a unique ocean-atmosphere facility160 show that biologicals exhibit heterogeneous reactivity towards HNO3.161 Using Raman microspectroscopy, Trueblood et al. proceed to show that lipopolysaccharide (LPS), a biomolecule found in the outer membrane of Gram-negative bacteria that has been detected in SSA, does react with HNO3 as shown in Fig. 6.162 This is manifested by the growth of nitrate peaks (ν4 724 cm−1, ν1 1066 cm−1, and ν4 1384 cm−1) following exposure of atomized LPS to HNO3 acid vapor. Highlighted in red, Fig. 7 shows the basic components of the complex structure of lipid A, one of the main components of LPS. The carboxylate and phosphate groups can effectively react with HNO3.
 | ||
| Fig. 6 Raman spectra showing reactivity of LPS before (blue) and after (red) exposure to HNO3 acid. The yellow highlighted peaks represent the nitrate vibrational modes. Adapted with permission from Trueblood et al., J. Phys. Chem. A, 2016, DOI: 10.1021/acs.jpca.6b07023. | ||
 | ||
| Fig. 7 Basic components of lipid A, one of the main components of LPS that could react with gas phase nitric acid. The phosphate and carboxylate sites are highlighted and are found in the lipid A and inner core oligosaccharide regions of LPS. | ||
Hydrogen peroxide (H2O2) is a powerful oxidant in anoxic environments. Stored in ice and glacier, it is released directly into the ocean and the atmosphere upon melting and could mediate global oxidation events.163 In addition, H2O2 and organic peroxides play important roles in the cycle of oxidants and the formation of SOA in the atmosphere.164 H2O2 in the presence of ferrous ion (Fe2+) produces the OH radical and ferryl iron (Fe3+), which is lethal to the cell and other bioaerosols.165 Notably, the H2O2 vapor finds its use in the decontamination of areas prone to accumulate biological pollutants. For instance, the indoor environment can be contaminated with bioaerosol such as viruses, bacteria, fungi, and their spores, either from natural occurring or outdoor environmental origins.166 It is also important to note that other oxidants exist in the atmosphere that can induce chemical aging of bioaerosols. Halogen atoms (i.e., Cl, Br, I) formed from marine environments167 and less reactive radicals such as HO2, ClO, and BrO can drive oxidation of bioaerosols.
Besides the presence of oxidants, there are other stimuli that can perturb the equilibrium chemical and physical state of bioaerosols. Sunlight promotes chemical processes in atmospheric molecular systems substantiated by the presence of photons of the correct wavelengths.168 Solar illumination can initiate photochemistry to specific radiation-absorbing chromophores present in the atmosphere169 and surface ocean.170 Subsequently, this reaction release different classes of organic compounds to the atmosphere. Studies done by Ciuraru et al. have demonstrated the production of gas phase functionalized and unsaturated organic compounds resulting from the degradation of surfactants by the photosensitized reactions involving dissolved organic matter.171–173 Irradiation with UV light can directly generate a series of biochemical events inside airborne cells like, for example, photo-oxidation of proteins that cause a complex cellular response involving DNA damage, cellular transformation, and eventually cell death.174 Indirectly, protein oxidation by irradiation is caused via the formation and subsequent reactions of singlet oxygen (1O2) generated by the transfer of energy to ground triplet state molecular oxygen by protein-bound chromophores.175 Sudden shifts in temperature and changes in pH can disrupt metabolic activity of bacteria killing some species and forcing others to adopt several survival mechanisms and adapt to a stressful environment.19
Summary and future outlook
Once airborne, aerosol particles transform rapidly from their freshly emitted states as they are exposed to atmospheric oxidant gases such as OH, NO3, and O3. Through heterogeneous and multiphase chemistry, these oxidants can significantly modify bioaerosol composition and properties. OH-initiated oxidation of bioaerosols leads to H-atom abstraction, carbon–carbon bond breakage and subsequent formation of smaller molecular species. The ozonolysis reaction proceeds through fragmentation reaction to form several products including gas phase precursors for SOA formation. Nitration of biomolecules scales the protein allergenic potential upwards. The presence of other trace gases can be a path to forming new atmospherically important species like organosulfates. Thus, there are large consequences of heterogeneous and multiphase chemistry of these biologically-derived aerosol particles. Heterogeneous and multiphase oxidation can result in a loss of biomarkers needed to track the formation and chemical transport like, for example levoglucosan and abietic acid which are both important molecular markers of biomass burning aerosol.176–178 Besides these chemical transformations, there are changes to the hygroscopicity, which can potentially affect the ability of bioaerosols to be effective cloud and ice nuclei and can thus alter cloud droplet number and size and modify radiative forcing. This can impact the lifetime of a particle in the atmosphere. A summary of select studies on the reaction of atmospheric oxidants with bioaerosols is summarized in Table 1.| Reaction | Reaction products and rate constant (k) | Ref. |
|---|---|---|
| OH + DPPC | (Top down) aldehydes, ketones, organic nitrate and nitrite ions; kOH+DPPC = (7 ± 1) × 10−14 cm3 per molecule per s | Dilbeck and Finlayson-Pitts (2013)83 |
| OH + POPC | (Top-down) aldehydes, ketones, organic nitrate and nitrite ions; kOH+POPC = (3 ± 1) × 10−13 cm3 per molecule per s | Dilbeck and Finlayson-Pitts (2013)83 |
| OH + OPPC | (Bottom-up) phospholipid aldehyde, organic nitrates and carbonyl compounds | Karagulian et al. (2008, 2009)84,86 |
| OH + GSH | Sulfenic acid (GSOH−), sulfinic acid (GSO2H−), sulfonic acid (GSO3H−) | Enami et al. (2015)78 |
| NO3 + aromatic dipeptides | Nitroaromatic compounds | Gamon et al. (2014)110 |
| NO3 + aromatic amino acids | Nitroaromatic compounds | Goeschen et al. (2011)109 |
| O3/NO2 + BSA | Nitrated tyrosine, protein dimers | Shiraiwa et al. (2012),105 Franze et al. (2005)89 |
| O3 + OPPC | Secondary ozonide then further decomposed to aldehydes, carboxylic acids and anhydrides; kO3+OPPC = (4.5 ± 0.6) × 10−16 cm3 per molecule per s | Karagulian et al. (2008)118 |
| O3 + POPC | Secondary ozonide, phospholipid aldehyde, carboxylic acid | Dilbeck and Finlayson-Pitts (2013)121 |
| O3 + BSA | Protein dimers, trimers, and higher oligomers | Kampf et al. (2015)130 |
| O3 + dipeptide (dileucine) | High molecular weight imides and amides | Geddes et al. (2009)134 |
| O3 + POPG | Hydroxyhydroperoxide and the secondary ozonide | Kim et al. (2010)148 |
| O3 + cysteine | Cysteine sulfenate (CySO−), cysteine sulfinate (CySO2−), and cysteine sulfonate (CySO3−) | Enami et al. (2009)151 |
| SO2 + oleic acid | Organosulfates | Shang et al. (2016)158 |
The field is complex and truly requires expertise and practitioners from a variety of fields including biology/microbiology, biochemistry/chemistry, atmospheric science, physics, proteomics and genomics. Furthermore, the strength of our current understanding of atmospheric aerosols builds on the collaborative efforts of atmospheric observations, laboratory investigations, and computer modeling. In particular, the increase in the number of observations in recent times in parallel with careful experiments in the laboratory shows a massive acceleration in the interest of probing the complex nature of primary bioaerosols and biologically derived particulate matter, so much so that the perspective given in this article only represents a select summary. Our understanding is far from complete and a significant effort is needed to guide this science from:
(i) A greater understanding of the production mechanisms of bioaerosols,
(ii) Understanding the composition, structure and reactive sites in bioaerosols,
(iii) Identifying reaction pathways involving trace gases with bioaerosols with a focus on oxidant chemistry, acid–base chemistry and,
(iv) Integrating this information and data to improve our quantitative understanding at the process level of how bioaerosols interact with trace gases and how this alters their impacts on health and climate.
As researchers proceed to define new reaction pathways for bioaerosols, it is equally important to investigate how solar radiation affects the chemistry of ocean surface water that drives the formation and release of new compounds to the atmosphere and to understand how the interaction with water vapor changes the size, chemical composition and water uptake of these compounds.
Looking ahead, the trajectory of bioaerosol studies in particular will continue to progress in ways we can only anticipate and will constantly pique the inquisitive minds of scientists because “particulate matter matters”.179 Atmospheric chemistry a well-established field of study now gives rise to atmospheric biochemistry which is in its infancy poised to open up a new set of scientific opportunities and challenges.
Acknowledgements
This work was funded by the Center for Aerosol Impacts on Climate and the Environment (CAICE), a National Science Foundation Center for Chemical Innovation program under grant number CHE1305427.References
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- M. Pósfai and P. R. Buseck, Annu. Rev. Earth Planet. Sci., 2010, 38, 17–43 CrossRef.
- V. Ramanathan, P. J. Crutzen, J. T. Kiehl and D. Rosenfeld, Science, 2001, 294, 2119–2124 CrossRef CAS PubMed.
- U. Lohmann and J. Feichter, Atmos. Chem. Phys., 2005, 5, 715–737 CrossRef CAS.
- N. Mahowald, D. S. Ward, S. Kloster, M. G. Flanner, C. L. Heald, N. G. Heavens, P. G. Hess, J.-F. Lamarque and P. Y. Chuang, Annu. Rev. Environ. Resour., 2011, 36, 45–74 CrossRef.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea., J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- B. Nozière, M. Kalberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgić, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahnt, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, Chem. Rev., 2015, 115, 3919–3983 CrossRef PubMed.
- M. W. Hallquist, J. C. Baltensperger, U. Rudich, Y. Simpson, D. Claeys, M. Dommen, J. Donahue, N. M. George, C. Goldstein, A. H. Hamilton, J. F. Herrmann, H. Hoffmann, T. Iinuma, Y. Jang, M. Jenkin, M. E. Jimenez, J. L. Kiendler-Scharr, A. Maenhaut, W. McFiggans, G. Mentel, T. F. Monod, A. Prévôt, A. S. H. Seinfeld, J. H. Surratt, J. D. Szmigielski and R. J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- G. D'Amato, C. E. Baena-Cagnani, L. Cecchi, I. Annesi-Maesano, C. Nunes, I. Ansotegui, M. D'Amato, G. Liccardi, M. Sofia and W. G. Canonica, Multidisciplinary Respiratory Medicine, 2013, 8, 12 CrossRef PubMed.
- J. A. Bernstein, N. Alexis, C. Barnes, I. L. Bernstein, A. Nel, D. Peden, D. Diaz-Sanchez, S. M. Tarlo and P. B. Williams, J. Allergy Clin. Immunol., 2004, 114, 1116–1123 CrossRef PubMed.
- M. Kampa and E. Castanas, Environ. Pollut., 2008, 151, 362–367 CrossRef CAS PubMed.
- A. Nel, Science, 2005, 308, 804–806 CrossRef CAS PubMed.
- M. Subramanian, Nature, 2016, 534, 166–169 CrossRef CAS PubMed.
- D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Chem. Rev., 2015, 115, 4199–4217 CrossRef CAS PubMed.
- V. R. Després, J. A. Huffman, S. M. Burrows, C. Hoose, A. S. Safatov, G. A. Buryak, J. Fröhlich-Nowoisky, W. Elbert, M. O. Andreae, U. Pöschl and R. Jaenicke, Tellus, Ser. B, 2012, 64, 15598 CrossRef.
- W. Elbert, P. E. Taylor, M. O. Andreae and U. Pöschl, Atmos. Chem. Phys., 2007, 7, 4569–4588 CrossRef CAS.
- J. Fröhlich-Nowoisky, D. A. Pickersgill, V. R. Despres and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12814–12819 CrossRef PubMed.
- R. M. Bowers, A. P. Sullivan, E. K. Costello, J. J. L. Collett, R. Knight and N. Fierer, Appl. Environ. Microbiol., 2011, 77, 6350–6356 CrossRef CAS PubMed.
- L. Deguillaume, M. Leriche, P. Amato, P. A. Ariya, A.-M. Delort, U. Pöschl, N. Chaumerliac, H. Bauer, A. I. Flossmann and C. E. Morris, Biogeosciences, 2008, 5, 1073–1084 CrossRef CAS.
- J. Douwes, P. Thorne, N. Pearce and D. Heederik, Ann. Occup. Hyg., 2003, 47, 187–200 CrossRef CAS PubMed.
- M. Shiraiwa, K. Selzle and U. Pöschl, Free Radical Res., 2012, 46, 927–939 CrossRef CAS PubMed.
- A. P. Alivisatos, M. J. Blaser, E. L. Brodie, M. Chun, J. L. Dangl, T. J. Donohue, J. A. Gilbert, J. L. Green, J. K. Jansson, R. Knight, M. E. Maxon, M. J. McFall-Ngai, J. F. Miller, K. S. Pollard, E. G. Ruby and S. S. Taha, Science, 2015, 350, 507–508 CrossRef CAS PubMed.
- J. S. Biteen, P. C. Blainey, Z. G. Cardon, M. Chun, G. M. Church, P. C. Dorrestein, S. E. Fraser, J. A. Gilbert, J. K. Jansson, R. Knight, J. F. Miller, A. Ozcan, K. A. Prather, S. R. Quake, E. G. Ruby, P. A. Silver, S. Taha, G. v. d. Engh, P. S. Weiss, G. C. L. Wong, A. T. Wright and T. D. Young, ACS Nano, 2016, 10, 6–37 CrossRef CAS PubMed.
- Y. Mazar, E. Cytryn, Y. Erel and Y. Rudich, Environ. Sci. Technol., 2016, 50, 4194–4202 CrossRef CAS PubMed.
- N. DeLeon-Rodriguez, T. L. Lathem, L. M. Rodriguez-R, J. M. Barazesh, B. E. Anderson, A. J. Beyersdorf, L. D. Ziemba, M. Bergin, A. Nenes and K. T. Konstantinidis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2575–2580 CrossRef CAS PubMed.
- A. G. Hallar, G. Chirokova, I. McCubbin, T. H. Painter, C. Wiedinmyer and C. Dodson, Geophys. Res. Lett., 2011, 38, L17801 CrossRef.
- J. P. Patterson, D. B. Collins, J. M. Michaud, J. L. Axson, C. M. Sultana, T. Moser, A. C. Dommer, J. Conner, V. H. Grassian, M. D. Stokes, G. B. Deane, J. E. Evans, M. D. Burkart, K. A. Prather and N. C. Gianneschi, ACS Cent. Sci., 2016, 2, 40–47 CrossRef CAS PubMed.
- C. A. Kellogg and D. W. Griffin, Trends in Ecology and Evolution, 2006, 21, 638–644 CrossRef PubMed.
- H. Tervahattu, K. Hartonen, V. M. Kerminen, K. Kupiainen, P. Aarnio, T. Koskentalo, A. F. Tuck and V. Vaida, J. Geophys. Res., 2002, 107, D74053 Search PubMed.
- K. Hara and D. Zhang, Atmos. Environ., 2012, 47, 20–25 CrossRef CAS.
- M. Kuznetsova, C. Lee and J. Aller, Mar. Chem., 2005, 96, 359–377 CrossRef CAS.
- K. Matsumoto and M. Uematsu, Atmos. Environ., 2005, 39, 2163–2170 CrossRef CAS.
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chem. Rev., 2015, 115, 4383–4399 CrossRef CAS PubMed.
- R. Jaenicke, Science, 2005, 308, 73 CrossRef CAS PubMed.
- A. G. Miguel, G. R. Cass, M. M. Glovsky and J. Weiss, Environ. Sci. Technol., 1999, 33, 4159–4168 CrossRef CAS.
- Q. Zhang and C. Anastasio, Atmos. Environ., 2003, 37, 2247–2258 CrossRef CAS.
- K. Mopper and R. G. Zika, Nature, 1987, 325, 246–249 CrossRef CAS.
- X. Wang, C. M. Sultana, J. Trueblood, T. C. J. Hill, F. Malfatti, C. Lee, O. Laskina, K. A. Moore, C. M. Beall, C. S. McCluskey, G. C. Cornwell, Y. Zhou, J. L. Cox, M. A. Pendergraft, M. V. Santander, T. H. Bertram, C. D. Cappa, F. Azam, P. J. DeMott, V. H. Grassian and K. A. Prather, ACS Cent. Sci., 2015, 1, 124–131 CrossRef CAS PubMed.
- C. Bozzetti, K. R. Daellenbach, C. Hueglin, P. Fermo, J. Sciarer, A. Kasper-Giebl, Y. Mazar, G. Abbaszade, M. E. Kazzi, R. Gonzalez, T. Shuster-Meiseles, M. Flasch, R. Wolf, A. Křepelová, F. Canonaco, J. Schnelle-Kreis, J. G. Slowik, R. Zimmermann, Y. Rudich, U. Baltensperger, I. E. Haddad and A. S. H. Prévôt, Environ. Sci. Technol., 2016, 50, 3425–3434 CrossRef CAS PubMed.
- J. M. Creamean, K. J. Suski, D. Rosenfeld, A. Cazorla, P. J. DeMott, R. C. Sullivan, A. B. White, F. M. Ralph, P. Minnis, J. M. Comstock, J. M. Tomlinson and K. A. Prather, Science, 2013, 339, 1572–1578 CrossRef CAS PubMed.
- R. Wolf, J. G. Slowik, C. Schaupp, P. Amato, H. Saathoff, O. Möhler, A. S. H. Prévôt and U. Baltensperger, J. Mass Spectrom., 2015, 50, 662–671 CrossRef CAS PubMed.
- J. Schneider, F. Freutel, S. R. Zorn, Q. Chen, D. K. Farmer, J. L. Jimenez, S. T. Martin, P. Artaxo, A. Wiedensohler and S. Borrmann, Atmos. Chem. Phys., 2011, 11, 11415–11429 CAS.
- S. E. Saari, M. J. Putkiranta and J. Keskinen, Atmos. Environ., 2013, 71, 202–209 CrossRef CAS.
- S. Saari, S. Järvinen, T. Reponen, J. Mensah-Attipoe, P. Pasanen, J. Toivonen and J. Keskinen, Aerosol Sci. Technol., 2016, 50, 126–132 CrossRef CAS.
- A. Tripathi, R. E. Jabbour, J. A. Guicheteau, S. D. Christesen, D. K. Emge, A. W. Fountain, J. R. Bottiger, E. D. Emmons and A. Peter, Anal. Chem., 2009, 81, 6981–6990 CrossRef CAS PubMed.
- A. Sengupta, N. Brar and E. J. Davis, J. Colloid Interface Sci., 2007, 309, 36–43 CrossRef CAS PubMed.
- D. J. Caruana, Analyst, 2011, 136, 4641–4652 RSC.
- J. Ho, Anal. Chim. Acta, 2002, 457, 125–148 CrossRef CAS.
- S. M. Burrows, W. Elbert, M. G. Lawrence and U. Pöschl, Atmos. Chem. Phys., 2009, 9, 9263–9280 CAS.
- A. R. Ravishankara, Science, 1997, 276, 1058–1065 CrossRef CAS.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- D. J. Donaldson and K. T. Valsaraj, Environ. Sci. Technol., 2010, 44, 865–873 CrossRef CAS PubMed.
- U. Pöschl and M. Shiraiwa, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- I. J. George and J. P. D. Abbatt, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- R. C. Chapleski, Y. Zhang, D. Troya and J. R. Morris, Chem. Soc. Rev., 2016, 45, 3731–3746 RSC.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- D. M. Cwiertny, M. A. Young and V. H. Grassian, Annu. Rev. Phys. Chem., 2008, 59, 27–51 CrossRef CAS PubMed.
- M. J. Rossi, Chem. Rev., 2003, 103, 4823–4882 CrossRef CAS PubMed.
- P. J. Ziemann and R. Atkinson, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- J. H. Kroll, C. Y. Lim, S. H. Kessler and K. R. Wilson, J. Phys. Chem. A, 2015, 119, 10767–10783 CrossRef CAS PubMed.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- P. A. Ariya, J. Sun, N. A. Eltouny, E. D. Hudson, C. T. Hayes and G. Kos, Int. Rev. Phys. Chem., 2009, 28, 1–32 CrossRef CAS.
- D. A. Knopf, P. A. Alpert, B. Wang and J. Y. Aller, Nat. Geosci., 2011, 4, 88–90 CrossRef CAS.
- S. M. Burrows, C. Hoose, U. Pöschl and M. G. Lawrence, Atmos. Chem. Phys., 2013, 13, 245–267 Search PubMed.
- N. Hiranuma, O. Möhler, K. Yamashita, T. Tajiri, A. Saito, A. Kiselev, N. Hoffmann, C. Hoose, E. Jantsch, T. Koop and M. Murakami, Nat. Geosci., 2015, 8, 273–277 CrossRef CAS.
- U. Pöschl, S. T. Martin, B. Sinha, Q. Q. Chen, S. S. Gunthe, J. A. Huffman, S. Borrmann, D. K. Farmer, R. M. Garland, G. Helas, J. L. Jimenez, S. M. King, A. Manzi, E. Mikhailov, T. Pauliquevis, M. D. Petters, A. J. Prenni, P. Roldin, D. Rose, J. Schneider, H. Su, S. R. Zorn, P. Artaxo and M. O. Andreae, Science, 2010, 329, 1513–1516 CrossRef PubMed.
- J. Sun and P. A. Ariya, Atmos. Environ., 2006, 40, 795–820 CrossRef CAS.
- T. W. Wilson, L. A. Ladino, P. A. Alpert, M. N. Breckels, I. M. Brooks, J. Browse, S. M. Burrows, K. S. Carslaw, J. A. Huffman, C. Judd, W. P. Kilthau, R. H. Mason, G. McFiggans, L. A. Miller, J. J. Nájera, E. Polishchuk, S. Rae, C. L. Schiller, M. Si, J. V. Temprado, T. F. Whale, J. P. S. Wong, O. Wurl, J. D. Yakobi-Hancock, J. P. D. Abbatt, J. Y. Aller, A. K. Bertram, D. A. Knopf and B. J. Murray, Nature, 2015, 525, 234–238 CrossRef CAS PubMed.
- R. Atkinson, Atmos. Chem. Phys., 2003, 3, 2233–2307 CrossRef CAS.
- Y. Yang, M. Shao, X. Wang, A. C. Nölscher, S. Kessel, A. Guenther and J. Williams, Atmos. Environ., 2016, 134, 147–161 CrossRef CAS.
- T. Hoffmann, R.-J. Huang and M. Kalberer, Anal. Chem., 2011, 83, 4649–4664 CrossRef CAS PubMed.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- S. A. Montzka, M. Krol, E. Dlugokencky, B. Hall, P. Jöckel and J. Lelieveld, Science, 2011, 331, 67–69 CrossRef CAS PubMed.
- J. M. Anglada, M. Martins-Costa, J. S. Francisco and M. F. Ruiz-López, Acc. Chem. Res., 2015, 48, 575–583 CrossRef CAS PubMed.
- M. C. Green, D. G. Fedorov, K. Kitaura, J. S. Francisco and L. V. Slipchenko, J. Chem. Phys., 2013, 138, 074111 CrossRef PubMed.
- M. C. G. S. Stelzleni and J. S. Francisco, J. Phys. Chem. A, 2013, 117, 550–565 CrossRef PubMed.
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2015, 6, 3935–3943 CrossRef CAS PubMed.
- H. Tervahattu, J. Juhanoja and K. Kupiainen, J. Geophys. Res., 2002, 107, D164319 Search PubMed.
- J. C. Marty, A. Saliot, P. Buat-Ménard, R. Chesselet and K. A. Hunter, J. Geophys. Res., 1979, 84, 5707–5716 CrossRef CAS.
- R. B. Gagosian, E. T. Peltzer and O. C. Zafiriou, Nature, 1981, 291, 312–315 CrossRef CAS.
- G. v. Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef PubMed.
- C. W. Dilbeck and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2013, 15, 9833–9844 RSC.
- F. Karagulian, C. W. Dilbeck and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2009, 113, 7205–7212 CrossRef CAS PubMed.
- G. B. Ellison, A. F. Tuck and V. Vaida, J. Geophys. Res., 1999, 104, 11633–11641 CrossRef CAS.
- F. Karagulian, C. W. Dilbeck and B. J. Finlayson-Pitts, J. Am. Chem. Soc., 2008, 130, 11272–11273 CrossRef CAS PubMed.
- J. M. Roberts, Atmos. Environ., 1990, 24, 243–287 CrossRef.
- K. R. Wilson, D. S. Peterka, M. Jimenez-Cruz, S. R. Leone and M. Ahmed, Phys. Chem. Chem. Phys., 2006, 8, 1884–1890 RSC.
- T. Franze, M. G. Weller, R. Niessner and U. Pöschl, Environ. Sci. Technol., 2005, 39, 1673–1678 CrossRef CAS PubMed.
- C. Ackaert, S. Kofler, J. Horejs-Hoeck, N. Zulehner, C. Asam, S. v. Grafenstein, J. E. Fuchs, P. Briza, K. R. Liedl, B. Bohle, F. Ferreira, H. Brandstetter, G. J. Oostingh and A. Duschl, PLoS One, 2014, 9, e104520 Search PubMed.
- P. Barzaghi and H. Herrmann, Phys. Chem. Chem. Phys., 2002, 4, 3669–3675 RSC.
- T. Umschlag, R. Zellner and H. Herrmann, Phys. Chem. Chem. Phys., 2002, 4, 2975–2982 RSC.
- R. Belloli, B. Barletta, E. Bolzacchini, S. Meinardi, M. Orlandi and B. Rindone, J. Chromatogr. A, 1999, 846, 277–281 CrossRef CAS.
- E. Bolzacchini, M. Bruschi, J. Hjorth, S. Meinardi, M. Orlandi, B. Rindone and E. Rosenbohm, Environ. Sci. Technol., 2001, 35, 1791–1797 CrossRef CAS PubMed.
- J. S. Beckman, H. Ischiropoulos, L. Zhu, M. v. d. Woerd, C. Smith, J. Chen, J. Harrison, J. C. Martin and M. Tsai, Arch. Biochem. Biophys., 1992, 298, 438–445 CrossRef CAS PubMed.
- A. Graham, N. Hogg, B. Kalyanaraman, V. O'Leary, V. Darley-Usmar and S. Moncada, FEBS Lett., 1993, 330, 181–185 CrossRef CAS PubMed.
- J. Goschnick and J. Schuricht, J. Aerosol Sci., 1996, 27, S229–S230 CrossRef.
- T. Franze, M. G. Weller, R. Niessner and U. Pöschl, Analyst, 2003, 128, 824–831 RSC.
- Y. K. Gruijthuijsen, I. Grieshuber, A. Stocklinger, U. Tischler, T. Fehrenbach, M. G. Weller, L. Vogel, S. Vieths, U. Pöschl and A. Duschl, Int. Arch. Allergy Immunol., 2006, 141, 265–275 CrossRef CAS PubMed.
- N. Lang-Yona, T. Shuster-Meiseles, Y. Mazar, O. Yarden and Y. Rudich, Sci. Total Environ., 2016, 541, 365–371 CrossRef CAS PubMed.
- H. Yang, Y. Zhang and U. Pöschl, Anal. Bioanal. Chem., 2010, 397, 879–886 CrossRef CAS PubMed.
- Y. Zhang, H. Yang and U. Pöschl, Anal. Bioanal. Chem., 2011, 399, 459–471 CrossRef CAS PubMed.
- K. Selzle, C. Ackaert, C. J. Kampf, A. T. Kunert, A. Duschl, G. J. Oostingh and U. Pöschl, Anal. Bioanal. Chem., 2013, 405, 8945–8949 CrossRef CAS PubMed.
- M. Shiraiwa, Y. Sosedova, A. Rouviere, H. Yang, Y. Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Poschl, Nat. Chem., 2011, 3, 291–295 CrossRef CAS PubMed.
- M. Shiraiwa, K. Selzle, H. Yang, Y. Sosedova, M. Ammann and U. Pöschl, Environ. Sci. Technol., 2012, 46, 6672–6680 CrossRef CAS PubMed.
- K. Reinmuth-Selzle, C. Ackaert, C. J. Kampf, M. Samonig, M. Shiraiwa, S. Kofler, H. Yang, G. Gadermaier, H. Brandstetter, C. G. Huber, A. Duschl, G. J. Oostingh and U. Pöschl, J. Proteome Res., 2014, 13, 1570–1577 CrossRef CAS PubMed.
- L. Sandhiya, P. Kolandaivel and K. Senthilkumar, J. Phys. Chem. B, 2014, 118, 3479–3490 CrossRef CAS PubMed.
- D. C. E. Sigmund and U. Wille, Chem. Commun., 2008, 2121–2123 RSC.
- C. Goeschen, N. Wibowo, J. M. White and U. Wille, Org. Biomol. Chem., 2011, 9, 3380–3385 CAS.
- L. F. Gamon, J. M. White and U. Wille, Org. Biomol. Chem., 2014, 12, 8280–8287 CAS.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. d. Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Phys. Chem. Chem. Phys., 2016, 18, 10241–10254 RSC.
- Y. Rudich, Chem. Rev., 2003, 103, 5097–5124 CrossRef CAS PubMed.
- C. Denjean, P. Formenti, B. Picquet-Varrault, M. Camredon, E. Pangui, P. Zapf, Y. Katrib, C. Giorio, A. Tapparo, B. Temime-Roussel, A. Monod, B. Aumont and J. F. Doussin, Atmos. Chem. Phys., 2015, 15, 883–897 CrossRef.
- K. Kristensen, T. Cui, H. Zhang, A. Gold, M. Glasius and J. D. Surratt, Atmos. Chem. Phys., 2014, 14, 4201–4218 Search PubMed.
- T. Berkemeier, M. Ammann, T. F. Mentel, U. Pöschl and M. Shiraiwa, Environ. Sci. Technol., 2016, 50, 6334–6342 CrossRef CAS PubMed.
- F. Karagulian, A. S. Lea, C. W. Dilbeck and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2008, 10, 528–541 RSC.
- O. Vesna, S. Sjogren, E. Weingartner, V. Samburova, M. Kalberer, H. W. Gäggeler and M. Ammann, Atmos. Chem. Phys., 2008, 8, 4683–4690 CrossRef CAS.
- O. Vesna, M. Sax, M. Kalberer, A. Gaschen and M. Ammann, Atmos. Environ., 2009, 43, 3662–3669 CrossRef CAS.
- C. W. Dilbeck and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2013, 15, 1990–2002 RSC.
- C. C. Lai, B. J. Finlayson-Pitts and W. V. Willis, Chem. Res. Toxicol., 1990, 3, 517–523 CrossRef CAS PubMed.
- C. C. Lai, S. H. Yang and B. J. Finlayson-Pitts, Langmuir, 1994, 10, 4637–4644 CrossRef CAS.
- Y. Wadia, D. J. Tobias, R. Stafford and B. J. Finlayson-Pitts, Langmuir, 2000, 16, 9321–9330 CrossRef CAS.
- L. Qiao, A. Ge, Y. Liang and S. Ye, J. Phys. Chem. B, 2015, 119, 14188–14199 CrossRef CAS PubMed.
- L. Qiao, A. Ge, M. Osawa and S. Ye, Phys. Chem. Chem. Phys., 2013, 15, 17775–17785 RSC.
- U. S. EPA, U.S. Environmental Protection Agency, Washington, DC, 2013, EPA/600/R-10/076F Search PubMed.
- M. Jerrett, R. T. Burnett, C. A. Pope, K. Ito, G. Thurston, D. Krewski, Y. Shi, E. Calle and M. Thun, N. Engl. J. Med., 2009, 360, 1085–1095 CrossRef CAS PubMed.
- M. Shiraiwa, M. Ammann, T. Koop and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS PubMed.
- C. J. Kampf, F. Liu, K. Reinmuth-Selzle, T. Berkemeier, H. Meusel, M. Shiraiwa and U. Pöschl, Environ. Sci. Technol., 2015, 49, 10859–10866 CrossRef CAS PubMed.
- E. Mikhailov, S. Vlasenko, S. T. Martin, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2009, 9, 9491–9522 CrossRef CAS.
- T. Koop, J. Bookhold, M. Shiraiwa and U. Pöschl, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC.
- T. Berkemeier, S. S. Steimer, U. K. Krieger, T. Peter, U. Pöschl, M. Ammann and M. Shiraiwa, Phys. Chem. Chem. Phys., 2016, 18, 12662–12674 RSC.
- S. Geddes, J. Zahardis and G. A. Petrucci, J. Atmos. Chem., 2009, 63, 187–202 CrossRef CAS.
- L. Fan, J. Song, P. D. Hildebrand and C. F. Forney, J. Appl. Microbiol., 2002, 93, 144–148 CrossRef CAS PubMed.
- A. V. Ignatenko, B. A. Tatarinov, N. N. Khovratovich and V. P. K. S. N. Cherenkevich, J. Appl. Spectrosc., 1982, 37, 781–784 CrossRef.
- A. V. Ignatenko, J. Appl. Spectrosc., 1988, 49, 691–695 CrossRef.
- E. Fujimori, Eur. J. Biochem., 1985, 152, 299–306 CrossRef CAS PubMed.
- V. V. Roshchina and E. V. Mel'nikova, Russ. J. Plant Physiol., 2001, 48, 74–83 CrossRef CAS.
- V. V. Roshchina, J. Fluoresc., 2003, 13, 403–420 CrossRef CAS.
- J. L. Santarpia, Y.-L. Pan, S. C. Hill, N. Baker, B. Cottrell, L. McKee, S. Ratnesar-Shumate and R. G. Pinnick, Opt. Express, 2012, 20, 29867–29881 CrossRef CAS PubMed.
- S. Ratnesar-Shumate, Y.-L. Pan, S. C. Hill, S. Kinahan, E. Corson, J. Eshbaugh and J. L. Santarpia, J. Quant. Spectrosc. Radiat. Transfer, 2015, 153, 13–28 CrossRef CAS.
- Y.-L. Pan, J. L. Santarpia, S. Ratnesar-Shumate, E. Corson, J. Eshbaugh, S. C. Hill, C. C. Williamson, M. Coleman, C. Bare and S. Kinahan, J. Quant. Spectrosc. Radiat. Transfer, 2014, 133, 538–550 CrossRef CAS.
- J. B. Mudd, R. Leavitt, A. Ongun and T. T. McManus, Atmos. Environ., 1969, 3, 669–681 CrossRef CAS PubMed.
- F. Cataldo, Int. J. Biol. Macromol., 2006, 38, 248–254 CrossRef CAS PubMed.
- J. M. Hemming, B. R. Hughes, A. R. Rennie, S. Tomas, R. A. Campbell, A. V. Hughes, T. Arnold, S. W. Botchway and K. C. Thompson, Biochemistry, 2015, 54, 5185–5197 CrossRef CAS PubMed.
- K. C. Thompson, S. H. Jones, A. R. Rennie, M. D. King, A. D. Ward, B. R. Hughes, C. O. M. Lucas, R. A. Campbell and A. V. Hughes, Langmuir, 2013, 29, 4594–4602 CrossRef CAS PubMed.
- H. I. Kim, H. Kim, Y. S. Shin, L. W. Beegle, W. A. Goddard, J. R. Heath, I. Kanik and J. L. Beauchamp, J. Phys. Chem. B, 2010, 114, 9496–9503 CrossRef CAS PubMed.
- H. I. Kim, H. Kim, Y. S. Shin, L. W. Beegle, S. S. Jang, E. L. Neidholdt, W. A. Goddard, J. R. Heath, I. Kanik and J. L. Beauchamp, J. Am. Chem. Soc., 2010, 132, 2254–2263 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Chem. Res. Toxicol., 2009, 22, 35–40 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2009, 113, 9356–9358 CrossRef CAS PubMed.
- C.-C. Su, Y. Yu, P.-C. Chang, Y.-W. Chen, I.-Y. Chen, Y.-Y. Lee and C. C. Wang, J. Phys. Chem. Lett., 2015, 6, 817–823 CrossRef CAS PubMed.
- F. Cataldo, Ozone: Sci. Eng., 2006, 28, 317–328 CrossRef CAS.
- F. Cataldo, Polym. Degrad. Stab., 2005, 89, 274–281 CrossRef CAS.
- J. W. Adams, D. Rodriguez and R. A. Cox, Atmos. Chem. Phys., 2005, 5, 2679–2689 CrossRef CAS.
- L. Y. Wu, S. R. Tong, W. G. Wang and M. F. Ge, Atmos. Chem. Phys., 2011, 11, 6593–6605 CAS.
- C. E. Nanayakkara, J. Pettibone and V. H. Grassian, Phys. Chem. Chem. Phys., 2012, 14, 6957–6966 RSC.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, Environ. Sci. Technol. Lett., 2016, 3, 67–72 CrossRef CAS.
- B. Graham, P. Guyon, P. E. Taylor, P. Artaxo, W. Maenhaut, M. M. Glovsky, R. C. Flagan and M. O. Andreae, J. Geophys. Res.: Atmos., 2003, 108, 4766 Search PubMed.
- K. A. Prather, T. H. Bertram, V. H. Grassian, G. B. Deane, M. D. Stokes, P. J. DeMott, L. I. Aluwihare, B. P. Palenik, F. Azam, J. H. Seinfeld, R. C. Moffet, M. J. Molina, C. D. Cappa, F. M. Geiger, G. C. Roberts, L. M. Russell, A. P. Ault, J. Baltrusaitis, D. B. Collins, C. E. Corrigan, L. A. Cuadra-Rodriguez, C. J. Ebben, S. D. Forestieri, T. L. Guasco, S. P. Hersey, M. J. Kim, W. F. Lambert, R. L. Modini, W. Mui, B. E. Pedler, M. J. Ruppel, O. S. Ryder, N. G. Schoepp, R. C. Sullivan and D. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7550–7555 CrossRef CAS PubMed.
- A. P. Ault, T. L. Guasco, J. Baltrusaitis, O. S. Ryder, J. V. Trueblood, D. B. Collins, M. J. Ruppel, L. A. Cuadra-Rodriguez, K. A. Prather and V. H. Grassian, J. Phys. Chem. Lett., 2014, 5, 2493–2500 CrossRef CAS PubMed.
- J. V. Trueblood, A. D. Estillore, C. Lee, J. A. Dowling, K. A. Prather and V. H. Grassian, J. Phys. Chem. A, 2016 DOI:10.1021/acs.jpca.6b07023.
- M.-C. Liang, H. Hartman, R. E. Kopp, J. L. Kirschvink and Y. L. Yung, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18896–18899 CrossRef CAS PubMed.
- Q. Q. Wu, L. B. Huang, H. Liang, Y. Zhao, D. Huang and Z. M. Chen, Atmos. Chem. Phys., 2015, 15, 6851–6866 CrossRef CAS.
- D. Touati, Arch. Biochem. Biophys., 2000, 373, 1–6 CrossRef CAS PubMed.
- W. A. Rutala and D. J. Weber, Am. J. Infect. Control, 2016, 44, e69–e76 CrossRef PubMed.
- C. B. Faxon and D. T. Allen, Environ. Chem., 2013, 10, 221–233 CrossRef CAS.
- V. Vaida, J. Phys. Chem. A, 2009, 113, 5–18 CrossRef CAS PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- L. J. Carpenter and P. D. Nightingale, Chem. Rev., 2015, 115, 4015–4034 CrossRef CAS PubMed.
- R. Ciuraru, L. Fine, M. v. Pinxteren, B. D'Anna, H. Herrmann and C. George, Environ. Sci. Technol., 2015, 49, 13199–13205 CrossRef CAS PubMed.
- R. Ciuraru, L. Fine, M. v. Pinxteren, B. D'Anna, H. Herrmann and C. George, Sci. Rep., 2015, 5, 12741 CrossRef CAS PubMed.
- H. Fu, R. Ciuraru, Y. Dupart, M. Passananti, L. Tinel, S. Rossignol, S. Perrier, D. J. Donaldson, J. Chen and C. George, J. Am. Chem. Soc., 2015, 137, 8348–8351 CrossRef CAS PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 CAS.
- M. J. Davies and R. J. W. Truscott, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS.
- M. Shiraiwa, U. Pöschl and D. A. Knopf, Environ. Sci. Technol., 2012, 46, 6630–6636 CrossRef CAS PubMed.
- A. M. Arangio, J. H. Slade, T. Berkemeier, U. Pöschl, D. A. Knopf and M. Shiraiwa, J. Phys. Chem. A, 2014, 119, 4533–4544 CrossRef PubMed.
- D. A. Knopf, S. M. Forrester and J. H. Slade, Phys. Chem. Chem. Phys., 2011, 13, 21050–21062 RSC.
- F. Dominici, M. Greenstone and C. R. Sunstein, Science, 2014, 344, 257–259 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |