
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of methanol with the oxygen-evolving complex: atomistic models, channel identification, species dependence, and mechanistic implications†
Marius
Retegan‡
 and
Dimitrios A.
Pantazis
*
and
Dimitrios A.
Pantazis
*
Max Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: dimitrios.pantazis@cec.mpg.de
First published on 5th July 2016
Abstract
Methanol has long being used as a substrate analogue to probe access pathways and investigate water delivery at the oxygen-evolving complex (OEC) of photosystem-II. In this contribution we study the interaction of methanol with the OEC by assembling available spectroscopic data into a quantum mechanical treatment that takes into account the local channel architecture of the active site. The effect on the magnetic energy levels of the Mn4Ca cluster in the S2 state of the catalytic cycle can be explained equally well by two models that involve either methanol binding to the calcium ion of the cluster, or a second-sphere interaction in the vicinity of the “dangler” Mn4 ion. However, consideration of the latest 13C hyperfine interaction data shows that only one model is fully consistent with experiment. In contrast to previous hypotheses, methanol is not a direct ligand to the OEC, but is situated at the end-point of a water channel associated with the O4 bridge. Its effect on magnetic properties of plant PS-II results from disruption of hydrogen bonding between O4 and proximal channel water molecules, thus enhancing superexchange (antiferromagnetic coupling) between the Mn3 and Mn4 ions. The same interaction mode applies to the dark-stable S1 state and possibly to all other states of the complex. Comparison of protein sequences from cyanobacteria and plants reveals a channel-altering substitution (D1-Asn87 versus D1-Ala87) in the proximity of the methanol binding pocket, explaining the species-dependence of the methanol effect. The water channel established as the methanol access pathway is the same that delivers ammonia to the Mn4 ion, supporting the notion that this is the only directly solvent-accessible manganese site of the OEC. The results support the pivot mechanism for water binding at a component of the S3 state and would be consistent with partial inhibition of water delivery by methanol. Mechanistic implications for enzymatic regulation and catalytic progression are discussed.
Introduction
A fundamental aspect of mechanistic regulation in metalloenzymes is how the active site, where catalytic transformation of substrates occurs, is connected to the protein environment. Connections may include solvent-permeable channels for the transport of substrates and products, pathways for the uptake or release of protons, and—in the case of redox transformations—electron transfer pathways to other sites of the enzyme or to peripheral cofactors. Such channels and pathways are not always easily identifiable from crystallographic models, and when they are (for example, in the form of ordered water chains) their function with respect to catalytic activity is not necessarily obvious.The water-oxidizing tetramanganese-calcium cluster of photosystem-II (PS-II) that forms the inorganic core of the oxygen-evolving complex (OEC, Fig. 1)1–6 is a prime example of a metallocofactor embedded in a membrane enzyme of such complexity7–10 that many structural aspects of substrate delivery, product evolution, and proton removal remain insufficiently defined. Driven by photo-induced charge separation at the chlorophyll complex P680+, the OEC cycles through five redox states denoted Si, where i indicates the number of accumulated oxidizing equivalents (i = 0–4).11,12 Stored in the dark the catalytic centers adopt predominantly the S1 state, whereas the S0, S2 and S3 states are metastable. In these intermediates the Mn oxidation states evolve from Mn(III)3Mn(IV) in S0 to Mn(IV)4 in S3 (for a recent review on oxidation state assignments, see Krewald et al.13). O–O bond formation14,15 occurs upon advancement to the reactive and still unobserved S4 state; following O2 release the cluster is reset to the most reduced S0 state. Strict control of accessibility and ordered substrate binding at the active site, buried into the membrane-embedded part of PS-II, have long been recognized to be functionally critical in regulating the complex chemistry of water oxidation and avoiding side reactions.16 A detailed understanding of the water oxidation process therefore requires a positive identification of how, when, and where precisely the substrate water binds at the cluster—a great challenge that requires the substrate to be differentiated from other water-derived ligands, solvent or structural water, and water involved in proton relays.
 | ||
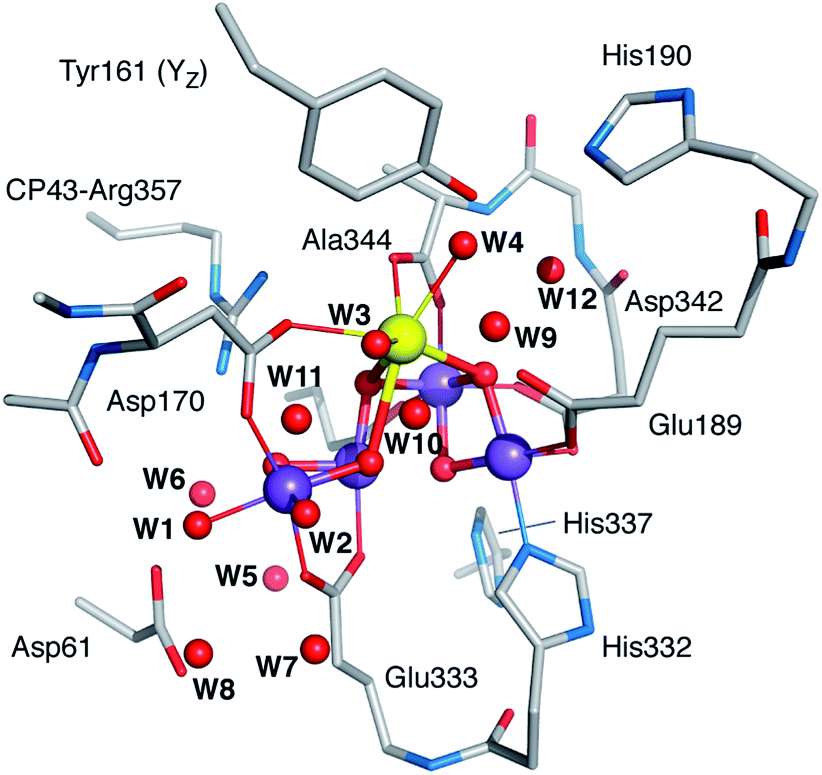
| Fig. 1 The OEC and the proximal branches of associated water channels. Selected residues are shown for orientation. Coordinates are taken from QM/MM models of the S2 state.17 Inset: schematic diagram and labelling of the inorganic core. | ||
Based on analysis of crystallographic models and molecular dynamics simulations,18–30 there is presently consensus on the existence of at least three well-defined water channels associated with the Mn4CaO5 cluster (Fig. 1).31 Two of them appear to make contact with the Mn4 ion or its immediate environment, while the third one with the calcium ion, although the presence of water molecules bridging between the two Mn4-bound water-derived terminal ligands and the two Ca2+-bound waters complicates the assignment of distinct contact points. The role of these channels in water delivery, proton removal,32 and dioxygen release remains undefined and contested,19–21,24,30,33–35 and so is the structural basis of these functionalities.
Substrate analogues such as ammonia and methanol can interact with the OEC or at least approach the cluster sufficiently to serve as spectroscopic probes.36–66 By perturbing the geometric and electronic structure of the manganese cluster, these molecules alter the spectroscopic signature of the complex compared to the native system. Direct structural interpretations of the observed effects are usually unattainable. However, when such data are combined with spectroscopy-oriented quantum chemical studies44,67–74 it is often possible to arrive at experimentally consistent atomistic models that reveal the mode of interaction and the role of specific channels, providing insight into the local enzymatic architecture as well as on regulatory and mechanistic aspects of water oxidation.
Methanol is a key molecule in this respect, because it affects the electronic structure and modifies the EPR signatures75 of all states.47–65 A major effect is that it increases the energy separation between the lowest magnetic levels of the OEC,48,51,60,62 stabilizing the S = 1/2 ground spin states of the S0 and S2 states and the diamagnetic ground state of S1. Other effects relate to the amplitude enhancement of specific spectral forms, for example of the g ≈ 2 multiline signal of the S2 state in plants over the g ≥ 4 component. It is not clear whether a common interaction mode is valid for all catalytic states. Interestingly, higher plants and cyanobacteria behave differently to addition of methanol,62,76,77 the latter appearing less sensitive in its spectroscopic response than the former, but the origin of this species dependence is not understood. Efforts to identify possible methanol binding modes employed deuterated methanol (CD3OH) in electron spin-echo envelope modulation (ESEEM) experiments for the S2 and S0 states.49,59,78 For the S2-state studies49,59 the inferred Mn–2H distances were interpreted as consistent with direct ligation of methanol to a Mn center, but the two ESEEM data sets and their interpretation in the context of a structural model of the OEC differ substantially, being consistent either with methanol displacing a water-derived ligand bound to Mn4, or with methanol displacing Glu189, an amino acid residue directly coordinated to Mn1.62 The concept of direct ligation to the Mn1 and Mn4 sites with different affinities was recently used also in the interpretation of D1-Tyr161 radical “split signals” that can be generated from the S2 state under different illumination conditions.63 On the other hand, the ESEEM study of the S0 state did not result in observation of specific interactions, disfavoring direct methanol binding to the OEC.78 It is clear that neither the data can be reconciled, nor any of the interpretations supported with confidence. Beyond the intrinsic limitations in the treatment of deuteron dipolar couplings, the involvement of Mn1 as a potential binding site49,59,63 conflicts with channel accessibility studies,20,25 while explanations that invoke drastic reorganizations of first-sphere ligands seem incompatible with the small observed spectroscopic perturbations and the retention of catalytic activity. Similar ambiguities pertain to structural interpretations of methanol effects on the miss parameter for S-state advancement.61
A significant advance in this line of study was achieved recently by Oyala et al.,65 who used 13C-labeled methanol in pulse EPR studies of spinach PS-II poised in the S2 state. This is the most direct study of methanol interaction with the OEC to date. The measured 13C hyperfine interactions, compared with a reference Mn(III,IV) complex, argued very strongly against direct binding of methanol to a Mn ion. Maps of the 13C dipolar hyperfine couplings identified two plausible regions for methanol interaction, either in the cluster of water molecules surrounding Ca2+, possibly by directly replacing one of the calcium-bound waters, or in the vicinity of Mn4 where water molecules can form hydrogen bonds with oxygen bridges, or a combination of the above. Two of the channels shown in Fig. 1 may therefore be implicated in methanol delivery, but no further differentiation can be made and no unique structural models can be proposed. This leaves still open the crucial question of substrate delivery and identification; for example, if MeOH indeed replaces the Ca-bound W3 and remains bound in later stages of the catalytic cycle, this would likely exclude the possibility of a calcium-bound water acting as a substrate in a nucleophilic attack scenario for O–O bond formation.79–82
In this study we use available experimental data, including the valuable dataset on 13C hyperfine interactions,65 to evaluate a series of quantum chemical models that include explicit binding of MeOH at different sites in either protonated or deprotonated form, as well as many second-sphere interaction modes. Based on the magnetic and spectroscopic properties of these models, we conclude that the experimental observations have a unique structural interpretation, identifying a second-sphere interaction site for MeOH along a water channel associated with the O4 bridge. The atomistic details of the model provide a novel explanation of how the energy level splittings are affected by modulation of the Mn3–O4–Mn4 superexchange pathway in the S2 and potentially in other S states. Comparison of PS-II amino acid sequences from plants and cyanobacteria reveal a conserved difference close to the proposed site of interaction, offering a structure-based explanation for the species-dependent response to methanol. By combining the present results with recent studies of ammonia interaction with the OEC44,64,66,83 it is possible to assign a unique channel as active in delivery of substrate analogues to the OEC. Taking into account the studies of the S2–S3 transition and the associated “pivot” mechanism for water binding,73 we conclude that the dangler manganese (Mn4) is the only directly accessible Mn ion of the cluster and that substrate inclusion to the OEC occurs through initial binding at this site.
Methodology
Generation of structural models
Our approach for creating atomistic models to describe the interaction of methanol with the OEC is based on systematically substituting first and second sphere water molecules by MeOH and exploring the conformational space for each substitution. The “open cubane” form of the cluster that corresponds to the multiline S = 1/2 signal of the S2 state70 was used as starting point. We employed our previously described QM/MM models of the S2 state17 for preliminary screening and set the size of the final QM-only models to sufficiently include all first-sphere hydrogen bonding interactions. The final models consist of 240–242 atoms. The formally “open” coordination site of the five-coordinate Mn(III) ion Mn1 was also considered as a possible interaction site. However, after multiple attempts we conclude that this site is both inaccessible, in line with previous studies of channel architecture,20 and unsuitable for methanol binding as it lies along the Jahn–Teller axis of the Mn(III) ion.Twelve water molecule positions were considered as candidates for MeOH substitution: four are direct Mn or Ca ligands (W1–W4) and eight are second-sphere crystallographically characterized water molecules (W5–W12) that participate in hydrogen-bonding interactions with first-sphere residues.7,8 The W1 and W2 ligands to Mn4 were considered as H2O and OH.69,72 The labeling used for the water molecules is shown in Fig. 2. The correspondence of these labels with the identification numbers of the most recent crystallographic models with PDB codes 3ARC, 3WU2, and 4UB6 is provided in Table S1 of the ESI.† For each of the twelve positions, after replacing the H2O for MeOH several conformations and protonation isomers were generated either by automatically scanning dihedral angles or by manually reorienting specific groups. Of the several tens of conformers thus generated and geometry-optimized, eventually we selected twenty “best-case” models that we consider representative of all conceivable MeOH arrangements. Given that MeOH is only mildly perturbing the spectroscopic signature of the OEC without significantly altering its electronic structure, any models in which the initial positioning of MeOH resulted in extensive structural rearrangements upon optimization such as decoordination of first-sphere residues with concomitant large changes in electronic structure, were discarded.
 | ||
| Fig. 2 Computational model of the OEC used in the present work, with labelling of the relevant water molecules (hydrogen atoms omitted for clarity). | ||
Geometries were optimized with the ORCA program package,84 using the BP86 functional85,86 combined with the D3 model for dispersion corrections,87 the COSMO model88 with a dielectric of 8 to simulate the effect of the protein environment, and the zeroth-order regular approximation (ZORA) Hamiltonian89–91 for scalar relativistic effects. ZORA-recontracted92 Karlsruhe basis sets of polarized valence triple-ζ (Mn, Ca, O, N) and double-ζ (C, H) quality were used,93,94 with increased integration grids (Grid6 and IntAcc 6.0 in ORCA notation) and tight SCF convergence criteria. Fully decontracted def2-TZVP/J basis sets95 were employed in density fitting.
Spin states and magnetic properties
For each model a complete set of broken-symmetry (BS) DFT calculations was carried out in terms of single and double spin flips to compute the six pairwise exchange coupling constants Jij that parameterize the magnetic coupling between the four Mn ions in an Ising-type Hamiltonian. These calculations employed the hybrid meta-GGA functional TPSSh,96 which has been shown to perform extremely well in the prediction of magnetic properties for high-valent Mn clusters.67,72,97–99 These BS solutions are not spin eigenfunctions and it is meaningless to attempt any direct comparison with experiment, either with respect to an observed ground spin state or with respect to measured energy differences between magnetic sublevels. It is unfortunate that this crucial point is missed in part of the literature. The exchange coupling constants obtained from the BS solutions serve to form and diagonalize the Heisenberg–Dirac–van Vleck (HDvV) Hamiltonianthereby obtaining the complete spectrum of spin eigenstates (e.g. 320 magnetic levels for the S2 state including spin multiplets) and their relative energies. These results constitute a valid basis for comparisons with experiment; we use the spin states and energy differences between HDvV states thus identified to evaluate all models.
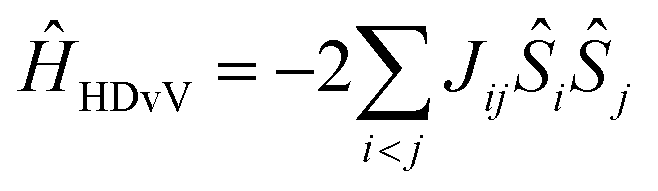
Spectroscopic properties
The hyperfine coupling results from the interaction of the spin of the unpaired electron(s) S and the magnetic nuclear moments I: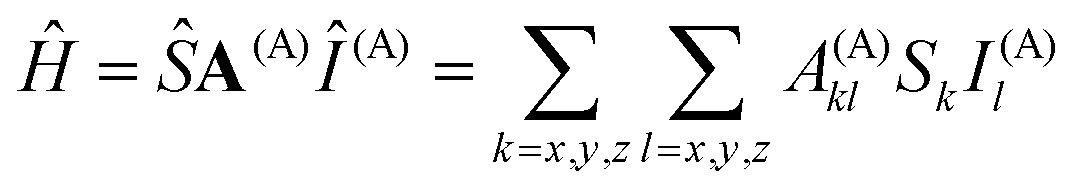
The tensor parameterizing the interaction, A(A), consists of three contributions: the Fermi contact, the spin dipolar, and the spin–orbit coupling contribution:
| A(A)kl = A(A;FC)kl + A(A;SD)kl + A(A;SO)kl |
In the framework of density functional methods analytical solutions for the three terms have been derived100 according to the following equations:
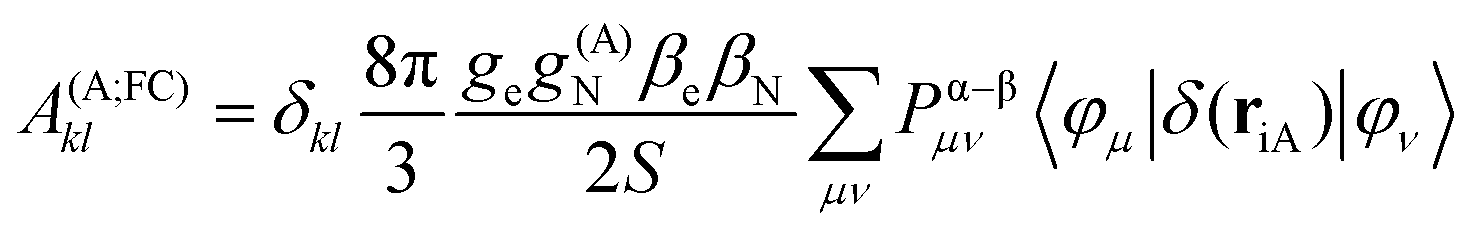
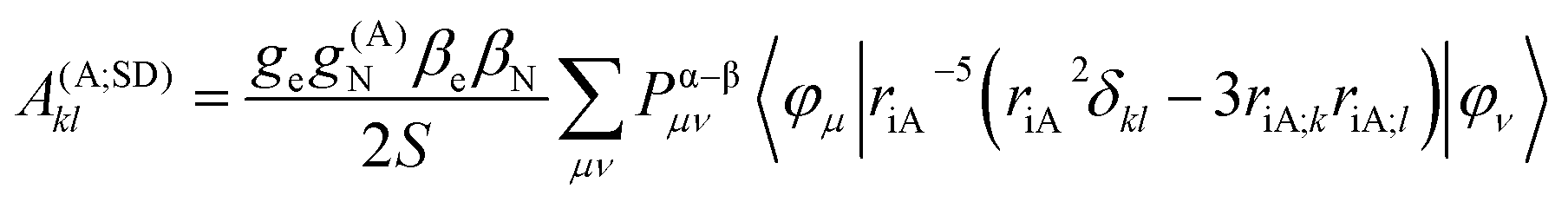
here, ge is the g-value of the free electron, g(A)N is the g-value of nucleus A, βe is the Bohr magneton, βN is the nuclear magneton, Pα–β is the spin-density matrix, I(A) is the nuclear spin operator of nucleus A, zSOMF is the spin–orbit coupling operator, and
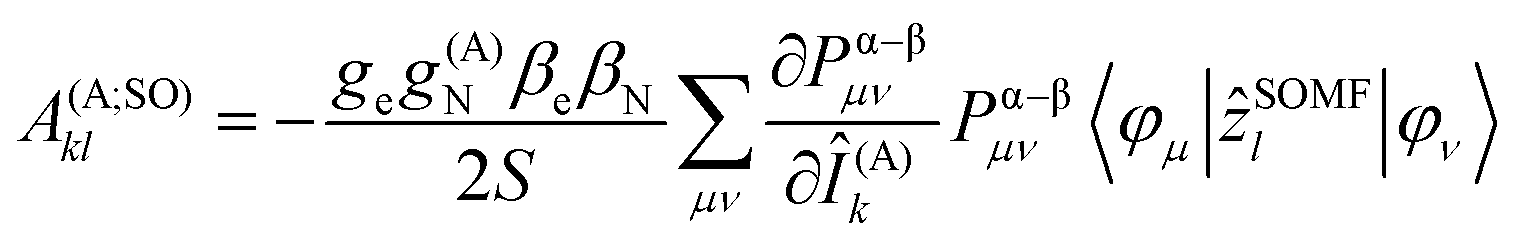
| riA = |riA| = |ri − RA| | (1) |
Sequence alignment
The sequences of the PS-II core proteins D1 (PsbA), D2 (PsbD), CP43 (PsbC) and CP47 (PsbB) from T. vulcanus, T. elongatus, and S. oleracea were aligned with ClustalW2.101,102Results and discussion
Atomistic models for methanol interaction
Among the many structural models that were optimized and evaluated in terms of their properties, we have selected twenty representative structures (Fig. 3) that fully cover the range of possibilities with respect to methanol interaction with the OEC. The models are labeled according to the water molecule that is substituted by methanol, followed by an additional number that refers to the conformational variant. The models include structures where methanol is deprotonated when it binds as a first-sphere ligand; these are indicated with the letter “d” as a label suffix.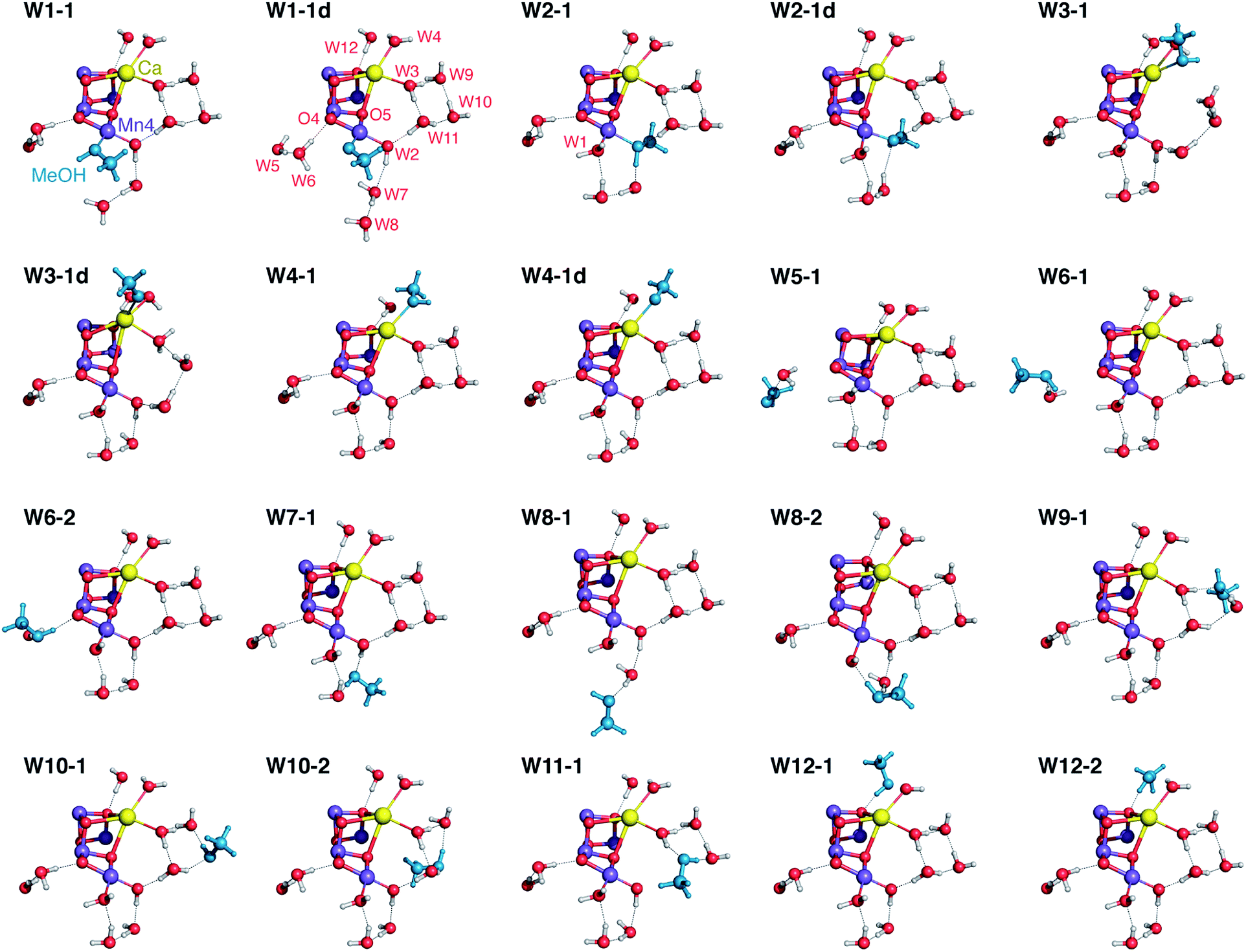 | ||
| Fig. 3 Position of methanol (in blue) and of water molecules around the OEC cluster in selected optimized models. All protein residues are omitted for clarity. | ||
In all cases there is minimal structural perturbation of the inorganic core, even in models where methanol ligates directly to a metal ion of the OEC. The relative energies of these models, computed with three different approaches, are provided in Table S2.† It is stressed however that comparison of these energies is only partially informative for four important reasons. First, a fundamental limitation is that not all models are isomers (e.g. W2-substituted models are unique, and a subset of models contain deprotonated methanol). Second, several models span a narrow energy range; this is particularly true for positions remote from the inorganic cluster that are not associated with large energetic perturbations (e.g. models W5-1, W7-1, and W10-1/2 are isoenergetic even though the access and interaction sites of methanol are fundamentally different). Third, for several structures the truncation of interactions that would propagate beyond the computational model employed in this study implies unknown uncertainties (for example, a high-energy model might be stabilized by hydrogen bonding or a low-energy model might be destabilized by steric interactions with parts of the protein missing from the QM models). And fourth, relative energies are irrelevant when comparing models that involve different access channels. To appreciate this, let us assume that methanol in reality can only access the calcium site of the cluster; then it is of no relevance that W4-1 is 3–4 kcal mol−1 higher in energy than W7-1 (Table S2†), because the latter would be impossible to exist. The only relevant energetic quantities in this case would be the relative permeation barriers through different channels of the enzyme, an aspect that is entirely missing from a cluster QM approach. Therefore, although relative energies are crucial in elucidating and comparing reaction pathways,14,15,103–107 they are of limited utility for the present study and make sense only when comparing closely related subsets, for example pairs of rotamers such as W6-1 and W6-2 (see Table S2†). A meaningful evaluation of models in the present case can be based only on comparison of the values of observable properties against experiment, so in the following we focus on the magnetism and spectroscopy of these twenty structures.
Magnetic properties
An important effect of methanol on the multiline signal of the S2 state is the increase of the energy gap ΔE between the ground (S = 1/2) and first excited (S = 3/2) states.62 For T. elongatus it is estimated that the native energy separation of ca. 13 cm−1 is increased to ca. 22 cm−1 upon addition of methanol, while for spinach the most recent study reported a change from ca. 3 cm−1 to ca. 25 cm−1.62 Older estimates in the literature place the native ΔE of spinach to ca. 6 cm−1,47 while for methanol-treated samples previous estimates included 12 cm−1,50 30 cm−1,47 and 36.5 cm−1.46 Therefore, our principal target here is to identify methanol interaction sites that correlate with an increase of ΔE.Table 1 lists all exchange coupling constants for all models of the present study. The spin of the ground and first excited states as well as their energy difference ΔE, obtained from diagonalization of the HDvV Hamiltonian, are also reported in Table 1 for each model. The results computed for the “native” S2 state model of the OEC are provided as reference. Before we proceed, it is important to clarify a methodological point: given that the present model of the unperturbed, “native” state of the OEC does not extend too far from the inorganic cluster, it cannot capture differences between species that originate in protein sequence variations at sites that are not included in the model. The structural nature of these remote differences is in any case undefined, as no atomic-resolution structure of higher plant PS-II is still available.108 In this sense, the unperturbed S2 state model used as the departure point in this study can be viewed in principle as “species-agnostic” and serves as a platform to study relative changes, i.e. the sign and magnitude of ΔΔE expected upon displacement of specific ligated or proximal waters by methanol.
| J 12 | J 13 | J 14 | J 23 | J 24 | J 34 | SGS | SES | ΔEGS–ES | ΔΔEGS–ES | |
|---|---|---|---|---|---|---|---|---|---|---|
| Native | −16.1 | 2.6 | 1.4 | 23.7 | 2.0 | −13.3 | 1/2 | 3/2 | 21.0 | |
| W1-1 | −15.9 | 3.0 | 1.1 | 23.5 | 2.0 | −13.5 | 1/2 | 3/2 | 20.8 | −1 |
| W1-1d | −16.4 | 6.4 | −0.4 | 24.5 | 2.1 | −12.4 | 1/2 | 3/2 | 17.3 | −18 |
| W2-1 | −14.4 | 1.1 | 1.9 | 23.7 | 1.7 | −14.7 | 1/2 | 3/2 | 22.0 | 5 |
| W2-1d | −15.6 | 2.5 | 2.0 | 23.8 | 1.5 | −12.9 | 1/2 | 3/2 | 20.4 | −3 |
| W3-1 | −15.7 | 2.8 | 1.6 | 23.1 | 2.1 | −13.9 | 1/2 | 3/2 | 21.0 | 0 |
| W3-1d | −16.1 | 3.4 | 5.1 | 29.0 | 1.5 | −20.2 | 1/2 | 3/2 | 25.0 | 19 |
| W4-1 | −17.1 | 2.8 | 1.6 | 20.2 | 1.8 | −11.6 | 1/2 | 3/2 | 20.2 | −4 |
| W4-1d | −10.6 | 4.6 | 2.1 | 1.5 | 0.5 | −18.6 | 1/2 | 3/2 | 28.8 | 37 |
| W5-1 | −17.2 | 3.2 | 1.1 | 18.5 | 1.8 | −15.6 | 1/2 | 3/2 | 24.4 | 16 |
| W6-1 | −16.8 | 2.8 | 1.4 | 19.7 | 2.0 | −21.2 | 1/2 | 3/2 | 28.7 | 37 |
| W6-2 | −15.8 | 4.7 | 0.5 | 28.5 | 1.4 | −8.6 | 1/2 | 3/2 | 14.3 | −32 |
| W7-1 | −16.1 | 3.4 | 1.7 | 22.6 | 2.0 | −11.3 | 1/2 | 3/2 | 18.5 | −12 |
| W8-1 | −16.2 | 2.6 | 1.3 | 24.3 | 1.9 | −13.9 | 1/2 | 3/2 | 21.6 | 3 |
| W8-2 | −16.2 | 4.4 | 0.8 | 25.8 | 1.5 | −16.0 | 1/2 | 3/2 | 21.5 | 2 |
| W9-1 | −14.6 | 1.7 | 1.9 | 24.7 | 1.9 | −11.2 | 1/2 | 3/2 | 18.5 | −12 |
| W10-1 | −16.2 | 2.8 | 1.3 | 23.3 | 2.0 | −12.7 | 1/2 | 3/2 | 20.5 | −2 |
| W10-2 | −15.3 | 2.6 | 1.8 | 23.5 | 2.1 | −12.8 | 1/2 | 3/2 | 19.9 | −5 |
| W11-1 | −15.9 | 2.9 | 1.7 | 23.8 | 2.1 | −15.4 | 1/2 | 3/2 | 22.4 | 7 |
| W12-1 | −16.2 | 2.8 | 1.2 | 22.3 | 2.0 | −12.6 | 1/2 | 3/2 | 20.4 | −3 |
| W12-2 | −15.3 | 2.8 | 1.4 | 23.6 | 2.0 | −12.8 | 1/2 | 3/2 | 19.8 | −6 |
In all cases the nature of the ground spin state (S = 1/2) and the first excited state (S = 3/2) remain the same, regardless of the type of methanol interaction. The exchange coupling constants and the energy splitting between the ground and the first excited states proved in general to be rather insensitive to water displacement by methanol, but there are also a few models where ΔE increases or decreases significantly. Focusing on the models that show a large increase as required for agreement with experiment, we can see that they belong to two groups: the first involves direct binding of methanol to calcium, but only in deprotonated (methoxy) form. These are the models W3-1d and W4-1d, the latter showing (together with W6-1) the largest increase in ΔE compared with the native system. The second group includes the two water sites that form part of the O4-related channel and is represented by models W5-1 and W6-1. Both groups correspond to two different channels, the calcium channel and the O4 channel of Fig. 1 respectively. By contrast, there is no water position associated with the third channel (W7, W8) that can reproduce to any extent the phenomenology when occupied by methanol. Direct ligation to Mn4 either has minimal effect on the predicted ΔE, or it has the opposite effect than what is experimentally observed, when MeOH binds as methoxy in the W1 position (W1-1d). Therefore, from the perspective of the effect on magnetism, direct ligation to Mn is disfavored and the two models W4-1d and W6-1 stand out from the rest as most consistent with experiment, even though they represent fundamentally different binding modes.
The fact that W4-1d reproduces the large positive change in ΔE is somewhat unexpected because there is no direct interaction with a magnetic site. Nevertheless, the values in Table 1 show that coordination of MeO− to Ca2+ does affect the exchange coupling constants, particularly within the cuboidal Mn3CaO4 unit. The major cause for the change in ΔE is the diminished value of the ferromagnetic coupling constant J23 compared to the native system. The changes in exchange coupling constants in this model as well as in W3-1d show that such alterations are not necessarily local and cannot always be properly analyzed as such. This serves as a warning that the interpretation of magnetic and spectroscopic responses under the assumption of locality of perturbations may sometimes be oversimplified.
The other model that emerged as a candidate for MeOH interaction is W6-1. Here the major change is observed for J34. To understand how the presence and orientation of methanol at the W6 site regulates the magnetic coupling between Mn3 and Mn4, it is necessary to examine both W6-1, which shows a shift in ΔE that agrees best with experiment, and W6-2, which behaves in exactly the opposite way and shows the poorest agreement among all models in terms of ΔE. In the native system W6 forms a hydrogen bond with O4. When methanol replaces W6 it can be oriented in a way that a hydrogen bond is also established between its hydroxy group and O4; this case is represented by W6-2. An additional hydrogen bonding interaction is established between W5 and MeOH. However, methanol can also be oriented in a way that abolishes hydrogen bonding with O4 and this is what happens in W6-1, where the OH group of methanol acts instead as a hydrogen bond donor to D1-Asp61 and as an acceptor from both W5 and CP43-Arg357 (Fig. 4). These differences have two implications. First, the orientation of methanol that is associated with a higher degree of hydrogen bonding, W6-1, is energetically favorable by ca. 7 kcal mol−1 compared to W6-2, because the positioning of methanol without hydrogen bonding to O4 but with three hydrogen-bonding interactions in total better stabilizes MeOH at the W6 site (Fig. 4, middle). Second, as expected99,109–112 the enhanced superexchange over O4 in the absence of the hydrogen bonding interaction compared to the native system leads to a more negative (antiferromagnetic) value for J34, stabilization of the low-spin S = 1/2 state of the OEC, and increase in ΔE. The differences in exchange coupling constants mirror the trend in optimized Mn3–Mn4 distances, which are 2.753 Å for the “native” model, 2.735 Å for W6-1, and 2.777 Å for W6-2.
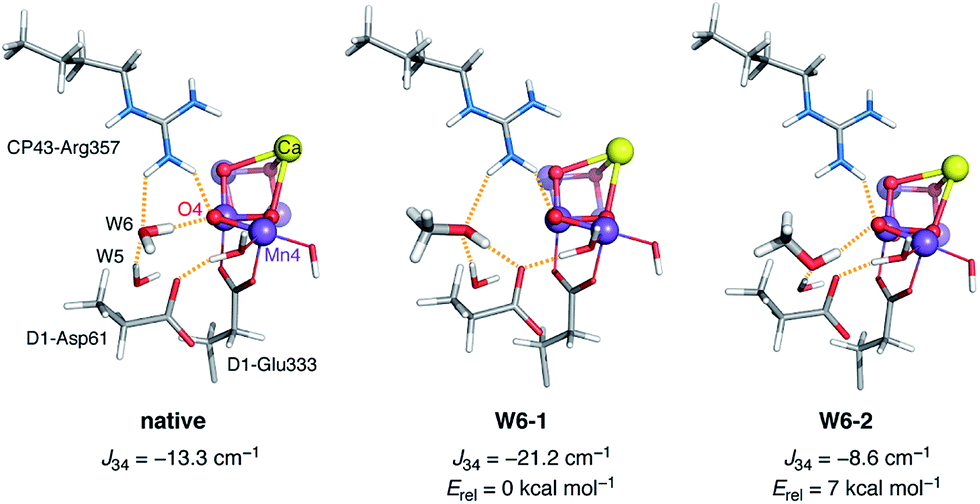 | ||
| Fig. 4 Comparison of the local hydrogen-bonding environment of O4 in the native system (left) and in the two models W6-1 and W6-2 with methanol occupation of the W6 site. | ||
This atomistic description of the methanol effect on the magnetic structure of the cluster in the case of model W6-1 is almost entirely local compared to W4-1d. As such, it has obvious parallels with our previous qualitative description of the methanol effect on the S2 state that was cast in terms of an effective interaction Jeff between the terminal Mn4 and the Mn3Ca subunit of the OEC in a monomer–trimer approximation of the magnetic topology.62 Model W6-1 provides for the first time a structural realization of this mode of action. As an aside, it is worth mentioning that the above analysis can clarify an interesting technical point. Earlier theoretical work employed smaller models that did not include the crystallographic water W6 which hydrogen-bonds to O4.69 It was already noticed that the computed ΔE values for these models compared well with experimental values from methanol-treated samples, even though methanol was obviously not included in the models. If we assume that W6-1 is a correct representation of how methanol interacts with the OEC, an explanation for this curious result becomes immediately obvious: a computational model that lacks W6 mimics the effect of methanol on the hydrogen bonding environment of O4.
Evaluation in terms of magnetic interactions allows us to eliminate most models, including all those that involve direct MeOH ligation to Mn. Still, this criterion is not sufficient to definitively distinguish between direct binding of methanol to Ca (model W4-1d) or second-sphere interaction in the vicinity of O4 (model W6-1). In the following section this distinction is achieved conclusively by comparison of the computed 13C hyperfine coupling parameters with the data of Oyala et al.65
13C hyperfine interactions
After confirming that our predictions for 13C hyperfine interactions are reliable by comparing with the same reference mixed-valence synthetic complex113 as the one used in the experimental study65 (Fig. S1†), we have computed the relevant parameters for all models of Fig. 3 and Table 2 lists the isotropic coupling Aiso and the dipolar coupling T for the 13C nucleus of methanol in all structures. Even though most models were already disfavored based on the energy level splittings in the previous section, it is important to discuss the 13C hyperfine parameters of the group of models that involve methanol ligation to Mn4, because the two properties lead to identical conclusions and strongly reinforce each other. The 13C Aiso values for MeO− binding are two orders of magnitude larger than the experimental values (5.73 MHz and 4.06 MHz for W1-1d and W2-2d), and even the “best-case” scenario represented by model W1-1 is off by an order of magnitude. The deviation of computed dipolar coupling T values from experiment is equally dramatic, with values ranging from 1.53 MHz (W2-1) up to 2.96 MHz (W1-1d). In view of the above, the conclusion that methanol does not coordinate to Mn4 is definitive.| Model | |Aiso| | |T| | Model | |Aiso| | |T| |
|---|---|---|---|---|---|
| W1-1 | 0.25 | 1.82 | W6-2 | 0.16 | 0.65 |
| W1-1d | 5.73 | 2.96 | W7-1 | 0.02 | 0.36 |
| W2-1 | 1.06 | 1.53 | W8-1 | 0.00 | 0.17 |
| W2-1d | 4.06 | 2.54 | W8-2 | 0.34 | 0.59 |
| W3-1 | 0.01 | 0.28 | W9-1 | 0.01 | 0.20 |
| W3-1d | 3.03 | 2.45 | W10-1 | 0.00 | 0.14 |
| W4-1 | 0.03 | 0.42 | W10-2 | 1.37 | 0.40 |
| W4-1d | 14.11 | 1.44 | W11-1 | 1.35 | 0.73 |
| W5-1 | 0.14 | 0.49 | W12-1 | 0.01 | 0.58 |
| W6-1 | 0.02 | 0.24 | W12-2 | 0.02 | 0.76 |
| Exp. | 0.05 ± 0.02 | 0.27 ± 0.05 | Exp. | 0.05 ± 0.02 | 0.27 ± 0.05 |
With respect to the calcium site, models W3-1 and W4-1 agree very well with the 13C hyperfine data, and this observation is in agreement with the conclusions of Oyala et al., who favored calcium as one of the likely methanol interaction sites.65W3-1, together with W6-1 discussed below, show the best overall agreement with the experimental values. On the other hand, both W3-1 and W4-1 were excluded in the previous section because they cannot explain the increase in the energy gap ΔE, thus these models cannot satisfy both constraints. The two models that were favored instead were those that involved binding of deprotonated methanol, W3-1d and W4-1d, the latter being one of the two strong candidates. However, the computed values are compelling: this type of methanol binding cannot possibly reproduce the experimental 13C hyperfine data, because both Aiso and T are orders of magnitude too large.
Combined with the results of the previous section this leaves only one candidate, model W6-1. In stark contrast to W4-1d, the agreement with experiment in this case is excellent, with Aiso = 0.02 versus the experimental value of 0.05 ± 0.02 and T = 0.24 versus the experimental value of 0.27 ± 0.05. Therefore, there is only one model, W6-1, requiring methanol delivery through the water channel associated with O4, that simultaneously accounts for the effect on the magnetic couplings of the OEC in the S2 state and reproduces the experimental 13C hyperfine data.
Possibility of a secondary binding site
Although the identification of W6 as the site occupied by methanol is definitive, one may consider whether a second molecule of methanol can be present at a secondary site, potentially depending on concentration. Based on the experimental data and our calculations, binding as a first-sphere ligand to Mn and as a methoxy to calcium are excluded, while occupation of the W6 site is required to explain simultaneously the effect on magnetism and the 13C hyperfine data, so if there is only one way that methanol interacts with the OEC, this is represented by model W6-1. On the other hand, the presence of a second molecule of methanol cannot be excluded if it occupies a site that satisfies the 13C hyperfine data while not adversely affecting the energy level splitting. A hypothesis regarding the presence of two methanol binding sites was previously advanced by Sjöholm et al. to account for the different methanol concentration dependence in the induction of “split” EPR signals of the OEC, that is, of states that contain a tyrosyl radical interacting with the manganese cluster.63 The postulated interaction modes (direct binding of methanol to Mn1 and/or Mn4) can no longer be considered viable, so it might be worth revisiting that analysis in light of the present results and of the required occupation of the W6 site.S-state invariance of the methanol binding pocket
The S1 state of the OEC is diamagnetic (S = 0). Parallel-mode EPR however probes the first excited state (S = 1)48,114,115 which is very low in energy, less than 2 cm−1 higher than the ground state.48 In terms of methanol interaction, it was shown that the parallel-mode S1 state signal at g ≈ 4.9 is no longer observable upon addition of methanol,48 suggesting that the triplet state is depopulated, i.e. the energy splitting ΔE between the ground and first excited states is increased. If we assume that the methanol binding pocket in the S1 state is the same as in S2 and is represented well by the S1 analogue of model W6-1, these observations obtain an obvious structure-based rationalization that mirrors the analysis we presented for the S2 state. The antiferromagnetic coupling between Mn3 and Mn4 in the S1 state72 would be enhanced by the presence of methanol via the same mechanism discussed above, stabilizing the S = 0 ground state and resulting in disappearance of the excited-state S = 1 signal.Given that the oxidation states of the manganese cluster cannot influence the channel architecture, methanol should access the OEC through the same channel regardless of the catalytic state of the cluster, so we also expect the same methanol site in the S0 and S3 states. The phenomenology of split EPR signals of both the S1YZ˙ and the S0YZ˙ states has been rationalized in terms of an increase in ΔE upon addition of methanol,60 so a similar mode of interaction as for the S1 and S2 states (operating on the Mn3–Mn4 antiferromagnetic coupling) would be valid for the S0 state as well. We prefer not to propose specific S0 models at this point because of the more complex EPR phenomenology of this state50,77,116,117 and remaining uncertainties in the protonation states of terminal water-derived ligands and oxo bridges.35,72,118–120 EPR studies of the S3 state reveal a rich and complex phenomenology.53,54,58,71,121–133 S3 is an integer spin state and interpretation of spectroscopic data is additionally complicated by heterogeneity, related at least in part to structural polymorphism that has only recently began to be understood.73,134 A more complete understanding of the S3 state is necessary before one can confidently discuss methanol interaction in terms of atomic structure. Nevertheless, the present work constrains the methanol interaction site to W6 and restricts the question only to how methanol might affect the different possible S3 forms, or stabilize one of these forms over the others, for example by inhibiting water delivery.
In conclusion, methanol likely occupies the same site in all states, adopts approximately the same orientation and, at least in the S0–S2 states, affects the electronic structure in the same fashion. Finally, we would like to point out that since direct ligation to a Mn ion is excluded, the apparent S state dependent sensitivity to methanol cannot be attributed to interaction with Mn ions in different oxidation states. An alternative and simpler interpretation of such observations emerges from the present work: if methanol can occupy the same site in all S states and have a similar effect on the electronic structure by modulating the Mn3–O4–Mn4 interaction, the apparent differences in sensitivity between catalytic states arise simply from the different weight of J34 in determining the magnetic properties of the cluster in each S state.
Effect on the two components of the S2 state
Two EPR signals are associated with the S2 state of the OEC in plants, a multiline signal centered at g ≈ 2 that corresponds to an S = 1/2 ground state species, and a signal at g ≈ 4.1 that arises from an S = 5/2 form of the cluster. In 2012 we demonstrated that these two signals correspond to two geometric and valence isomers that are interconvertible and lie very close in energy, within 1 kcal mol−1 or less.70 The S = 1/2 signal arises from the most stable open-cubane form of the cluster (with respect to the position of the O5 bridge) that has the oxidation state distribution III–IV–IV–IV, i.e. the unique Mn(III) ion is Mn1. The S = 5/2 species adopts instead a closed-cubane form with the Mn(III) ion located at the “dangler” Mn4 site (Scheme 1).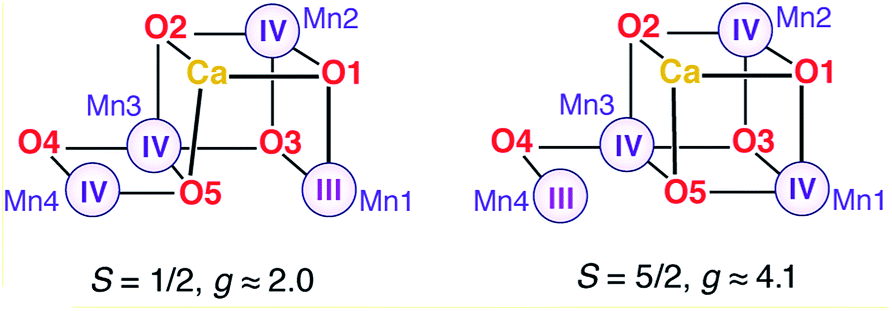 | ||
| Scheme 1 | ||
A crucial experimental observation is that in PS-II from plants, methanol prevents formation of the g ≈ 4.1 signal, that is, it destabilizes the valence isomer that contains a Mn(III) at the Mn4 site. The reorganization of the hydrogen bonding described above for model W6-1 provides a possible structure-based explanation of the phenomenology: the energetically favorable removal of the H-bonding interaction with O4 when methanol occupies the W6 site results in a more negatively charged O4 bridge, which in turn favors the +IV oxidation state for the directly ligated Mn4, shifting the equilibrium from the closed-cubane isomer that contains a Mn4(III) towards the open-cubane, S = 1/2, Mn4(IV) isomer associated with the multiline signal.
Species dependence
A historically persistent question that relates to the methanol effect is the species-dependent response. Specifically, the influence of methanol on the EPR spectra and the magnetic coupling is reported to be more pronounced in higher plants than in cyanobacteria.62 The present work converges to an atomistic model that is consistent with observations (particularly those from spinach samples), but cannot directly explain why the methanol effect is less pronounced in cyanobacteria. No species-dependent substitutions are known for any of the amino acid residues included in our QM model and hence it is reasonable to expect that model W6-1 should apply equally well to all organisms. It is improbable that fundamentally different access modes exists between species, therefore we consider the following explanation as most likely: the model favored in the present work must be equally valid for both cyanobacteria and higher plants, if methanol can reach this site and adopt this position in all organisms. In the following we identify the structural origin of the species-dependent response and we demonstrate that it strongly supports the proposed delivery channel.Sequence alignment for the four core proteins of T. vulcanus and S. oleracea (spinach) PS-II reveals in total 41 differences in D1 (PsbA), 34 in D2 (PsbD), 73 in CP43 (PsbC), and 114 in CP47 (PsbB). The vast majority are conservative substitutions; non-conservative substitutions are found mostly in the CP proteins (detailed sequence alignments are provided in the ESI†). Overall homology is approximately 88% and 91% for the D1 and D2 chains, while if conservative substitutions are not taken into account, the sequence similarity rises to 96%. Inspection of the sites with respect to the crystallographic model of PS-II from T. vulcanus7,8 shows that most differences occur in the flexible outer loops of the D1/D2 proteins, or in the periphery of transmembrane helices of the CP43/CP47 proteins and the lumenal side of CP47 (Fig. S2†). Consistent with the strictly conserved nature of the OEC and the identical chemistry across all oxygenic photosynthetic organisms, no variants exist in the first coordination sphere (associated with the D1 and CP43 proteins), in the environment of the redox active tyrosine YZ (D1-Y161), or in any parts of the protein that either bond covalently or interact via direct or water-mediated hydrogen bonding with the immediate environment of the OEC.
Within a sphere of 10 Å radius around any atom of the Mn4CaO5 cluster, we find two differences, both in the D1 protein: cyanobacterial Ser85 is threonine in spinach PS-II (a conservative substitution that has no relevance to any of the methanol interaction models), and cyanobacterial Asn87 is alanine in spinach (note that T. vulcanus and T. elongatus are identical in this respect). The latter substitution is distinctly non-conservative: it involves side chains of markedly different size and properties. No other such difference between the two organisms could be found along any of the channels in a radius of ca. 20 Å around the OEC for the core proteins. The unique importance of the N87A substitution is that it occurs right at the final turn of the O4-related channel we identified as the delivery channel for methanol.
Focusing on the local structure of this site (Fig. 5), it is apparent that cyanobacterial D1-Asn87 forms a hydrogen bond with the backbone of CP43-Glu354 (the residue that bridges Mn2 and Mn3), whereas in spinach D1-Ala87 cannot form such a bond. The difference in the formation of this hydrogen bond and the difference in the size of the side chains means that the diameter and local architecture of the channel at this point are also different: in the D1-Ala87 analogue methanol can access the interaction site unimpeded and adopt the energetically favorable conformation that corresponds to model W6-1. In this sense, W6-1 most probably reflects better how methanol interacts in spinach, something that explains why our QM models for methanol interaction agree better with experimental data from spinach than from cyanobacteria. By contrast, in the D1-Asn87 analogue the larger side chain of asparagine may sterically interact with the W6 pocket, while the formation of a hydrogen bond with CP43-Glu354 results in a reduced channel diameter and restricted accessibility. This might impose a different orientation of methanol than in model W6-1, or most probably prohibit access of methanol to the W6 site altogether. Therefore, W6-1 is a good model for methanol interaction in plants, while a model with a more remote placement of methanol along the same water channel (e.g. a variant of W5-1 or a site even more remote than W5) would better represent methanol interaction in cyanobacteria.
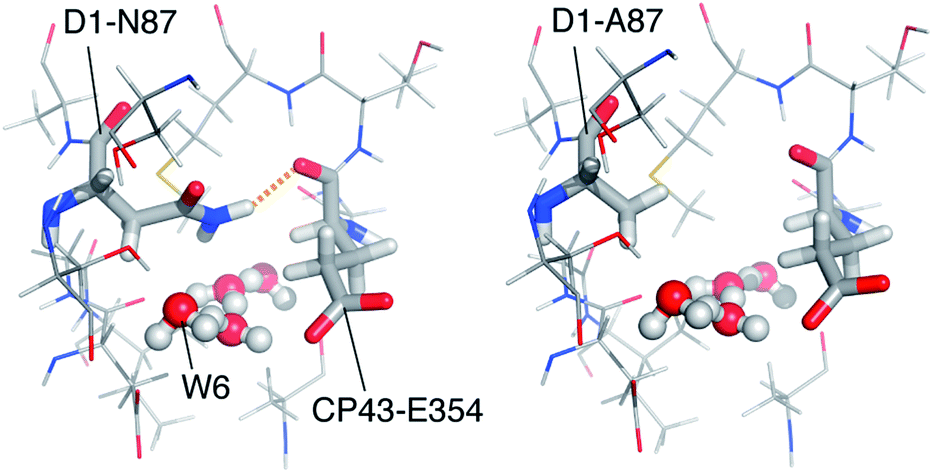 | ||
| Fig. 5 The water channel with the critical N87A substitution, viewed from the OEC. Coordinates for the cyanobacterial model (left) are taken from a QM/MM model17 that was based on the crystallographic model of PS-II from T. vulcanus.8 The model for spinach (right) was constructed by local side chain replacement to demonstrate the expected structural effect. | ||
Computational studies using multiscale approaches that involve dynamics simulations with extended models of PS-II would be necessary to determine the intrinsic differences between the two species with atomic detail; such models are currently under development in our group. Within the context of the present work, the above analysis is sufficient to establish that: (a) the only relevant difference in protein sequences between cyanobacteria and spinach is the D1-N87A substitution at the channel identified as responsible for delivery of methanol and at a position close to the W6 site, and (b) the presence of D1-Ala87 in spinach should result in increased accessibility of the W6 site to methanol compared to cyanobacterial PS-II, where access to the W6 site may be severely restricted or impossible.
Implications for regulation and catalysis
The model for methanol interaction proposed here requires access from the water channel associated with the O4 bridge of the cluster. In the literature this channel is referred to as the “narrow” channel by Ho and Styring,20 “E, F” in the work of Gabdulkhakov et al.,22 “channel 2” by Vassiliev et al.,25 and “Path 3” by Ogata et al.30 Consistent with the present findings, Ho and Styring had already suggested by analysis of a low-resolution static crystallographic model of PS-II that this channel would be the most permeable to methanol.20 Molecular dynamics simulations by Vassiliev et al. subsequently demonstrated that: (a) this channel connects the OEC directly with the lumenal surface of PS-II at the cavity formed by the extrinsic PsbO and PsbU proteins, and (b) this channel has the lowest free energy barrier for water permeation (peak activation energy ca. 9 kcal mol−1) than all other channels identified.25 Moreover, recent experimental and theoretical work has demonstrated that NH3 binds to Mn4 in the place of W1 in the S2 state of the OEC.44,64,66,83 Although the interaction modes of NH3 and MeOH are fundamentally different—the first one acting as a direct ligand to Mn4, the latter interacting from the second coordination sphere—access of methanol coincides with access of ammonia. This is consistent with competition studies that showed interrelations between the binding of these substrate analogues56 and reinforces the assignment of the O4 channel as the one involved in delivery of substrate analogues, and, by extension, of substrate water.The O4 water channel was recently proposed instead to be involved in proton transfer, at least in the S0–S1 transition, if the critical assumption is made that O4 is protonated in the S0 state.35,135 Although this idea is not implausible,135 it seems to us improbable for three reasons: (a) explicit calculation of protonation patterns by a wide variety of methods72,118,119 suggests that O4 protonation is unfavorable in S0, therefore the basis for these computational constructs is questionable; (b) the involvement of this channel in the delivery of methanol (identified here), of ammonia (as a ligand to Mn4),44,64,66,83 as well as of water in the S3 state73 would necessitate an unlikely dual role for this channel; and (c) residue D1-Asn87 would have to be involved in the hypothetical proton transfer, so models that might be valid for T. vulcanus would not be transferable to D1-Ala87 organisms, which is implausible for a function as critical as proton removal from the water oxidation site. Proton egress instead is most likely to occur via the other channel associated with D1-Asp61, as previously assigned by Knapp and co-workers based on the monotonic increase of calculated pKa values of titratable residues,34 and by FTIR and mutation studies.136,137 The H2O molecule occupying the W1 site in the S2 state has been identified as the Mn-bound ligand that is deprotonated (by D1-Asp61)10,34,138–141 in order to enable oxidation of Mn4 and allow transition to the S3 state.73,132,142–144 In this case, the influence of MeOH on proton release from W1 would be a second-order effect, acting through perturbation of the proton acceptor. This may result in modulation of the relative stabilities of intermediates such as tyrosyl radical states53,57,58,63,122,145,146 in a species-dependent manner and is a subject that deserves further study.
The lack of experimental or theoretical support for either NH3 or MeOH interacting with Ca2+ suggests that the calcium channel system may not be involved in small molecule access to the OEC. It has been proposed instead that the channels at this side of the cluster are more hydrophobic and hence better optimized for removal of dioxygen,20,26 and that calcium itself may play a role in facilitating O2 release from the cluster.147 In this case, Ca2+ is not a likely site of substrate binding and by extension the water ligands of Ca2+ (W3, W4) are not likely to be substrates in O–O bond formation. Substrate analogues are instead found either on Mn4 or at the endpoint of the O4 channel, implying that this channel may be the only one involved in substrate delivery and that Mn4 may be the only site involved in substrate binding. This is consistent with recent studies of the S2–S3 progression,73,134,142,148 which showed that the first component of the S3 state is a “closed-cubane” structure with a five-coordinate Mn4(IV) center.73 The approximately trigonal bipyramidal geometry of the Mn4(IV) site of this species results in properties such as absorption in the near-IR and high zero-field splitting,73 similarly to analogous synthetic Mn(IV) complexes.149–151 These studies established that water binding can only occur at this S3-state species: water is delivered via the O4 channel to the Mn4 center and binds via a low-barrier transition state,73,148 allowing access to the other components of the S3 state.71 Based on the above, we propose that occupation of the W6 site by methanol may enhance the S3 population with the five-coordinate Mn(IV) ion by inhibiting water delivery to the Mn4 site.73,148
Water binding to the Mn4 ion73,148 and internal structural rearrangement134,152 would be necessary to reach the structural form of the S3 state attributed to an all-octahedral Mn(IV) species71 that has been proposed by Siegbahn to be the active form of the OEC in O–O bond formation,15,143,153 in an oxo–oxyl coupling scenario.14,104,154,155 Given that neither methanol nor ammonia are inhibitors and that the kinetics of substrate exchange show both substrates to be already bound in the S2 state,156–159 we believe that further evidence is required to conclusively establish whether water binding in the S3 state71,160,161 is catalytically required for progression to the S3YZ˙ state and its subsequent deprotonation and transition to S4. A better characterization of the S3 and S3YZ˙ states is needed to clarify this point.133,145,146,162–167
Conclusions
Precise structural characterization of how substrate analogues interact with the oxygen-evolving complex of photosystem-II is important for understanding regulatory and mechanistic aspects of water oxidation. We studied the interaction of methanol with the S2 state of the OEC using extensive quantum mechanical models evaluated against experimental data. The two main criteria used were: (a) the change in the energy splitting between the S = 1/2 ground-state and the S = 3/2 first excited state of the complex,62 and (b) the recently reported 13C hyperfine coupling parameters of isotopically labeled methanol.65 Our results definitively exclude direct binding of methanol to manganese and strongly disfavor direct interaction with calcium. The only interaction mode that is consistent with observations involves displacement of W6, the final water molecule of the water channel associated with the O4 bridge of the cluster, proximal to the “dangler” Mn4 ion. This site reproduces the isotropic and dipolar 13C hyperfine couplings for methanol and provides a physically transparent explanation for the observed ground-state stabilization: the preferred methanol orientation removes the hydrogen bond between O4 and W6, amplifying the antiferromagnetic coupling between Mn3 and Mn4. The same interaction mode can be expected in other states. The effect of removing the hydrogen bond to O4 can also explain the stabilization of the S = 1/2 form of the S2 state in plant PS-II. Sequence alignments of PS-II core proteins from cyanobacteria and spinach reveal an important difference at the suggested delivery channel close to the methanol binding pocket, where D1-Asn87 of cyanobacteria is D1-Ala87 in spinach, a substitution that affects the accessibility of the site to methanol and can account for the species-dependence of the methanol effect. This result presents an obvious target for future research, the introduction of the D1-N87A mutation in cyanobacterial PS-II. The structural characterization of the methanol interaction site, the identification of the delivery channel and the correlation with other experimental and computational results are in line with the pivot mechanism for delivery and binding of water to the five-coordinate Mn4(IV) ion of an S3-state component,73 and support the idea that the O4 channel is uniquely responsible for delivery of water and substrate analogues to the OEC, with Mn4 being the only directly solvent-accessible site of the cluster.Acknowledgements
The authors gratefully acknowledge computing time granted by the John von Neumann Institute for Computing (NIC) and provided on the supercomputer JUROPA at Jülich Supercomputing Centre (JSC) (NIC project No. 7056). This work is supported by the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft. Network support by the COST action CM1305 “Explicit Control Over Spin-states in Technology and Biochemistry (ECOSTBio)” is gratefully acknowledged.Notes and references
- V. Krewald, M. Retegan and D. A. Pantazis, Top. Curr. Chem., 2016, 371, 23–48 CrossRef PubMed.
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- J. Messinger, T. Noguchi and J. Yano, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, The Royal Society of Chemistry, Cambridge, 2012, pp. 163–207 Search PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2014, 517, 99–103 CrossRef PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- P. Joliot, G. Barbieri and R. Chabaud, Photochem. Photobiol., 1969, 10, 309–329 CrossRef CAS.
- V. Krewald, F. Neese and D. A. Pantazis, Isr. J. Chem., 2015, 55, 1219–1232 CrossRef CAS.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- T. Wydrzynski, W. Hillier and J. Messinger, Physiol. Plant., 1996, 96, 342–350 CrossRef CAS.
- M. Retegan, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2013, 9, 3832–3842 CrossRef CAS PubMed.
- J. W. Murray and J. Barber, J. Struct. Biol., 2007, 159, 228–237 CrossRef CAS PubMed.
- F. M. Ho, Photosynth. Res., 2008, 98, 503–522 CrossRef CAS PubMed.
- F. M. Ho and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 140–153 CrossRef CAS PubMed.
- J. Murray and J. Barber, in Photosynthesis. Energy from the Sun, ed. J. Allen, E. Gantt, J. Golbeck and B. Osmond, Springer Netherlands, 2008, pp. 467–470 Search PubMed.
- A. Gabdulkhakov, A. Guskov, M. Broser, J. Kern, F. Muh, W. Saenger and A. Zouni, Structure, 2009, 17, 1223–1234 CrossRef CAS PubMed.
- S. Vassiliev, P. Comte, A. Mahboob and D. Bruce, Biochemistry, 2010, 49, 1873–1881 CrossRef CAS PubMed.
- F. M. Ho, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, The Royal Society of Chemistry, Cambridge, 2012, pp. 208–248 Search PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1671–1678 CrossRef CAS PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1148–1155 CrossRef CAS PubMed.
- L. Vogt, D. J. Vinyard, S. Khan and G. W. Brudvig, Curr. Opin. Chem. Biol., 2015, 25, 152–158 CrossRef CAS PubMed.
- A.-N. Bondar and H. Dau, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1177–1190 CrossRef CAS PubMed.
- K. Linke and F. M. Ho, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 14–32 CrossRef CAS PubMed.
- K. Ogata, T. Yuki, M. Hatakeyama, W. Uchida and S. Nakamura, J. Am. Chem. Soc., 2013, 135, 15670–15673 CrossRef CAS PubMed.
- These three channels may split into more branches or intersect with branches of other channels at other regions of the enzyme, having different exit/entry points. The channel architecture of PS-II as a whole is still an open field of study; in the present work we are only concerned with the well-defined immediate vicinity of the active site.
- H. Bao, P. Dilbeck and R. Burnap, Photosynth. Res., 2013, 116, 215–229 CrossRef CAS PubMed.
- I. L. McConnell, Photosynth. Res., 2008, 98, 261–276 CrossRef CAS PubMed.
- H. Ishikita, W. Saenger, B. Loll, J. Biesiadka and E.-W. Knapp, Biochemistry, 2006, 45, 2063–2071 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- W. F. Beck, J. C. De Paula and G. W. Brudvig, J. Am. Chem. Soc., 1986, 108, 4018–4022 CrossRef CAS.
- L.-E. Andréasson, Ö. Hansson and K. von Schenck, Biochim. Biophys. Acta, Bioenerg., 1988, 936, 351–360 CrossRef.
- R. D. Britt, J. L. Zimmermann, K. Sauer and M. P. Klein, J. Am. Chem. Soc., 1989, 111, 3522–3532 CrossRef CAS.
- A. Boussac, A. W. Rutherford and S. Styring, Biochemistry, 1990, 29, 24–32 CrossRef CAS PubMed.
- H. Dau, J. C. Andrews, T. A. Roelofs, M. J. Latimer, W. Liang, V. K. Yachandra, K. Sauer and M. P. Klein, Biochemistry, 1995, 34, 5274–5287 CrossRef CAS PubMed.
- H.-A. Chu, Y.-W. Feng, C.-M. Wang, K.-A. Chiang and S.-C. Ke, Biochemistry, 2004, 43, 10877–10885 CrossRef CAS PubMed.
- C.-H. Fang, K.-A. Chiang, C.-H. Hung, K. Chang, S.-C. Ke and H.-A. Chu, Biochemistry, 2005, 44, 9758–9765 CrossRef CAS PubMed.
- L.-H. Hou, C.-M. Wu, H.-H. Huang and H.-A. Chu, Biochemistry, 2011, 50, 9248–9254 CrossRef CAS PubMed.
- M. Pérez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- J. L. Zimmermann and A. W. Rutherford, Biochemistry, 1986, 25, 4609–4615 CrossRef CAS.
- G. A. Lorigan and R. D. Britt, Biochemistry, 1994, 33, 12072–12076 CrossRef CAS PubMed.
- R. J. Pace, P. Smith, R. Bramley and D. Stehlik, Biochim. Biophys. Acta, Bioenerg., 1991, 1058, 161–170 CrossRef CAS.
- T. Yamauchi, H. Mino, T. Matsukawa, A. Kawamori and T.-a. Ono, Biochemistry, 1997, 36, 7520–7526 CrossRef CAS PubMed.
- D. A. Force, D. W. Randall, G. A. Lorigan, K. L. Clemens and R. D. Britt, J. Am. Chem. Soc., 1998, 120, 13321–13333 CrossRef CAS.
- K. A. Åhrling, S. Peterson and S. Styring, Biochemistry, 1998, 37, 8115–8120 CrossRef PubMed.
- Z. Deák, S. Peterson, P. Geijer, K. A. Åhrling and S. Styring, Biochim. Biophys. Acta, Bioenerg., 1999, 1412, 240–249 CrossRef.
- G. A. Lorigan and R. David Britt, Photosynth. Res., 2000, 66, 189–198 CrossRef CAS PubMed.
- N. Ioannidis and V. Petrouleas, Biochemistry, 2000, 39, 5246–5254 CrossRef CAS PubMed.
- N. Ioannidis and V. Petrouleas, Biochemistry, 2002, 41, 9580–9588 CrossRef CAS PubMed.
- K. A. Åhrling, M. C. W. Evans, J. H. A. Nugent and R. J. Pace, Biochim. Biophys. Acta, Bioenerg., 2004, 1656, 66–77 CrossRef PubMed.
- M. C. W. Evans, R. J. Ball and J. H. A. Nugent, FEBS Lett., 2005, 579, 3081–3084 CrossRef CAS PubMed.
- V. Petrouleas, D. Koulougliotis and N. Ioannidis, Biochemistry, 2005, 44, 6723–6728 CrossRef CAS PubMed.
- N. Ioannidis, G. Zahariou and V. Petrouleas, Biochemistry, 2006, 45, 6252–6259 CrossRef CAS PubMed.
- K. A. Åhrling, M. C. W. Evans, J. H. A. Nugent, R. J. Ball and R. J. Pace, Biochemistry, 2006, 45, 7069–7082 CrossRef PubMed.
- J.-H. Su, K. G. V. Havelius, F. Mamedov, F. M. Ho and S. Styring, Biochemistry, 2006, 45, 7617–7627 CrossRef CAS PubMed.
- B. Nöring, D. Shevela, G. Renger and J. Messinger, Photosynth. Res., 2008, 98, 251–260 CrossRef PubMed.
- J.-H. Su, N. Cox, W. Ames, D. A. Pantazis, L. Rapatskiy, T. Lohmiller, L. V. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 829–840 CrossRef CAS PubMed.
- J. Sjöholm, G. Chen, F. Ho, F. Mamedov and S. Styring, Biochemistry, 2013, 52, 3669–3677 CrossRef PubMed.
- T. Lohmiller, V. Krewald, M. Pérez Navarro, M. Retegan, L. Rapatskiy, M. M. Nowaczyk, A. Boussac, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Phys. Chem. Chem. Phys., 2014, 16, 11877–11892 RSC.
- P. H. Oyala, T. A. Stich, J. A. Stull, F. Yu, V. L. Pecoraro and R. D. Britt, Biochemistry, 2014, 53, 7914–7928 CrossRef CAS PubMed.
- P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed.
- D. A. Pantazis, M. Orio, T. Petrenko, S. Zein, E. Bill, W. Lubitz, J. Messinger and F. Neese, Chem.–Eur. J., 2009, 15, 5108–5123 CrossRef CAS PubMed.
- D. A. Pantazis, M. Orio, T. Petrenko, S. Zein, W. Lubitz, J. Messinger and F. Neese, Phys. Chem. Chem. Phys., 2009, 11, 6788–6798 RSC.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, Chem. Sci., 2016, 7, 72–84 RSC.
- V. Krewald, M. Retegan, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Inorg. Chem., 2016, 55, 488–501 CrossRef CAS PubMed.
- A. Haddy, Photosynth. Res., 2007, 92, 357–368 CrossRef CAS PubMed.
- A. Boussac, H. Kuhl, S. Un, M. Rögner and A. W. Rutherford, Biochemistry, 1998, 37, 8995–9000 CrossRef CAS PubMed.
- A. Boussac, H. Kuhl, E. Ghibaudi, M. Rögner and A. W. Rutherford, Biochemistry, 1999, 38, 11942–11948 CrossRef CAS PubMed.
- A. Boussac, Y. Deligiannakis and A. W. Rutherford, in Photosynthesis: Mechanisms and Effects, ed. G. Garab, Kluwer Academic Publishers, Dordrecht, 1998, vol. II, pp. 1233–1240 Search PubMed.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, W.-Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925–929 CrossRef CAS.
- J. S. Vrettos, J. Limburg and G. W. Brudvig, Biochim. Biophys. Acta, Bioenerg., 2001, 1503, 229–245 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Phys. Chem. Chem. Phys., 2004, 6, 4754–4763 RSC.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 3428–3442 CrossRef CAS PubMed.
- J. Schraut and M. Kaupp, Chem.–Eur. J., 2014, 20, 7300–7308 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and D. Schüürman, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- D. A. Pantazis, X. Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- M. Orio, D. A. Pantazis and F. Neese, Photosynth. Res., 2009, 102, 443–453 CrossRef CAS PubMed.
- M. Orio, D. A. Pantazis, T. Petrenko and F. Neese, Inorg. Chem., 2009, 48, 7251–7260 CrossRef CAS PubMed.
- V. Krewald, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2013, 135, 5726–5739 CrossRef CAS PubMed.
- F. Neese, J. Chem. Phys., 2003, 118, 3939–3948 CrossRef CAS.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS PubMed.
- M. Goujon, H. McWilliam, W. Li, F. Valentin, S. Squizzato, J. Paern and R. Lopez, Nucleic Acids Res., 2010, 38, W695–W699 CrossRef CAS PubMed.
- P. E. M. Siegbahn and T. Borowski, Acc. Chem. Res., 2006, 39, 729–738 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem.–Eur. J., 2008, 14, 8290–8302 CrossRef CAS PubMed.
- P. E. M. Siegbahn and F. Himo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 323–336 CrossRef CAS.
- P. E. M. Siegbahn and M. R. A. Blomberg, J. Chem. Theory Comput., 2014, 10, 268–272 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2014, 16, 11893–11900 RSC.
- X. Wei, X. Su, P. Cao, X. Liu, W. Chang, M. Li, X. Zhang and Z. Liu, Nature, 2016, 534, 69–74 CrossRef CAS PubMed.
- M. J. Baldwin, T. L. Stemmler, P. J. Riggs-Gelasco, M. L. Kirk, J. E. Penner-Hahn and V. L. Pecoraro, J. Am. Chem. Soc., 1994, 116, 11349–11356 CrossRef CAS.
- M. J. Baldwin, N. A. Law, T. L. Stemmler, J. W. Kampf, J. E. Penner-Hahn and V. L. Pecoraro, Inorg. Chem., 1999, 38, 4801–4809 CrossRef CAS PubMed.
- D. A. Pantazis, V. Krewald, M. Orio and F. Neese, Dalton Trans., 2010, 39, 4959–4967 RSC.
- V. Krewald, B. Lassalle-Kaiser, T. T. Boron, C. J. Pollock, J. Kern, M. A. Beckwith, V. K. Yachandra, V. L. Pecoraro, J. Yano, F. Neese and S. DeBeer, Inorg. Chem., 2013, 52, 12904–12914 CrossRef CAS PubMed.
- E. Larson, A. Haddy, M. L. Kirk, R. H. Sands, W. E. Hatfield and V. L. Pecoraro, J. Am. Chem. Soc., 1992, 114, 6263–6265 CrossRef CAS.
- S. L. Dexheimer and M. P. Klein, J. Am. Chem. Soc., 1992, 114, 2821–2826 CrossRef CAS.
- D. Koulougliotis, C. Teutloff, Y. Sanakis, W. Lubitz and V. Petrouleas, Phys. Chem. Chem. Phys., 2004, 6, 4859–4863 RSC.
- J. Messinger, J. H. A. Nugent and M. C. W. Evans, Biochemistry, 1997, 36, 11055–11060 CrossRef CAS PubMed.
- J. Messinger, J. H. Robblee, W. O. Yu, K. Sauer, V. K. Yachandra and M. P. Klein, J. Am. Chem. Soc., 1997, 119, 11349–11350 CrossRef CAS PubMed.
- R. Pal, C. F. A. Negre, L. Vogt, R. Pokhrel, M. Z. Ertem, G. W. Brudvig and V. S. Batista, Biochemistry, 2013, 52, 7703–7706 CrossRef CAS PubMed.
- M. Amin, L. Vogt, W. Szejgis, S. Vassiliev, G. W. Brudvig, D. Bruce and M. R. Gunner, J. Phys. Chem. B, 2015, 119, 7366–7377 CrossRef CAS PubMed.
- A. Galstyan, A. Robertazzi and E. W. Knapp, J. Am. Chem. Soc., 2012, 134, 7442–7449 CrossRef CAS PubMed.
- T. Matsukawa, H. Mino, D. Yoneda and A. Kawamori, Biochemistry, 1999, 38, 4072–4077 CrossRef CAS PubMed.
- N. Ioannidis, J. H. A. Nugent and V. Petrouleas, Biochemistry, 2002, 41, 9589–9600 CrossRef CAS PubMed.
- A. Boussac, M. Sugiura, Y. Inoue and A. W. Rutherford, Biochemistry, 2000, 39, 13788–13799 CrossRef CAS PubMed.
- Y. Sanakis, J. Sarrou, G. Zahariou and V. Petrouleas, in Photosynthesis: Energy from the Sun, ed. J. F. Allen, E. Gantt, J. H. Golbeck and B. Osmond, Springer, Dordrecht, 2008, pp. 479–482 Search PubMed.
- J.-H. Su, K. G. V. Havelius, F. M. Ho, G. Han, F. Mamedov and S. Styring, Biochemistry, 2007, 46, 10703–10712 CrossRef CAS PubMed.
- G. Han, F. M. Ho, K. G. V. Havelius, S. F. Morvaridi, F. Mamedov and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 496–503 CrossRef CAS PubMed.
- A. Boussac, M. Sugiura, A. W. Rutherford and P. Dorlet, J. Am. Chem. Soc., 2009, 131, 5050–5051 CrossRef CAS PubMed.
- M. Chrysina, G. Zahariou, N. Ioannidis and V. Petrouleas, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 487–493 CrossRef CAS PubMed.
- M. Chrysina, G. Zahariou, Y. Sanakis, N. Ioannidis and V. Petrouleas, J. Photochem. Photobiol., B, 2011, 104, 72–79 CrossRef CAS PubMed.
- K. G. V. Havelius, J.-H. Su, G. Han, F. Mamedov, F. M. Ho and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 11–21 CrossRef CAS PubMed.
- G. Chen, G. Han, E. Göransson, F. Mamedov and S. Styring, Biochemistry, 2012, 51, 138–148 CrossRef CAS PubMed.
- M. Retegan, N. Cox, W. Lubitz, F. Neese and D. A. Pantazis, Phys. Chem. Chem. Phys., 2014, 16, 11901–11910 RSC.
- A. Boussac, A. W. Rutherford and M. Sugiura, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 576–586 CrossRef CAS PubMed.
- M. Capone, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 6439–6442 CrossRef CAS PubMed.
- T. Takaoka, N. Sakashita, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2016, 7, 1925–1932 CrossRef CAS PubMed.
- R. J. Service, W. Hillier and R. J. Debus, Biochemistry, 2014, 53, 1001–1017 CrossRef CAS PubMed.
- R. J. Debus, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 19–34 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- I. Rivalta, M. Amin, S. Luber, S. Vassiliev, R. Pokhrel, Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, D. Bruce, G. W. Brudvig, M. R. Gunner and V. S. Batista, Biochemistry, 2011, 50, 6312–6315 CrossRef CAS PubMed.
- R. J. Debus, Biochemistry, 2014, 53, 2941–2955 CrossRef CAS PubMed.
- P. L. Dilbeck, H. J. Hwang, I. Zaharieva, L. Gerencser, H. Dau and R. L. Burnap, Biochemistry, 2012, 51, 1079–1091 CrossRef CAS PubMed.
- D. Narzi, D. Bovi and L. Guidoni, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8723–8728 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2012, 14, 4849–4856 RSC.
- A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef CAS PubMed.
- G. Zahariou, M. Chrysina, V. Petrouleas and N. Ioannidis, FEBS Lett., 2014, 588, 1827–1831 CrossRef CAS PubMed.
- G. Zahariou and N. Ioannidis, Photosynth. Res., 2016 DOI:10.1007/s11120-11016-10274-11126.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- M. Capone, D. Narzi, D. Bovi and L. Guidoni, J. Phys. Chem. Lett., 2016, 7, 592–596 CrossRef CAS PubMed.
- S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, A. S. Borovik and M. P. Hendrich, Inorg. Chem., 2013, 52, 12568–12575 CrossRef CAS PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2015, 636, 172–179 CrossRef CAS.
- P. E. M. Siegbahn, ChemPhysChem, 2011, 12, 3274–3280 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2015, 17, 12168–12174 RSC.
- P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 117–127 CrossRef CAS.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- H. Nilsson, T. Krupnik, J. Kargul and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1257–1262 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Phys. Chem. Chem. Phys., 2004, 6, 4882–4889 RSC.
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS.
- T. Noguchi, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1189–1195 CrossRef CAS PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2008, 47, 11024–11030 CrossRef CAS PubMed.
- H. Bao and R. L. Burnap, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6139–E6147 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, J. Phys. Chem. B, 2015, 119, 2677–2689 CrossRef CAS PubMed.
- T. Noguchi, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 35–45 CrossRef CAS PubMed.
- F. Rappaport, N. Ishida, M. Sugiura and A. Boussac, Energy Environ. Sci., 2011, 4, 2520–2524 CAS.
- L. Gerencsér and H. Dau, Biochemistry, 2010, 49, 10098–10106 CrossRef PubMed.
- H. Nilsson, F. Rappaport, A. Boussac and J. Messinger, Nat. Commun., 2014, 5, 4305 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional geometric and energetic data, sequence alignments, and Cartesian coordinates of all models. See DOI: 10.1039/c6sc02340a |
| ‡ Present address: European Synchrotron Radiation Facility, 71 avenue des Martyrs, 38000 Grenoble, France. |
| This journal is © The Royal Society of Chemistry 2016 |