
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral iodotriazoles as scaffolds for stable halogen-bonded assemblies in solution†
Leonardo
Maugeri
 a,
Julia
Asencio-Hernández
b,
Tomáš
Lébl
a,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Marc-André
Delsuc
b and
Douglas
Philp
*a
a,
Julia
Asencio-Hernández
b,
Tomáš
Lébl
a,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Marc-André
Delsuc
b and
Douglas
Philp
*a
aSchool of Chemistry and EaStCHEM, University of St Andrews, North Haugh St Andrews, Fife KY16 9ST, UK. E-mail: d.philp@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 467264
bInstitut de Génétique et de Biologie Moléculaire et Cellulaire, INSERM U596, CNRS UMR 7104, Université de Strasbourg, 1 rue Laurent Fries, 67404 Illkirch-Graffenstaden, France
First published on 23rd June 2016
Abstract
The halogen bond (XB) donor properties of neutral 1,4-diaryl-5-iodo-1,2,3-triazoles are explored using a combination of computational and experimental results and are shown to be competitive in halogen bonding efficiency with the classic pentafluoroiodobenzene XB donor. The SNAr reactivity of these donors permits the facile assembly of an iodotriazole functionalised with a 3-oxypyridine XB acceptor, thus generating a molecular scaffold capable of undergoing dimerisation through the formation of two halogen bonds. The formation of this halogen-bonded dimer is demonstrated by 1H and DOSY NMR experiments and a plausible structure generated using DFT calculations.
Introduction
The non-covalent interaction between an electron deficient halogen atom and an electron donor has found1 a clear definition in the term halogen bonding. Halogen bonds (XBs) have proven to be a powerful tool in a wide range of chemistries. The contribution of Metrangolo, Resnati and co-workers has revolutionised2 the conception of XB, making it a valuable tool in all the areas of chemistry where molecular recognition plays a central role.3 A significant effort has also been invested in unravelling the fundamental structural and energetic features of XBs. Computational4 and experimental5 studies made significant contribution to the understanding of the so-called “σ-hole”4b – a zone of low electron density displaying a positive electrostatic potential. Simplifying XBs to merely electrostatic interactions is, however, reductive: the factors contributing to the stability of a XB are a convolution of electrostatic,4a polarisation,6 charge transfer7 and dispersion8 forces and the role played by each of these factors is influenced significantly by the molecular components involved and the medium where the interaction takes place. It is generally recognised though that the ability of a halogen atom to participate in a XB increases with the electron withdrawing effect of the group to which the halogen is attached. The plethora of halogenated molecules constitutes a huge set of XB donors. Among these, a simple demarcation in XB ability can be made between cationic and neutral XB donors. Cationic XB donors are generally halogenated (mainly Br and I) five or six membered nitrogen heterocycles, where one of the nitrogen atoms is quaternarised with an alkyl group. The interaction of such XB donors with XB acceptors is in fact defined9 as ‘charge-assisted XB’ and these interactions are significantly stronger than those between two neutral XB partners. Beer and co-workers, for example, have designed and synthesised10 a variety of macrocycles, catenanes and rotaxanes incorporating halotriazolium and haloimidazolium units able to perform anion sensing in organic and aqueous media via charge-assisted XB. Pyridinium11 and imidazolium12,13 XB donors have been used extensively by the group of Huber to catalyse C–Br bond cleavage reactions in organic media.On the other hand, when a polarisable halogen atom is bonded to a neutral organic backbone, its ability to act as a XB donor depends greatly on the electronic properties of the organic residue. It is, therefore, not surprising that the most common neutral XB donors are perfluorohalocarbons (PFHCs). Whether aromatic or aliphatic, these compounds are now considered ‘iconic’ XB donors. In 2002, Metrangolo, Resnati et al. reported14 the first semiquantitative evaluation of XB in solution using 1,2-dibromo and 1,2-diiodo tetrafluoroethanes as the XB donors. Taylor et al. have successfully measured15 the strength of XBs formed by a set of variously substituted iodoperfluorobenzenes XC6F4I in solution using 19F NMR spectroscopy. The association strength could be correlated to the σ Hammett parameter for the X substituent. More recently multivalent PFHC-based XB donors have been successfully exploited in molecular recognition16 and catalysis.17
The quest for new and less conventional organic XB donors has been tackled18 by several research groups and haloalkenes,19 haloalkynes,20N-haloimides21 and halogenated metallic complexes22 have been found to be strong XB donors in the solid state. Iodoalkynes have been shown23 to behave as XB donors in solution as well as in the solid state.
In this paper, we describe the development of an underexplored24 class of iodinated scaffolds, 1,4-diaryl-5-iodo-1,2,3-triazoles, as a progenitor for assemblies supported by XBs. The XB properties have been examined computationally, in the solid state and in solution. During the course of these studies, a serendipitous discovery led us to design a system containing both a diaryliodotriazole and a Lewis base, thus generating a molecule capable of undergoing dimerisation through self-complementary XBs. A detailed NMR study of the self-assembled dimer in solution has allowed us to evaluate the efficiency of chelate cooperativity on the dimerisation process.
Results and discussion
Iodotriazole 1 can be prepared readily and in high yield through the Cu-catalysed reaction of pentafluorophenyl azide and iodophenylacetylene. The solid state structure (Fig. 1) of compound 1, determined by single crystal X-ray diffraction, exhibits antiparallel tapes in which the molecules are connected by a series of short, nitrogen to iodine contacts (r(N⋯I) = 2.973 Å) indicative of the presence of a halogen bond between the iodotriazole rings. | ||
| Fig. 1 Solid state structure of diaryl-5-iodo-1,2,3-triazole 1 determined from single crystal X-ray diffraction data. Potential halogen bonds are marked in red (r(N⋯I) = 2.973 Å; ∠C–I⋯N = 168.9°). Atom colouring: C atoms = grey, N atoms = blue, F atoms = light green, I atoms = purple. Hydrogen atoms are omitted for clarity. | ||
The presence of these close contacts in the solid state structure of 1 suggested that appropriately designed diaryl-5-iodo-1,2,3-triazoles could function as viable XB donors.
In order to gain some insight into the potential interactions between this class of compounds and XB acceptors, we performed a series of calculations that compared the interaction of iodotriazole 1 and pentafluoroiodobenzene 2 with a series of pyridine-based XB acceptors (Fig. 2). These calculations were performed at the TPSSh/def2-TZVP level of theory.
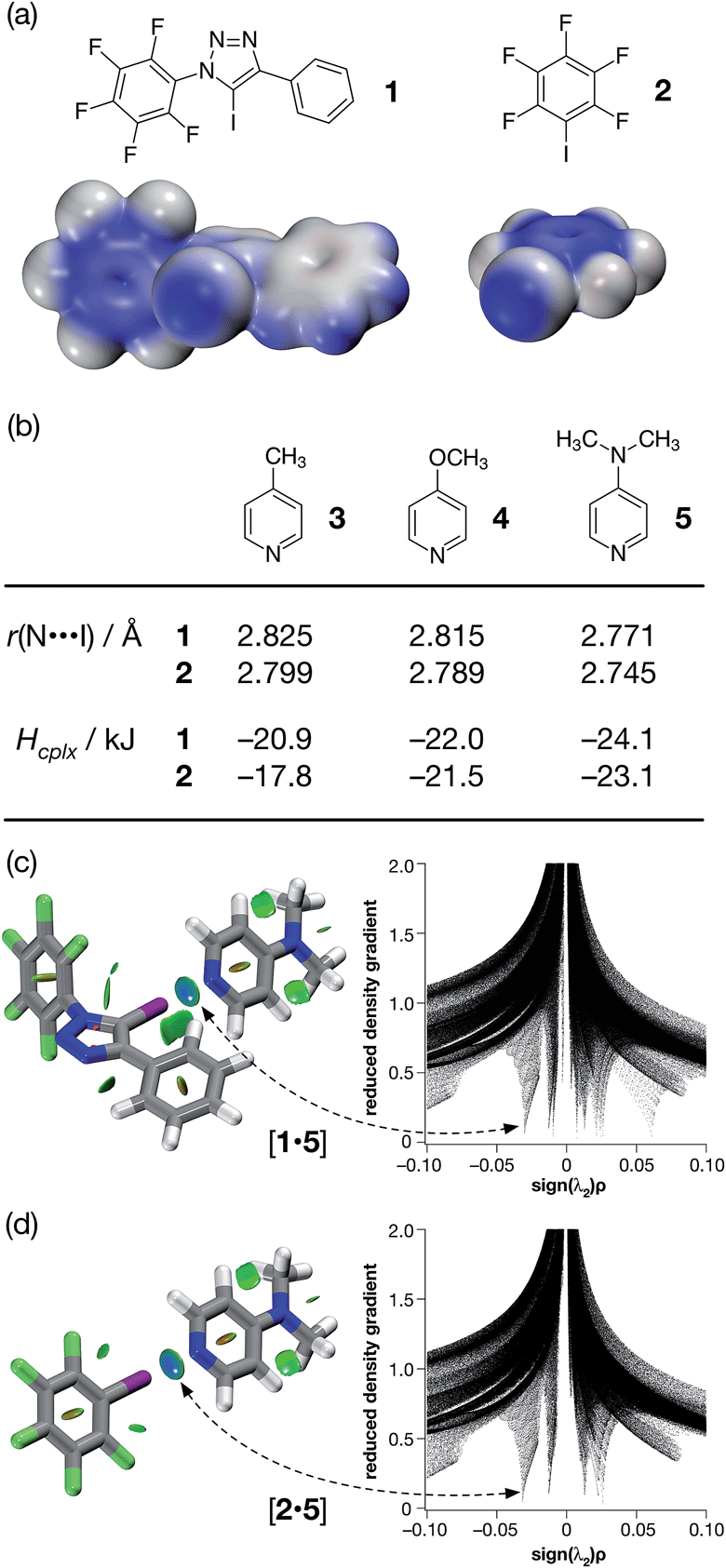 | ||
| Fig. 2 (a) Calculated electrostatic potential surfaces for compounds 1 and 2 (TPSSh/def2-TZVP). Colour scale: positive (blue = +0.08) → neutral (white) → negative (red = −0.08). (b) Calculated pyridine ring N⋯I distances and interaction energies (Hcplx, TPSSh/def2-TZVP enthalpies of complexation at 298 K, see ESI† for details) for a series of complexes between halogen bond donors 1 and 2 and pyridine-based acceptors. Visualisation of the interaction between 4-(dimethyl amino)pyridine 5 and (c) compound 1 and (d) compound 2. Left: intermolecular interaction isosurfaces generated by NCIPLOT26 for s = 0.5 and −0.05 < sign(λ2)ρ < 0.05 (colour scale: attractive (blue) → repulsive (red)). Right: plots of sign(λ2)ρ vs. reduced density gradient highlighting the favourable interaction corresponding to the halogen bond at sign(λ2)ρ ∼ −0.035. Atom colouring: C atoms = grey, N atoms = blue, O atoms = red, F atoms = light green, I atoms = purple. | ||
Examination of the electrostatic potential of 1 (Fig. 2a) shows that this compound possesses an area of significant positive potential associated with the iodine atom. This area of positive potential is similar in magnitude to that calculated for pentafluoroiodobenzene 2 – the iconic25 XB donor. These results suggest that iodotriazole 1 should interact strongly with suitable electron donors. Indeed, the calculated geometries for the complexes formed between the three pyridines shown in Fig. 2b and compound 1 all possess N⋯I distances much shorter than the sum of the van der Waals radii and significantly negative enthalpies of complexation at 298 K (Fig. 2b). These structural and energetic parameters are all similar to those for the complexes formed between the well-known halogen bond donor 2 and the same set of pyridines. Natural bond order (NBO) analyses (see ESI† for details) demonstrated that, in all six complexes, there are significant interactions between the nitrogen lone pair and the σ* orbital associated with the C–I bond present in the donor. Intermolecular interaction isosurfaces, generated26 through an analysis of the reduced gradient of the electron density, have been used27 extensively to identify non-covalent interactions that stabilise intermolecular complexes. Analyses of the complexes [1·5] (Fig. 2c) and [2·5] (Fig. 2d) using this method (see ESI† for details) reveal significant low density, low gradient regions that are consistent with the presence of a halogen bond between the interacting partners in both complexes. The similarities between the results for [1·5] and [2·5] suggest that the neutral iodotriazole might be used interchangeably with the perfluorinated iodobenzene as a halogen bond donor.
Encouraged by these computational results, we examined the interaction of iodotriazole 1 and the pyridine-based XB acceptors 3 and 5 in solution. Initially, we examined the interaction of 1 and 4-methylpyridine 3 in d8-toluene. Titration of increasing amounts of 3 into a solution of 1 in d8-toluene did not result in any significant chemical shift changes in the 19F NMR spectrum of 1. However, when the titration was performed again, this time titrating increasing amounts of 1 into a solution of 3 in d8-toluene at 293 K, a series of 1H–15N HMBC experiments revealed significant upfield 15N chemical shift changes for the pyridine ring nitrogen atom. The 15N chemical shift data were fitted28 to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model for the complex [1·3] affording (see ESI† for details) a stability constant for this complex in d8-toluene at room temperature of 1.67 ± 0.55 M−1. For comparison purposes, we repeated this analysis, this time using pentafluoroiodobenzene 2 as the halogen bond donor. The 15N chemical shift data from this experiment were once again fitted28 to a 1:1 binding model for the complex [2·3] (see ESI† for details) affording a stability constant for this complex in d8-toluene at 293 K of 2.67 ± 0.69 M−1. These results confirm experimentally the outcome of our calculations – iodotriazole 1 and pentafluoroiodobenzene 2 have similar halogen bond donor abilities towards pyridine acceptors.
:1 binding model for the complex [1·3] affording (see ESI† for details) a stability constant for this complex in d8-toluene at room temperature of 1.67 ± 0.55 M−1. For comparison purposes, we repeated this analysis, this time using pentafluoroiodobenzene 2 as the halogen bond donor. The 15N chemical shift data from this experiment were once again fitted28 to a 1:1 binding model for the complex [2·3] (see ESI† for details) affording a stability constant for this complex in d8-toluene at 293 K of 2.67 ± 0.69 M−1. These results confirm experimentally the outcome of our calculations – iodotriazole 1 and pentafluoroiodobenzene 2 have similar halogen bond donor abilities towards pyridine acceptors.
From the set of pyridine XB acceptors studied computationally, DMAP 5 was predicted to form the most stable complexes. When we performed an experiment where increasing amounts of 5 were titrated into a solution of 1 in d8-toluene, significant chemical shift changes in the 376.4 MHz 19F NMR spectrum of 1 were observed (Fig. 3a). In particular, the resonance for the fluorine atom para to the iodotriazole ring exhibited a significant upfield shift (Fig. 3a, dotted line) from δ −148.7 to δ −149.6, consistent with the formation of the [1·5] complex. However, this chemical shift data could not be fitted to a 1:1 binding model for the [1·5] complex and close examination of the NMR samples revealed that a precipitate had formed in all samples. Clearly, the formation of the [1·5] complex was accompanied by a concomitant chemical transformation.
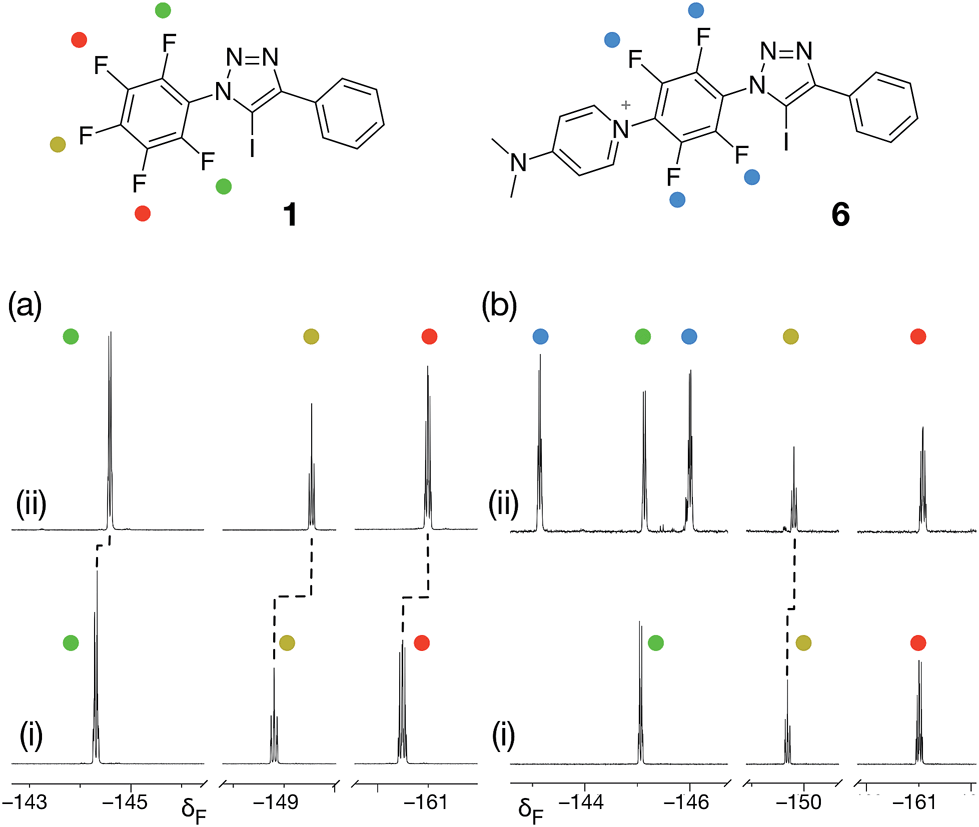 | ||
| Fig. 3 (a) Partial 376.4 MHz 19F NMR spectra of (i) a solution of 1 in d8-toluene and (ii) the same solution after the addition of 10 equivalents of DMAP 5. (b) Partial 376.4 MHz 19F NMR spectra of (i) a solution of 1 in CD3CN and (ii) the same solution after the addition of 10 equivalents of DMAP 5. In both case, dashed lines indicate resonances that show chemical shift changes between (i) and (ii). | ||
Intrigued by the presence of this precipitate, we repeated this titration experiment using CD3CN as the solvent. In this case, no precipitate was formed. However, the 376.4 MHz 19F NMR spectrum revealed a new set of resonances at δ −143.1 and δ −145.9 (Fig. 3b, blue circles) that were consistent with the presence of a 1,4-disubstituted tetrafluorobenzene ring. We attributed these resonances to the presence of the insoluble pyridinium salt 6. This product arises from the nucleophilic aromatic substitution29,30 (SNAr) of the fluorine para to the triazole ring in 1 by DMAP 5 and analysis of the precipitate formed in d8-toluene confirmed its identity as the pyridinium salt 6.
Although DMAP 5 is potentially the best halogen bond acceptor, it is clearly much too reactive towards the perfluorinated aromatic ring present in 1. We therefore turned to alkoxypyridines as halogen bond acceptors. We performed a series of calculations examining the interaction of iodotriazole 1 with 3-methoxypyridine at the TPSSh/def2-TZVP level of theory. These calculations reveal (Fig. 4a, left) a complex that is very similar in structure (r(N⋯I) = 2.836 Å, ∠C–I⋯N = 179.5°) and with a similar calculated enthalpy of complexation at 298 K (−23.6 kJ mol−1) to that formed between 1 and 4-methylpyridine 3. Titration of 7, a more soluble variant of 1, into a solution of 3-pentyloxypyridine 8 in d8-toluene at 298 K resulted in small, but significant, chemical shift changes in the 700.1 MHz 1H NMR spectrum for the resonances arising from the pyridine ring protons of 8. A series of 1H–15N HMBC experiments (Fig. 4b), performed on the same sample set, also reveal significant 15N chemical shift changes for the pyridine ring nitrogen atom. Both the 1H and the 15N chemical shift data were fitted28 to a 1:1 binding model for the complex [7·8] affording (see ESI† for details) a stability constant for this complex in d8-toluene at 293 K of 1.44 ± 0.24 M−1.
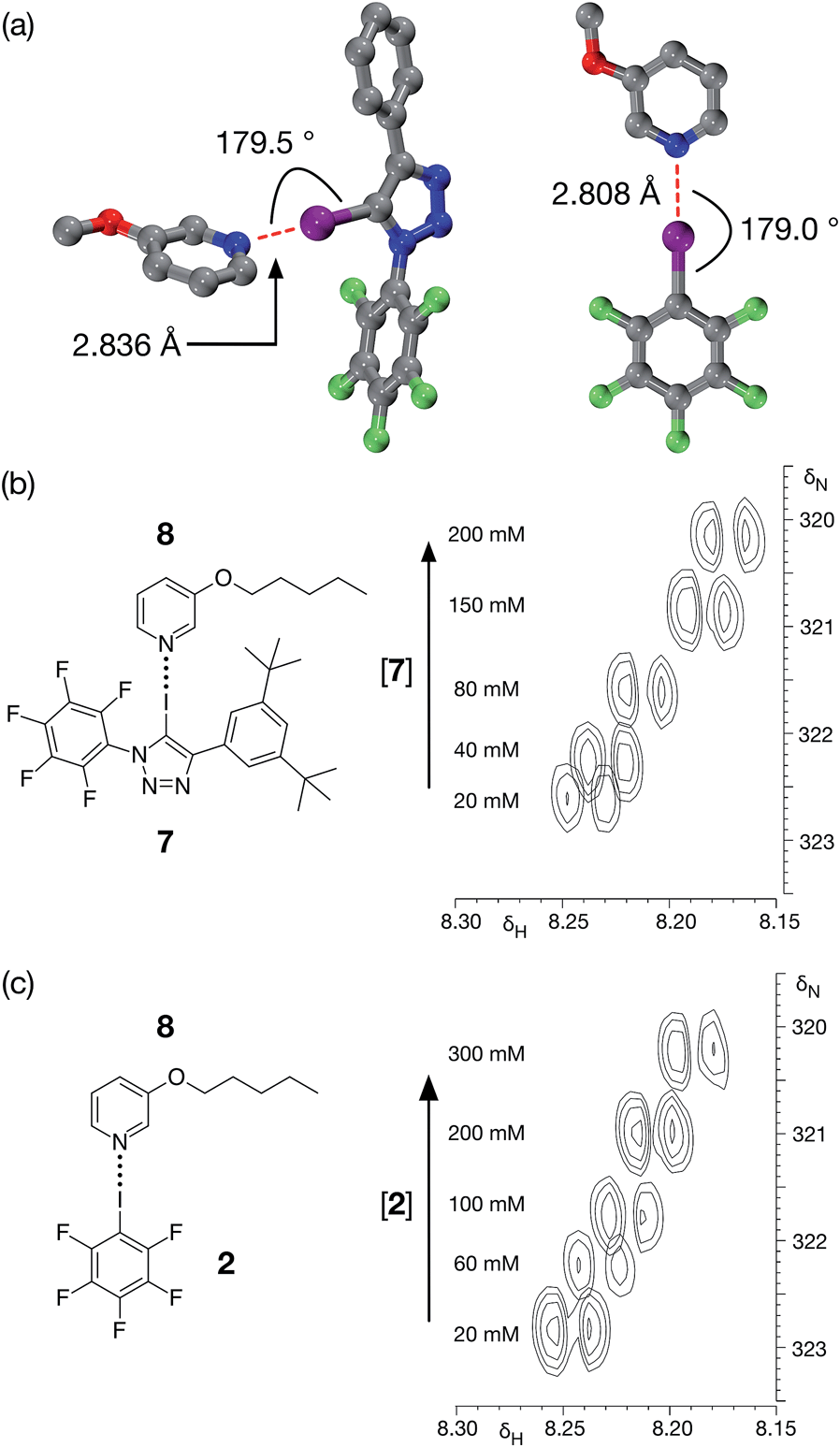 | ||
| Fig. 4 (a) Calculated structure (TPSSh/def2-TZVP) for the complexes formed between 1 and 3-methoxypyridine (left) and between 2 and 3-methoxypyridine (right). Halogen bonds are marked in red. For the complex of 3-methoxypyridine with 1: (r(N⋯I) = 2.836 Å; ∠C–I⋯N = 180.0°). For the complex of 3-methoxypyridine with 2: (r(N⋯I) = 2.808 Å; ∠C–I⋯N = 179.0°). Carbon atoms are coloured grey, nitrogen atoms are coloured blue, fluorine atoms are coloured green and iodine atoms are coloured purple. Hydrogen atoms are not shown. (b) Partial 700.1 MHz 1H–15N HMBC correlations of a 20 mM solution of pyridine 8 in d8-toluene with increasing amounts (0 to 200 mM) of iodotriazole 7 added to the solution. (c) Partial 700.1 MHz 1H–15N HMBC correlations of a 20 mM solution of pyridine 8 in d8-toluene with increasing amounts (0 to 200 mM) of pentafluoroiodobenzene 2 added to the solution. | ||
For comparison purposes, we also evaluated the stability of the complex formed between pentafluoroiodobenzene 2 and 3-methoxypyridine at the same level of theory. These calculations reveal (Fig. 4a, right) a complex with a halogen bond that is similar in geometry (r(N⋯I) = 2.808 Å, ∠C–I⋯N = 179.0°) and a similar calculated enthalpy of complexation at 298 K (−19.6 kJ mol−1) to the complex formed between 3-methoxypyridine and iodotriazole 1. Analysis of a titration of 2 into a solution of 3-pentyloxypyridine 8 in d8-toluene using a series of 700.1 MHz 1H–15N HMBC experiments (Fig. 4c) reveal significant 1H and 15N chemical shift changes in the pyridine ring. These data were fitted28 to a 1:1 binding model for the complex [2·8] affording a stability constant for this complex in d8-toluene at room temperature of 1.40 ± 0.17 M−1 (see ESI† for details).
It is clear from these data that the interactions between both 2 and 8 and 7 and 8 are is not particularly strong in d8-toluene at 298 K. However, we reasoned that the reactivity observed between 1 and nucleophiles, such as an amino- or hydroxypyridine, should allow us to construct rapidly a molecule that possessed both a pyridine ring and an iodotriazole. Such a molecule would be self-complementary and could, potentially, benefit from cooperative binding31 between the self-complementary recognition sites, thus forming a halogen-bonded dimer.
Accordingly, we designed compound 9a (Fig. 5a), which was prepared in two steps, in high yield, from 5-(2-iodoethynyl)-1,3-bis(tert-butyl)benzene and 1-azido-2,3,4,5,6-pentafluoro benzene (see ESI† for details). We envisaged that 9a might be able to form a halogen-bonded dimer in which the pyridine ring of one molecule interacts with the iodotriazole of a second molecule. This expectation was supported by calculations (Fig. 5b) at the TPSSh/def2-TZVP level of theory on compound 9b – identical to 9a save for the replacement of the tert-butyl groups by hydrogen atoms in the interests of computational efficiency.
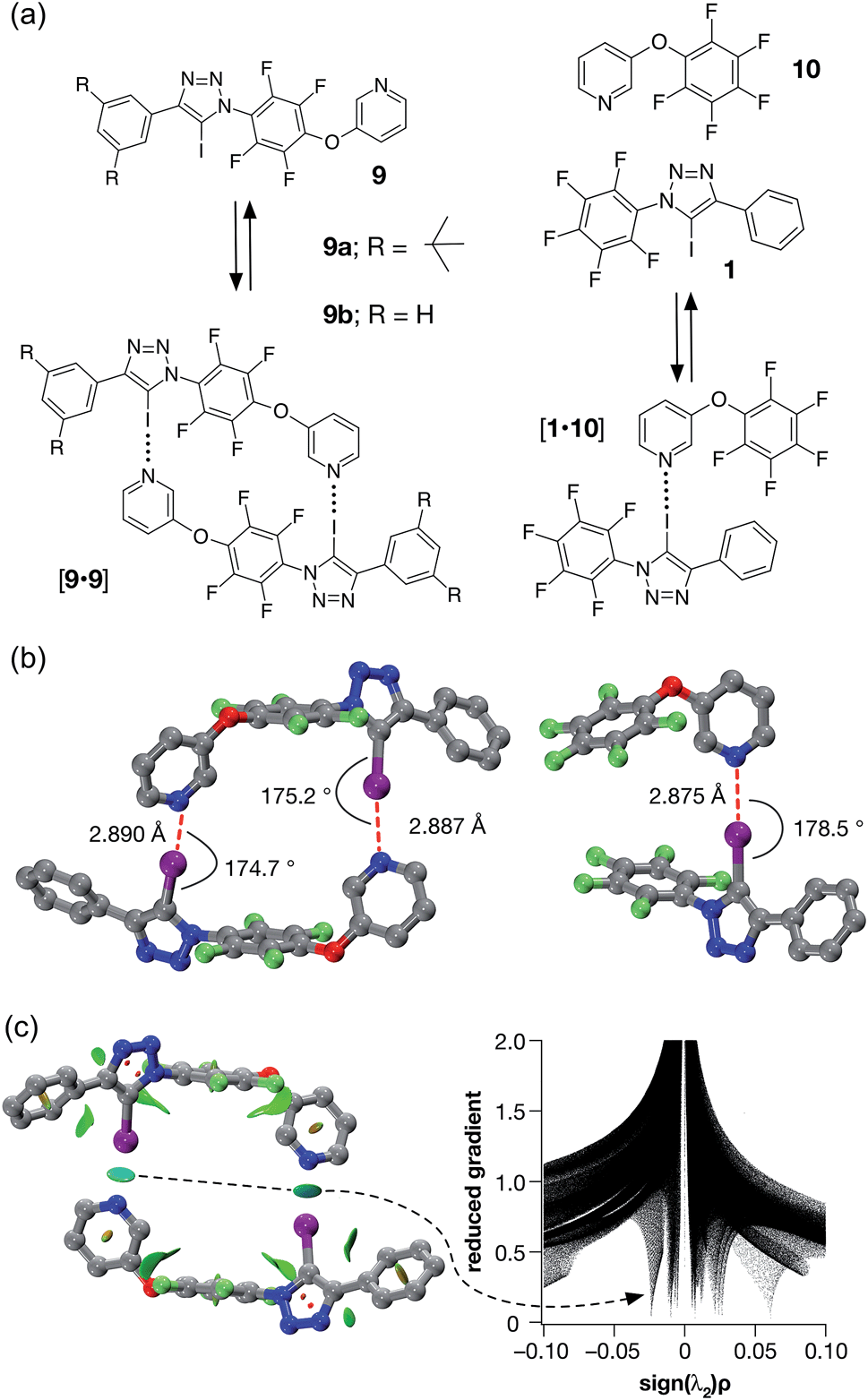 | ||
| Fig. 5 (a) Iodotriazole 9b provides a computational model for 9a, which was prepared synthetically. Both iodotriazoles can dimerise through two halogen bonds to form homodimers. Complex [1·10] provides a computational model for the halogen bonds found in the dimers. (b) Comparison of the calculated structures (TPSSh/def2-TZVP) of the [9b·9b] dimer and the complex [1·10]. (c) Left: intermolecular interaction isosurfaces for the [9b·9b] dimer, generated by NCIPLOT,26 for s = 0.5 and −0.05 < sign(λ2)ρ < 0.05 (colour scale: attractive (blue) → repulsive (red)). Right: plot of sign(λ2)ρ vs. reduced gradient highlighting the favourable interaction corresponding to the halogen bonds at sign(λ2)ρ ∼ −0.035. Atom colouring in molecular structures: C atoms = grey, N atoms = blue, O atoms = red, F atoms = light green, I atoms = purple. H atoms are omitted for clarity. | ||
A doubly halogen-bonded homodimeric structure was located that possessed approximate C2 symmetry and in which the tetrafluoroaromatic rings in the two molecules of 9b are rotated by around 120° with respect to each other. The two halogen bonds showed almost identical lengths and geometries (r(N⋯I) = 2.887 Å; ∠C–I⋯N = 175.2° and r(N⋯I) = 2.890 Å; ∠C–I⋯N = 174.7°). The calculated enthalpy of dimerisation at 298 K for [9b·9b] is −30.6 kJ at this level of theory. Comparison of [9b·9b] with the corresponding monodentate interaction – as represented by the calculated structure of the complex formed between 1 and pyridine 10 at the same level of theory (Fig. 5b, left) – is instructive. The calculated geometry of the halogen bond in [1·10] (r(N⋯I) = 2.875 Å; ∠C–I⋯N = 178.5°) reveals an interaction that is marginally shorter and more linear than those in [9b·9b]. The geometry of the [1·10] complex, together with the calculated enthalpy of dimerisation at 298 K (−17.9 kJ) – more than half the total for [9b·9b], suggested that a slight structural mismatch may be present in the [9b·9b] complex, preventing it from taking full advantage of both halogen bonds.
Single crystals of 9a, suitable for analysis by X-ray diffraction, were grown by slow evaporation of a solution of 9a in toluene. The solid-state structure of 9a (Fig. 6) reveals antiparallel chains of molecules connected by halogen bonds between the pyridine of one molecule and the iodotriazole of the next. The geometry of these close contacts between the pyridine ring nitrogen atoms and the iodotriazole rings are suggestive of strong halogen bonds – r(N⋯I) = 2.767 Å; ∠C–I⋯N = 176.5°.
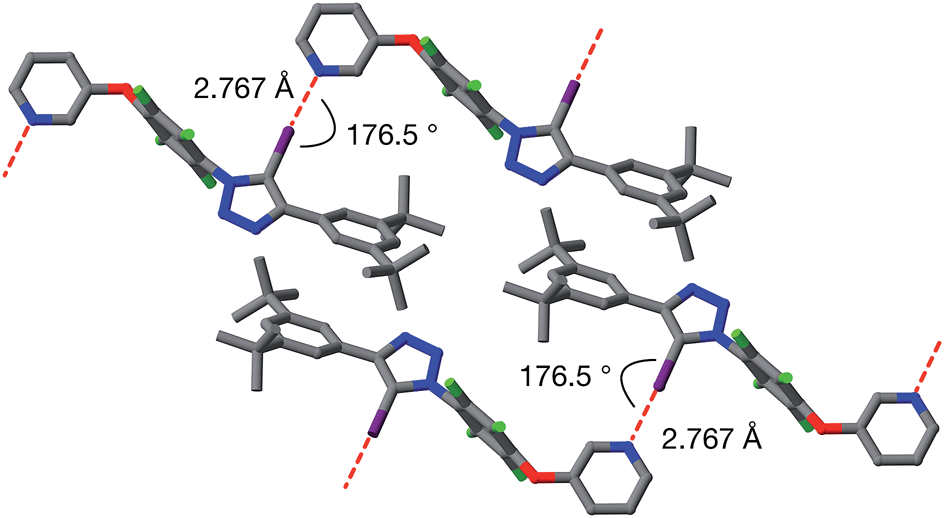 | ||
| Fig. 6 Solid state structure of 9a determined from single crystal X-ray diffraction data. Potential halogen bonds are marked in red (r(N⋯I) = 2.767 Å; ∠C–I⋯N = 176.5°). Atom colouring: C atoms = grey, N atoms = blue, O atoms = red, F atoms = light green, I atoms = purple. Hydrogen atoms are omitted for clarity. | ||
Despite the absence of homodimers in the solid state structure of 9a, we wished to characterise the stability of the [9a·9a] complex in solution. Accordingly, we performed a dilution experiment in order to assess the stability of the [9a·9a] dimer in C6D6 solution. From a starting concentration of 200 mM, progressive dilution of a solution of 9a resulted in chemical shift changes in the 1H NMR spectrum for the resonances associated with the pyridine ring. These chemical shift changes were fitted28 (see ESI† for details) to a dimerisation binding model, affording a stability constant for the [9a·9a] dimer in C6D6 of 3.4 ± 0.7 M−1. This value was disappointingly low and indicated that the [9a·9a] dimer benefits from little, if any, cooperativity31 arising from the connection of the halogen bond donor and acceptor within the same molecule.
In order to characterise the association of 9a in C6D6 solution further, we turned to DOSY NMR experiments to assess the nature of the assembly formed. A series of DOSY experiments were performed (Fig. 7a) on sample 9a in C6D6, at concentrations ranging from 200 mM down to 1.0 mM. The diffusion coefficients of 9a and that of the solvent were measured at each concentration. The observed variations of the diffusion coefficient of the solvent were interpreted as a variation in the viscosity of the sample.
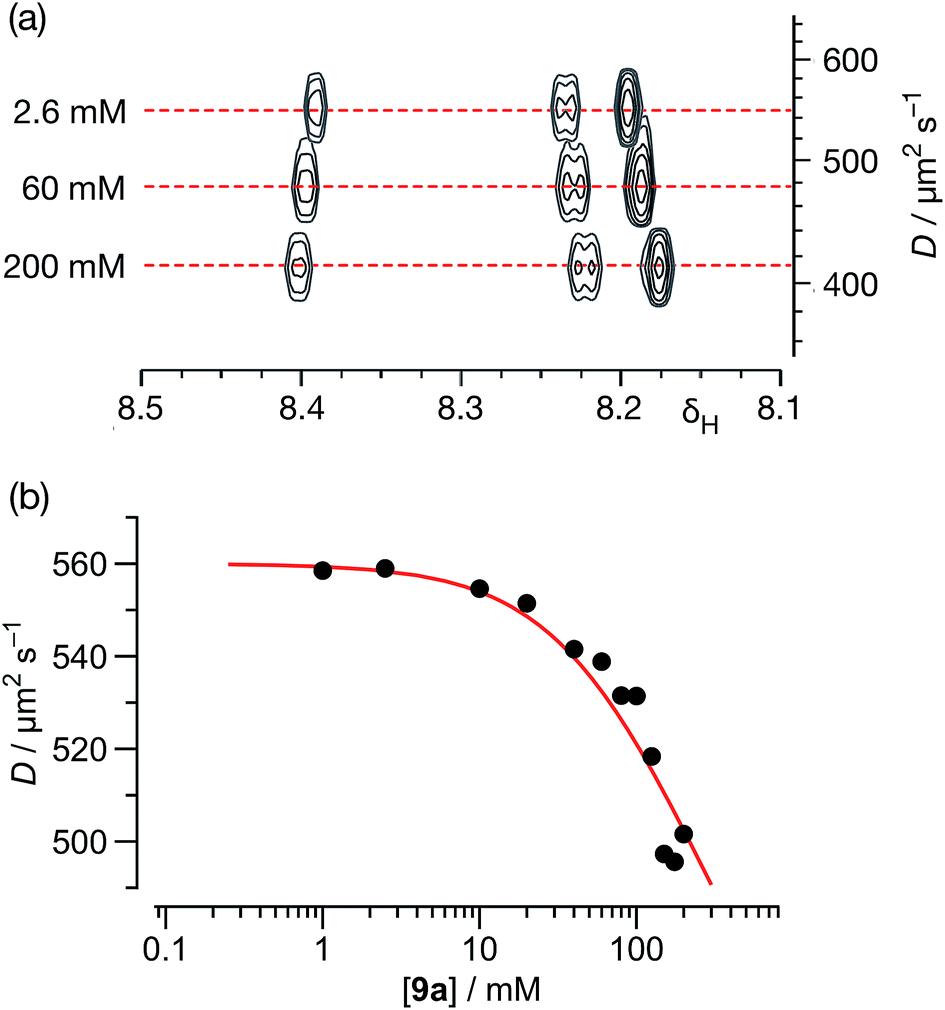 | ||
| Fig. 7 (a) A series of 500.1 MHz 1H DOSY NMR experiments, performed on solutions of 9a in C6D6 at 298 K, illustrating the decrease in the measured diffusion coefficient (D) for 9a with increasing concentration. (b) Variation of diffusion coefficients (D), corrected for viscosity, with respect to the concentration of 9a in C6D6 solution at 298 K. The red line shows the best fit of these data to a monomer ⇆ dimer equilibrium model with a Ka for the [9a·9a] homodimer of 2.1 ± 0.4 M−1 (see ESI† for details). | ||
Using this data, the diffusion coefficients of solute 9a were corrected for viscosity changes using the diffusion coefficient for the solvent (C6D6) measured on the same samples. The variation of these corrected diffusion coefficients for 9a across the concentration range studied were then fitted (Fig. 7b, see ESI† for further details) to a simple dimerisation model, using the model32 of Morris and co-workers for estimating the diffusion of the dimer. The results of this fitting procedure confirm the presence of dimer [9a·9a] in solution, and the estimated value of the stability constant for [9a·9a] at 298 K – 2.1 ± 0.4 M−1 – was in good agreement with that obtained using the more conventional NMR titration method.
Conclusions
We have demonstrated that 1,4-diaryl-5-iodo-1,2,3-triazoles possess XB properties that make them reliable, neutral XB donors in organic solvents, able to interact with pyridine XB acceptors with efficiencies similar to those displayed by the iconic XB donor iodopentafluorobenzene. The synthetic versatility of these molecular scaffolds allowed the facile construction a self-complementary molecular module, incorporating both an XB donor and an XB acceptor, that was capable of forming a homodimer through the formation of two neutral XB interactions. The stability of this dimeric assembly was evaluated by means of DFT calculations and in C6D6 solutions using 1H NMR and DOSY experiments. The results of these investigations showed that, despite the increased stability of the dimeric assembly, full exploitation of the chelate effect could not be achieved as a result of a partial structural mismatch between the two monomeric units. Nevertheless, the application of chelate cooperativity represents a valid strategy to reinforce XB interactions between two neutral partners in solution and further studies directed towards the optimisation of the monomer design are currently underway in our laboratory.Acknowledgements
We thank Marie Curie ITN ReAd and the University of St Andrews for financial support.Notes and references
- G. G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- (a) A. Lunghi, P. Cardillo, M. T. Messina, P. Metrangolo, W. Panzeri and G. Resnati, J. Fluorine Chem., 1998, 91, 191–194 CrossRef CAS; (b) A. Farina, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati and G. Vecchio, Angew. Chem., Int. Ed., 1999, 38, 2433–2436 CrossRef CAS; (c) P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed; (d) A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed.
- (a) R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- (a) T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44, 57–64 CrossRef; (b) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed; (c) M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed.
- (a) G. R. Desiraju and R. L. Harlow, J. Am. Chem. Soc., 1989, 111, 6757–6764 CrossRef CAS; (b) J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS; (c) A. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- M. Hennemann, J. S. Murray, K. E. Riley, P. Politzer and T. Clark, J. Mol. Model., 2012, 18, 2461–2469 CrossRef CAS PubMed.
- M. Palusiak, J. Mol. Struct., 2010, 945, 89–92 CrossRef CAS.
- K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2008, 4, 232–242 CrossRef CAS PubMed.
- (a) S. M. Walter, F. Kniep, L. Rout, F. P. Schmidtchen, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2012, 134, 8507–8512 CrossRef CAS PubMed; (b) F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 51, 11533–11541 CrossRef PubMed; (c) J. Lieffrig, O. Jeannin, A. Frąckowiak, I. Olejniczak, R. Świetlik, S. Dahaoui, E. Aubert, E. Espinosa, P. Auban-Senzier and M. Fourmigué, Chem.–Eur. J., 2013, 19, 14804–14813 CrossRef CAS PubMed.
- (a) A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed; (b) L. C. Gilday, T. Lang, A. Caballero, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2013, 52, 4356–4360 CrossRef CAS PubMed; (c) J. M. Mercurio, R. C. Knighton, J. Cookson and P. D. Beer, Chem.–Eur. J., 2014, 20, 11740–11749 CrossRef CAS PubMed; (d) M. G. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- S. M. Walter, S. H. Jungbauer, F. Kniep, S. Schindler, E. Herdtweck and S. M. Huber, J. Fluorine Chem., 2013, 150, 14–20 CrossRef CAS.
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- M. T. Messina, P. Metrangolo, W. Panzeri, E. Ragg and G. Resnati, Tetrahedron Lett., 1998, 39, 9069–9072 CrossRef CAS.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
- (a) S. H. Jungbauer, D. Bul, F. Kniep, C. W. Lehmann, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2014, 136, 16740–16743 CrossRef CAS PubMed; (b) O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef CAS PubMed.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed.
- R. W. Troff, T. Mäkelä, F. Topić, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS.
- C. W. Padgett, R. D. Walsh, G. W. Drake, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2005, 5, 745–753 Search PubMed.
- (a) K. Gao and N. S. Goroff, J. Am. Chem. Soc., 2000, 122, 9320–9321 CrossRef CAS; (b) A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030–1034 CrossRef CAS PubMed; (c) H. Jin, C. N. Young, G. P. Halada, B. L. Philipps and N. S. Goroff, Angew. Chem., Int. Ed., 2015, 54, 14690–14695 CrossRef CAS PubMed.
- (a) K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC; (b) R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- (a) C. Laurence, Can. J. Chem., 1983, 61, 135–138 CrossRef CAS; (b) P. D. Rege, O. L. Malkina and N. S. Goroff, J. Am. Chem. Soc., 2002, 124, 370–371 CrossRef CAS PubMed; (c) O. Dumele, D. Wu, N. Trapp, N. S. Goroff and F. Diederich, Org. Lett., 2014, 16, 4722–4725 CrossRef CAS PubMed.
- (a) L. C. Gilday, N. G. White and P. D. Beer, Dalton Trans., 2013, 42, 15766–15773 RSC; (b) T. K. Mole, W. E. Arter, I. Marques, V. Félix and P. D. Beer, J. Organomet. Chem., 2015, 792, 206–210 CrossRef CAS; (c) R. Tepper, B. Schulze, H. Görls, P. Bellstedt, M. Jäger and U. S. Schubert, Org. Lett., 2015, 17, 5740–5743 CrossRef CAS PubMed.
- (a) L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CrossRef CAS; (b) S. Tsuzuki, A. Wakisaka, T. Ono and T. Sonoda, Chem.–Eur. J., 2012, 18, 951–960 CrossRef CAS PubMed; (c) S. Tsuzuki, T. Uchimaru, A. Wakisaka, T. Ono and T. Sonoda, Phys. Chem. Chem. Phys., 2013, 15, 6088–6096 RSC.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- (a) S. Kozuch and J. M. L. Martin, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS PubMed; (b) B. Pinter, N. Nagels, W. A. Herrebout and F. De Proft, Chem.–Eur. J., 2013, 19, 519–530 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- J. M. Birchall, R. N. Haszeldine and M. E. Jones, J. Chem. Soc., 1971, 1343–1348 Search PubMed.
- J. F. W. Keana and S. Xiong Cai, J. Org. Chem., 1990, 55, 3640–3647 CrossRef CAS.
- (a) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS; (b) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed; (c) G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762–1768 CrossRef CAS PubMed.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational methods, synthesis and characterisation data for all compounds are provided. CCDC 1442418–1442421. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01974a |
| This journal is © The Royal Society of Chemistry 2016 |